

Abstract
In mouse cerebellar granule neurons (CGN) the marine neurotoxin domoic acid (DomA) induces neuronal cell death, either by apoptosis or by necrosis, depending on its concentration, with apoptotic damage predominating in response to low concentrations (100 nM). DomA-induced apoptosis is due to selective activation of AMPA/kainate receptors, and is mediated by DomA-induced oxidative stress, leading to mitochondrial dysfunction and activation of caspase-3. The p38 MAP kinase and the c-Jun NH2-terminal protein kinase (JNK) have been shown to be preferentially activated by oxidative stress. Here we report that DomA increases p38 MAP kinase and JNK phosphorylation, and that this effect is more pronounced in CGNs from Gclm (−/−) mice, which lack the modifier subunit of glutamate-cysteine ligase, have very low glutathione (GSH) levels, and are more sensitive to DomA-induced apoptosis than CGNs from wild-type mice. The increased phosphorylation of JNK and p38 kinase was paralleled by a decreased phosphorylation of Erk 1/2. The AMPA/kainate receptor antagonist NBQX, but not the NMDA receptor antagonist MK-801, prevents DomA-induced activation of p38 and JNK kinases. Several antioxidants (GSH ethyl ester, catalase, phenylbutylnitrone) also prevent DomA-induced phosphorylation of JNK and p38 MAP kinases. Inhibitors of p38 (SB203580) and of JNK (SP600125) antagonize DomA-induced apoptosis. These results indicate the importance of oxidative stress-activated JNK and p38 MAP kinase pathways in DomA-induced apoptosis in CGNs.
Keywords: domoic acid, neurotoxicity, apoptosis, oxidative stress, p38 kinase, JNK
Mitogen-activated protein kinases (MAPK) are a family of kinases that regulate diverse cellular functions such as embryogenesis, proliferation, differentiation and apoptotic death (Raman et al. 2007). The best known MAPK are the extracellular signal-regulated kinases 1 and 2 (Erk 1/2), the Jun N-terminal kinases [JNK (1–3)], also known as stress-activated protein kinases (SAPK), and the p38 kinases (α, β, γ, δ) (Raman et al. 2007).
Xia et al. (1995) found that Erk 1/2, JNK and p38, exert opposing effects on apoptosis. Upon nerve growth factor withdrawal, PC12 cells undergo apoptotic cell death; this is preceded by a decrease in phosphorylated Erk 1/2, and an increase in the activation (by phosphorylation) of JNK and p38. Convincing evidence has since emerged that sustained activation of JNK and/or p38 has a pro-apoptotic role (Yang et al. 1997; Kawasaki et al. 1997; Luo et al. 1998). JNK and p38 are preferentially activated by cell stress-inducing signals, such as oxidative stress, environmental stress, and toxic chemical insults (Davis, 2000; Kyriakis and Avruch, 2001; Raman et al. 2007). Additionally, a number of neurotoxic chemicals have been shown to cause apoptosis in various neuronal cell preparations that is mediated by activation of JNK and/or p38 (Namnung and Xia, 2000; Caughlan et al. 2004; Newhouse et al. 2004; Klintworth et al. 2007).
We have recently shown that the marine neurotoxin domoic acid (DomA) causes apoptotic cell death in mouse cerebellar granule neurons (CGNs) (Giordano et al. 2007). DomA is a potent neurotoxin, that was identified as the toxic agent responsible for an outbreak of poisoning due to consumption of contaminated shellfish (Perl et al. 1990). Neuronal loss, particularly in the hippocampus and the amygdala, was observed in the four individuals who died in this incident (Teitelbaum et al. 1990), and similar brain lesions have been seen in primates, rats and mice following administration of DomA (Tryphonas et al. 1990; Strain and Tasker, 1991; Scallet et al. 1993; Sobotka et al. 1996). The pattern of brain damage observed in humans and animals following exposure to DomA resembles that observed after administration of kainic acid (Teitelbaum, 1990). DomA is indeed a structural analog of kainic acid, and exerts is toxicity by activating the AMPA/kainate subtype of glutamate receptors (Hampson and Manalo, 1998). In vitro studies have shown that in mouse CGNs DomA causes necrotic or apoptotic cell death, depending on its concentration (Giordano et al. 2006, 2007). Necrosis is observed at higher concentrations of DomA (10 uM) and involves activation of both AMPA/kainate and of NMDA receptors, the latter activated by DomA-induced glutamate release (Berman and Murray, 1997; Giordano et al. 2006). In contrast, apoptotic damage predominates at lower concentrations of DomA (100 nM); DomA –induced apoptosis is solely due to activation of AMPA/kainate receptors (Giordano et al. 2007).
Both necrosis and apoptosis have been shown to be mediated by oxidative stress, as evidenced by the following observations (Giordano et al. 2006, 2007): 1) DomA causes a concentration-dependent increase of reactive oxygen species (ROS); lipid peroxidation is also increased; 2) Various antioxidants inhibit DomA-induced neurotoxicity; 3) Glutathione (GSH) levels modulate the necrotic and apoptotic effects of DomA. In particular, neurotoxicity of DomA is enhanced in CGNs from Gclm (−/−) mice, which lack the modifier subunit of glutamate-cysteine ligase, the first and rate limiting step in the synthesis of GSH, and as a consequence display very low levels of GSH (Yang et al. 2002). Depletion of GSH in CGNs from wild-type mice also significantly increases DomA neurotoxicity (Giordano et al. 2006).
Given that activation of JNK and p38 has been associated with oxidative stress generated by ultraviolet or gamma radiation, or by direct exposure to hydrogen peroxide (Torres and Forman, 2003; McCubrey et al. 2006), the present study was aimed at determining whether DomA-induced apoptosis would involve activation of one or both of these two kinases in mouse CGNs.
Experimental procedures
Materials
Domoic acid, poly-D-lysine, (+)-5-methyl-10,11-dihydro-5H-dibenzo[a.d] cyclohepten-5,10-imine-maleate (MK-801), catalase (CAT), glutathione ethyl ester (GSHEE), N-tert-butyl-alpha-2-sulfophenyl-nitrone (PBN), horseradish peroxidase-conjugated anti-mouse, horseradish peroxidase-conjugated anti-rabbit IgG and dimethylsulfoxide (DMSO) were from Sigma Chemical Co. (St. Louis, MO, USA). Mouse anti-β-actin antibody was from ABCAM (Cambridge, MA, USA). 2,3-Dihydroxy-6-nitro-sulfamoylbenzo(f) quinoxaline (NBQX) was from Tocris Cookson (Ellisville, MO, USA). The reagents for enhanced chemiluminescence were from Amersham (Arlington Heights, IL, USA). Neurobasal, Hibernate A and B27 MinusAO media, Hank’s balanced salt solution, GlutaMax, dispase, fetal bovine serum and gentamycin were from Invitrogen (Carlsbad, CA, USA). Phospho 44/42 MAP Kinase, phospho p38 MAP Kinase and phospho SAPK/JNK antibodies were from Cell Signaling (Danvers, MA). The inhibitors 4-(4-Fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1H-imidazole (SB 203580) and 1,9-pyrazoloanthrone (SP600125) were purchased from Calbiochem (San Diego, CA). OxyBurstR Green H2HFF BSA was from Molecular Probes (Eugene, OR, USA).
Generation of Gclm-null mice and genotyping
All procedures for animal use were in accordance with the National Institute of Health Guide for the Use and Care of Laboratory Animals, and were approved by the University of Washington Animal Care and Use Committee. Gclm-null [Gclm (−/−)] mice were derived by homologous recombination techniques in mouse embryonic stem cells, as previously described in Giordano et al (2006). Pups were genotyped as described by Giordano et al (2006).
Cultures of cerebellar granule neurons and cell treatments
Cultures of cerebellar granule neurons (CGN) were prepared from 7 day-old mice, as described by Giordano et al. (2006). Neurons were grown for 10–12 days before treatments. All compounds were dissolved in Locke’s solution, with the exception of PBN which was dissolved in DMSO.
Incubations with DomA were for 1 h, followed by washout, and cells were harvested at different times, depending on the end-point measured, as specified in Results.
Measurements of apoptosis
To visualize nuclear morphology following toxin treatment, cells were fixed with methanol and stained in 10 μg/mL Hoechst DNA binding dye for 15 min. All cells exhibiting either classical apoptotic bodies or shrunken, rounded nuclei were scored as being apoptotic. Five fields across a diameter were counted per well at 20×, using a fluorescence microscope.
Immunoblotting analysis
Neurons were scraped in lysis buffer (Tris 50 mM pH 7.5, 2 mM EDTA, 0.5 mM EGTA, 0.5mM dithiothreitol, 0.5 mM phenylmethylsulfonylfluoride (PMSF), 10 ug/ml leupeptin, and 2 ug/ml aprotinin, 1mM sodium orthovanadate, 1 mM NaF, 0.25% SDS). Whole homogenates were subjected to SDS-PAGE and immunoblotting as previously described (Giordano et al., 2006), using rabbit antibodies against pSAPK/JNK, phospho-p38, p44/42 MAPK (1:1000) or mouse anti-β-actin antibody (1:2000). After electrophoresis, proteins were transferred to PDVF membranes that were incubated with the above antibodies. Membranes were rinsed in TBS-T and incubated with horseradish peroxidase-conjugated anti-rabbit IgG for pSAPK/JNK, phosphor-p38, p44/42 MAPK, or with horseradish peroxidase-conjugated anti-mouse IgG for actin at the appropriate dilutions (1:1000 and 1:5000 respectively). Densitometric quantification of the immunoblot bands was performed using Epson Perfection high-resolution flatbed scanner and NIH image J densitometry software. The values of phosphorylated forms were normalized according to the values of the actin.
Statistical Analysis
Data are expressed as the mean ± SD of three independent experiments. Statistical analysis was performed by Student’s t-test for paired samples or by one way ANOVA followed by a Bonferroni post-test.
Results
In CGNs from Gclm (+/+) mice DomA (100 nM) caused a time-dependent increase in the phosphorylation of JNK (Fig. 1) and of p38 (Fig. 2) kinases, with a maximal effect at 3 h. At this time point, DomA caused a 2.4 +/− 0.5 and 2.2 +/− 0.1-fold increase in phosphorylation of JNK and p38, respectively. In CGN from Gclm (−/−) mice, the effect of DomA was much more pronounced, with a 8.2 +/− 1.0 and a 6.7 +/− 0.8-fold induction of JNK and p38, respectively, at 3 h (Fig. 1 and Fig. 2). Such increases were not due to an up-regulation of kinase expression, since the total levels of these proteins were unchanged. Furthermore, in agreement with the findings of Xia et al. (1995), we found that DomA caused a decrease in the phosphorylation of Erk 1/2, without altering Erk 1/2 expression (Fig. 3). The higher levels of phosphorylation of JNK and p38 kinase induced by DomA in CGN from Gclm (−/−) mice, which have a reduced level of GSH, suggest that activation of these kinases involves oxidative stress.
Figure 1.
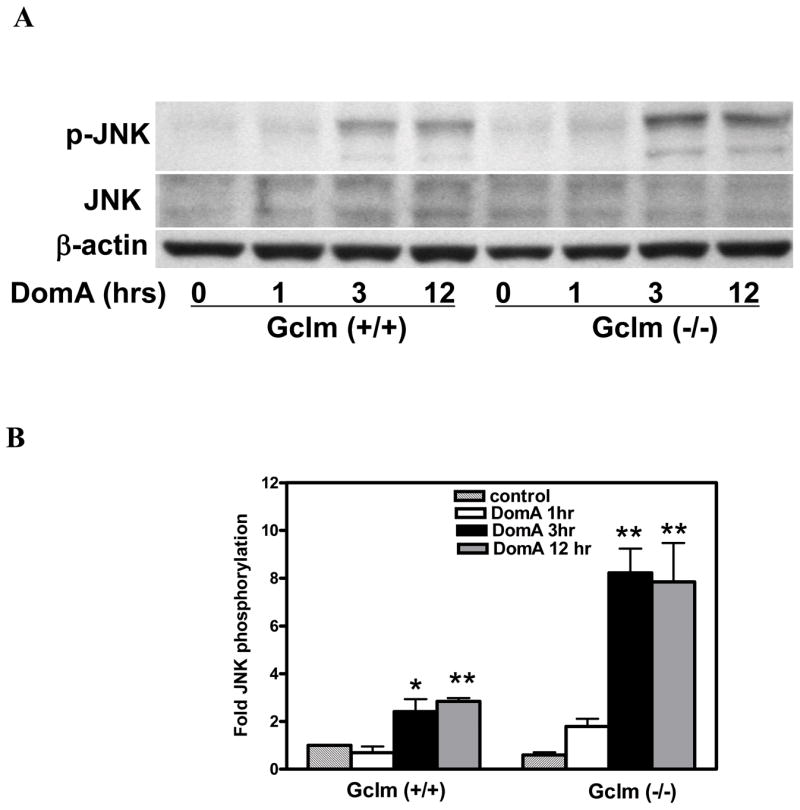
DomA induces JNK phosphorylation in mouse CGNs. A. CGNs from Gclm (+/+) and Gclm (−/−) mice were incubated with 0.1 uM DomA for 1 h, and JNK activation was determined at the indicated times by Western analysis using an antibody recognizing phosphorylated JNK. The anti beta-actin antibody was used to confirm equal protein loading in each gel lane, while an anti-JNK antibody was used to determine levels of JNK protein. B. Quantitation of the experiments shown in panel A, representing the mean (+/− SD) of three independent experiments from three separate cultures. *Significantly different from control, p < 0.05; **Significantly different from control, p<0.01.
Figure 2.

DomA induces p38 phosphorylation in mouse CGNs. A. CGNs from Gclm (+/+) and Gclm (−/−) mice were incubated with 0.1 uM DomA for 1 h, and p38 activation was determined at the indicated times by Western analysis using an antibody recognizing phosphorylated p38. The anti beta-actin antibody was used to confirm equal protein loading in each gel lane, while an anti-p38 antibody was used to determine levels of p38 protein. B. Quantitation of the experiments shown in panel A, representing the mean (+/− SD) of three independent experiments from three separate cultures. *Significantly different from control, p < 0.05; **Significantly different from control, p<0.01.
Figure 3.

DomA decreases the phosphorylation of Erk 1/2 (p42/p44) in mouse CGNs. A. CGNs from Gclm (+/+) and Gclm (−/−) mice were incubated with 0.1 uM DomA for 1 h, and Erk 1/2 activation was determined at the indicated times by Western analysis using an antibody recognizing phosphorylated Erk 1/2. An anti-Erk 1/2 antibody was used to determine levels of Erk 1/2 proteins. B. Quantitation of the experiments shown in panel A, representing the mean (+/− SD) of three independent experiments from three separate cultures. *Significantly different from control, p < 0.05; **Significantly different from control, p<0.01.
DomA neurotoxicity has been ascribed to activation of both non NMDA (AMPA/kainate) and NMDA ionotropic receptors. At higher concentrations (e.g. 10 uM), DomA neurotoxicity is mediated by both AMPA/kainate and NMDA receptors [the latter activated by DomA-induced glutamate release (Berman and Murray, 1997; Giordano et al. 2006)], while apoptosis induced by 100 nM DomA is antagonized only by inhibitors of AMPA/kainate receptors (Giordano et al. 2007). As shown in Fig. 4A and 4B, the AMPA/kainate receptor antagonist NBQX, but not the NMDA receptor antagonist MK-801, prevented the increases in phosphorylation of both kinases, again suggesting that p38 and JNK kinases may play an important part in Dom-A mediated apoptosis.
Figure 4.

A. Activation of JNK and p38 activation by 0.1 uM DomA in mouse CGNs is inhibited by the AMPA/kainate receptor antagonist NBQX (10 uM) but not by the NMDA receptor antagonist MK-801 (5 uM). CGNs were incubated with DomA for 1 h, antagonists were added 1 h before DomA, and JNK and p38 phosphorylation was assessed at 3 h. B. Quantitation of the experiments shown in panel A, representing the mean (+/− SD) of three separate experiments. ** Significantly different from DomA, p<0.01.
DomA-induced apoptosis is more pronounced in CGNs from Gclm (−/−) mice which have a reduced ability to respond to oxidative stress due to their low levels of GSH (Giordano et al. 2007). In addition, the antioxidant melatonin and glutathione ethyl ester (GSHEE) completely prevented the apoptosis caused by DomA, and apoptotic concentrations of DomA (100 nM) caused an increase in intracellular reactive oxygen species (Giordano et al. 2007). This evidence suggests that apoptosis induced by DomA is mediated by oxidative stress. Since induction of p38 and of JNK kinases by DomA was also higher in CGNs from Gclm (−/−) mice, it is conceivable that oxidative stress may activate phosphorylation of both kinases. Fig. 5C and 5D show that catalase, GSHEE, and the spin trapping agent N-tert-butyl alpha nitrone (PBN) prevented phosphorylation of both p38 and JNK.
Figure 5.

A. Antioxidants prevent DomA-induced JNK and p38 phosphorylation. CGNs from Gclm (+/+) mice were exposed for 1 hr to 0.1 uM DomA alone or after 1 h pre-treatment with 100 U/ml catalase (CAT), 2.5 mM GSH ethylester (GSHEE), or 100 uM PBN. JNK and p38 phosphorylation was assessed at 3 h. B. Quantitation of the experiments shown in panel A, representing the mean (+/− SD) of three separate experiments. ** Significantly different from DomA, p<0.01.
To examine the functional consequence of p38 and JNK activation in DomA-induced neuronal apoptosis, we utilized inhibitors of both signaling pathways, the p38 inhibitor SB203580 ( Lal et al., 1999; Gould et al., 1995), and the JNK inhibitor SP600125 (Wang et al., 2004). DomA-induced apoptosis was significantly reduced when cells were treated with these inhibitors alone, and even more when they were used in combination (Fig. 6).
Figure 6.
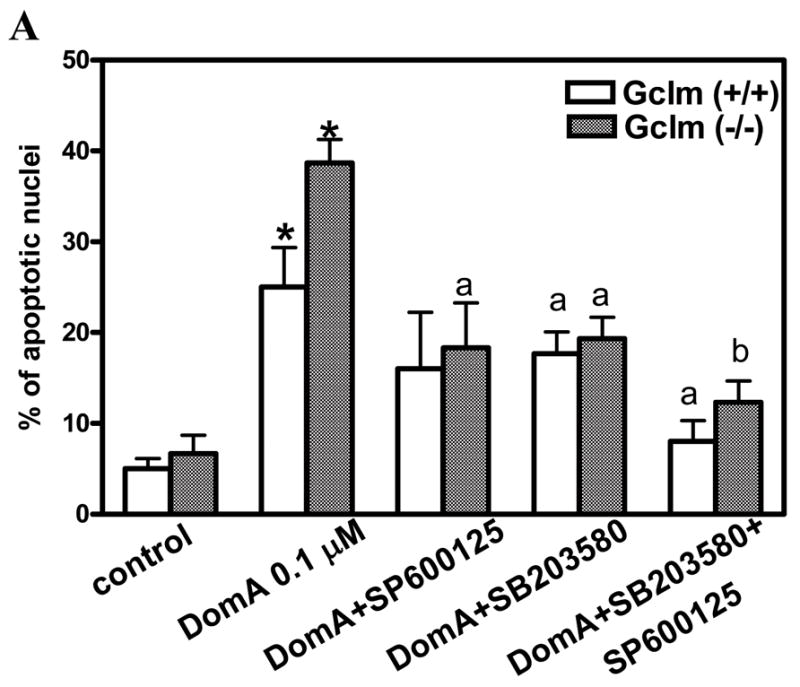
Antagonists of the p38 and JNK signaling pathways prevent DomA-induced apoptosis. CGNs from mice of both genotype were pretreated for 30 min with SB203580 (10 uM) or SP600125 (5 uM), and then exposed to 0.1 uM DomA for 1h. Apoptotic cells were determined at 24 h.
Discussion
The present study indicates that a concentration of DomA causing apoptotic cell death of mouse CGNs causes sustained activation of both JNK and p38. Such activation is blocked by an AMPA/kainate receptor antagonist, but not by an antagonist of NMDA receptors. The same pharmacological profile has been previously seen in regard to DomA-induced apoptosis (Giordano et al. 2007). Activation of both kinases is inhibited by the antioxidants catalase and PBN, and by GSHEE, which significantly increases intracellular GSH levels (Giordano et al. 2006). Furthermore, DomA-induced activation of JNK and p38 is significantly more pronounced in CGNs from Gclm (−/−) mice, which have very low GSH levels (2.4 nmol/mg protein vs. 12.4 nmol/mg protein in CGNs from wild-type mouse; Giordano et al. 2006). Finally, inhibition of JNK or p38 antagonizes the apoptotic effect of DomA, confirming the involvement of these two kinases in the apoptotic process.
There are no prior reports on the involvement of JNK and p38 in DomA-induced apoptosis. Limited evidence is, however, available for kainic acid. In rat CGNs, the p38 inhibitor SB203580 was found to partially attenuate kainic acid-induced apoptosis (Giardina and Beart, 2002). In addition, activation of p38 by kainic acid, and protection against excitotoxicity by a p38 inhibitor, have also been observed in vivo (Kim et al. 2004; Segura-Torres et al. 2006). With regard to JNK, it has been reported that JNK-3 knockout mice are more sensitive than wild-type animals to kainic acid neurotoxicity (Yang et al. 1997; Brecht et al. 2005).
JNKs are activated in response to inflammatory cytokines, heat shock, ionizing radiation, oxidative stress, and DNA damage (Kyriakis and Avruch, 2001), and phosphorylate various transcription factors such as c-Jun, p53, Elk-1, which in turn regulate the expression of genes involved in cell proliferation or apoptosis (Weston and Davis, 2007; Raman et al. 2007). Interestingly, both a transient (< 1 h) and a sustained activation of JNK contribute to induction of gene expression; however, only prolonged activation of JNK promotes apoptosis (Ventura et al. 2006; Weston and Davis, 2007). As shown in Fig. 1, DomA caused a sustained activation of JNK. As for all MAPKs, JNKs are activated by a cascade of mitogen-activated protein kinase kinases (MAPKKs), and the predominant MAP2Ks upstream of JNK are MKK4 and MKK7, in turn activated by various MAPKKKs (Raman et al. 2007). The p38 kinases are activated by the same stimuli as JNK; however, upstream MAPKKs are MKK3 and MKK6. JNK and p38 MAPKs share a large number of common substrates including transcriptional factors and cytoskeletal proteins (Raman et al. 2007).
While both JNK and p38 kinase appear to be involved in DomA-induced apoptosis in CGNs, in some cases only one kinase is involved. For example, apoptosis induced by the herbicide paraquat in dopaminergic neurons requires JNK, but not p38 (Peng et al. 2004; Klintworth et al. 2007). JNK has also been shown to be involved in caspase-independent, non-apoptotic cell death (Zhang et al. 2007). The activation of JNK and p38 kinase by DomA was paralleled by a decrease in the phosphorylation of Erk 1/2 (Fig. 3), thus substantiating the findings of Xia et al. (1995) suggesting that these three MAPKs exert opposing effects on apoptosis.
In summary, the present findings indicate that a relatively low concentration of DomA (100 nM) causes activation of both JNK and p38 kinase, and that this activation is involved in the ability of this neurotoxin to induce a caspase 3-dependent apoptosis in mouse CGNs (Giordano et al. 2007). As an increase of ROS was shown to mediate DomA-induced apoptosis (Giordano et al. 2007), the signals for JNK and p38 kinase activation are most likely ROS. Indeed, antioxidants antagonize their phosphorylation (see Fig. 5); phosphorylation of JNK and p38 is also 3–4-fold more pronounced in CGNs from Gclm (−/−) mice, which display low levels of GSH (Giordano et al. 2006), and in which levels of ROS induced by DomA are much higher than in CGNs from wild-type mice (Giordano et al. 2007). The results provide another model indicating a role for sustained induction of JNK and p38 kinase in mediating apoptosis, and their opposing effect to Erk 1/2 in modulating the apoptotic process.
Supplementary Material
Acknowledgments
This study was supported in part by grants ES012762/NSF-OCE-0434087 and ES07033.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Berman FW, Murray TF. Domoic acid neurotoxicity in cultured cerebellar granule neurons is mediated predominantly by NMDA receptors that are activated as a consequence of excitatory amino acid release. J Neurochem. 1997;69:693–703. doi: 10.1046/j.1471-4159.1997.69020693.x. [DOI] [PubMed] [Google Scholar]
- Brecht S, Kirchhof R, Chromik A, Willesen M, Nicolaus T, Raivich G, Wessig J, Waetzig V, Goetz M, Claussen M, Pearse D, Kuan CY, Vaudano E, Behrens A, Wagner E, Flavell RA, Davis RJ, Herdegen T. Specific pathophysiological functions of JNK isoforms in the brain. Eur J Neurosci. 2005;21:363–377. doi: 10.1111/j.1460-9568.2005.03857.x. [DOI] [PubMed] [Google Scholar]
- Caughlan A, Newhouse K, Namgung U, Xia Z. Chlorpyrifos induces apoptosis in rat cortical neurons that is regulated by a balance between p38 and ERK/JNK MAP kinases. Toxicol Sci. 2004;78:125–134. doi: 10.1093/toxsci/kfh038. [DOI] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Felipo V, Miñana MD, Azorín I, Grisolía S. Induction of rat brain tubulin following ammonium ingestion. J Neurochem. 1988;51:1041–1045. doi: 10.1111/j.1471-4159.1988.tb03065.x. [DOI] [PubMed] [Google Scholar]
- Giardina SF, Beart PM. Kainate receptor-mediated apoptosis in primary cultures of cerebellar granule cells is attenuated by mitogen-activated protein and cyclin-dependent kinase inhibitors. Br J Pharmacol. 2002;135:1733–1742. doi: 10.1038/sj.bjp.0704636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano G, White CC, McConnachie LA, Fernandez C, Kavanagh TJ, Costa LG. Neurotoxicity of domoic Acid in cerebellar granule neurons in a genetic model of glutathione deficiency. Mol Pharmacol. 2006;70:2116–2126. doi: 10.1124/mol.106.027748. [DOI] [PubMed] [Google Scholar]
- Giordano G, White CC, Mohar I, Kavanagh TJ, Costa LG. Glutathione levels modulate domoic acid-induced apoptosis in mouse cerebellar granule cells. Toxicol Sci. 2007 doi: 10.1093/toxsci/kfm236. (in press) [DOI] [PubMed] [Google Scholar]
- Gould GW, Cuenda A, Thomson FJ, Cohen P. The activation of distinct mitogen-activated protein kinase cascades is required for the stimulation of 2-deoxyglucose uptake by interleukin-1 and insulin-like growth factor-1 in KB cells. Biochem J. 1995;311:735–738. doi: 10.1042/bj3110735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson DR, Manalo JL. The activation of glutamate receptors by kainic acid and domoic acid. Nat Toxins. 1998;6:153–158. doi: 10.1002/(sici)1522-7189(199805/08)6:3/4<153::aid-nt16>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Morooka T, Shimohama S, Kimura J, Hirano T, Gotoh Y, Nishida E. Activation and involvement of p38 mitogen-activated protein kinase in glutamate-induced apoptosis in rat cerebellar granule cells. J Biol Chem. 1997;272:18518–18521. doi: 10.1074/jbc.272.30.18518. [DOI] [PubMed] [Google Scholar]
- Kim SW, Yu YM, Piao CS, Kim JB, Lee JK. Inhibition of delayed induction of p38 mitogen-activated protein kinase attenuates kainic acid-induced neuronal loss in the hippocampus. Brain Res. 2004;1007:188–191. doi: 10.1016/j.brainres.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Klintworth H, Newhouse K, Li T, Choi WS, Faigle R, Xia Z. activation of c-Jun N-terminal protein kinase is a common mechanism underlying paraquat-and rotenone-induced dopaminergic cell apoptosis. Toxicol Sci. 2007;97:149–162. doi: 10.1093/toxsci/kfm029. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- Lal AS, Clifton AD, Rouse J, Segal AW, Cohen P. Activation of the neutrophil NADPH oxidase is inhibited by SB 203580, a specific inhibitor of SAPK2/p38. Biochem Biophys Res Commun. 1999;259:465–470. doi: 10.1006/bbrc.1999.0759. [DOI] [PubMed] [Google Scholar]
- Luo YQ, Umegaki H, Wang XT, Abe R, Roth GS. Dopamine induces apoptosis through an oxidation-involved SAPK/JNK activation pathway. J Biol Chem. 1998;273:3756–3764. doi: 10.1074/jbc.273.6.3756. [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal. 2006;8:1775–1789. doi: 10.1089/ars.2006.8.1775. [DOI] [PubMed] [Google Scholar]
- Namgung U, Xia Z. Arsenite-induced apoptosis in cortical neurons is mediated by c-Jun N-terminal protein kinase 3 and p38 mitogen-activated protein kinase. J Neurosci. 2000;20:6442–6451. doi: 10.1523/JNEUROSCI.20-17-06442.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newhouse K, Hsuan SL, Chang SH, Cai B, Wang Y, Xia Z. Rotenone-induced apoptosis is mediated by p38 and JNK MAP kinases in human dopaminergic SH-SY5Y cells. Toxicol Sci. 2004;79:137–146. doi: 10.1093/toxsci/kfh089. [DOI] [PubMed] [Google Scholar]
- Peng J, Mao XO, Stevenson FF, Hsu M, Andersen JK. The herbicide paraquat induces dopaminergic nigral apoptosis through sustained activation of the JNK pathway. J Biol Chem. 2004;279:32626–32632. doi: 10.1074/jbc.M404596200. [DOI] [PubMed] [Google Scholar]
- Perl TM, Bedard L, Kosatsky T, Hockin JC, Todd EC, Remis RS. An outbreak of toxic encephalopathy caused by eating mussels contaminated with domoic acid. N Engl J Med. 1990;322:1775–1780. doi: 10.1056/NEJM199006213222504. [DOI] [PubMed] [Google Scholar]
- Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26:3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- Scallet AC, Binienda Z, Caputo FA, Hall S, Paule MG, Rountree RL, Schmued L, Sobotka T, Slikker W. Domoic acid-treated cynomolgus monkeys (M. fascicularis): effects of dose on hippocampal neurons and terminal degeneration. Brain Res. 1993;627:307–313. doi: 10.1016/0006-8993(93)90335-k. [DOI] [PubMed] [Google Scholar]
- Segura Torres JE, Chaparro-Huerta V, Rivera Cervantes MC, Montes-Gonzales R, Flores Soto ME, Beas-Zerate C. Neuronal cell death due to glutamate excitotoxicity is mediated by p38 activation in the rat cerebral cortex. Neurosci Lett. 2006;403:233–238. doi: 10.1016/j.neulet.2006.04.063. [DOI] [PubMed] [Google Scholar]
- Sobotka TJ, Brown R, Quander DY, Jackson R, Smith M, Long SA, Barton CN, Rountree RL, Hall S, Eilers P, Johannesen JN, Scallet AC. Domoic acid: neurobehavioral and neurohistological effects of low-dose exposure in adult rats. Neurotoxicol Teratol. 1996;18:659–670. doi: 10.1016/s0892-0362(96)00120-1. [DOI] [PubMed] [Google Scholar]
- Strain SM, Tasker RA. Hippocampal damage produced by systemic injections of domoic acid in mice. Neuroscience. 1991;44:343–352. doi: 10.1016/0306-4522(91)90059-w. [DOI] [PubMed] [Google Scholar]
- Teitelbaum JS, Zatorre RJ, Carpenter S, Gendron D, Evans AC, Gjedde A, Cashman NR. Neurologic sequelae of domoic acid intoxication due to ingestion of contaminated mussels. N Engl J Med. 1990;322:1781–1787. doi: 10.1056/NEJM199006213222505. [DOI] [PubMed] [Google Scholar]
- Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. Biofactors. 2003;17:287–296. doi: 10.1002/biof.5520170128. [DOI] [PubMed] [Google Scholar]
- Tryphonas L, Truelove J, Todd EC, Nera E, Iverson F. Experimental oral toxicity of domoic acid in cynomolgous monkeys (Macaca fascicularis) and rats. Preliminary investigations. Food Chem Toxicol. 1990;28:707–715. doi: 10.1016/0278-6915(90)90147-f. [DOI] [PubMed] [Google Scholar]
- Ventura JJ, Hubner A, Zhang C, Flavell RA, Shokat KM, Davis RJ. Chemical genetic analysis of the time course of signal transduction by JNK. Mol Cell. 2006;21:710–710. doi: 10.1016/j.molcel.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Wang W, Shi L, Xie Y, Ma C, Li W, Su X, Xiang S, Chen R, Zhu Z, Mao Z, Han Y, Li M. SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Neurosci Res. 2004;48:195–202. doi: 10.1016/j.neures.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Op Cell Biol. 2007;19:142–149. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- Yang Y, Dieter MZ, Chen Y, Shertzer HG, Nebert DW, Dalton TP. Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm (−/−) knockout mouse. Novel model system for a severely compromised oxidative stress response. J Biol Chem. 2002;277:49446–49452. doi: 10.1074/jbc.M209372200. [DOI] [PubMed] [Google Scholar]
- Zhang S, Lin Y, Kim YS, Hande MP, Liu ZG, Shen HM. c-Jun N-terminal kinase mediates hydrogen peroxide-induced cell death via sustained poly (ADP-ribose) polymerase-1 activation. Cell Death Differ. 2007;14:1001–1010. doi: 10.1038/sj.cdd.4402088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.