

Abstract
The intercalated disk protein Xin was originally discovered in chicken striated muscle and implicated in cardiac morphogenesis. In the mouse, there are two homologous genes, mXinα and mXinβ. The human homolog of mXinα, Cmya1, maps to chromosomal region 3p21.2–21.3, near a dilated cardiomyopathy with conduction defect-2 locus. Here we report that mXinα-null mouse hearts are hypertrophied and exhibit fibrosis, indicative of cardiomyopathy. A significant upregulation of mXinβ likely provides partial compensation and accounts for the viability of the mXinα-null mice. Ultrastructural studies of mXinα-null mouse hearts reveal intercalated disk disruption and myofilament disarray. In mXinα-null mice, there is a significant decrease in the expression level of p120-catenin, β-catenin, N-cadherin, and desmoplakin, which could compromise the integrity of the intercalated disks and functionally weaken adhesion, leading to cardiac defects. Additionally, altered localization and decreased expression of connexin 43 are observed in the mXinα-null mouse heart, which, together with previously observed abnormal electrophysiological properties of mXinα-deficient mouse ventricular myocytes, could potentially lead to conduction defects. Indeed, ECG recordings on isolated, perfused hearts (Langendorff preparations) show a significantly prolonged QT interval in mXinα-deficient hearts. Thus mXinα functions in regulating the hypertrophic response and maintaining the structural integrity of the intercalated disk in normal mice, likely through its association with adherens junctional components and actin cytoskeleton. The mXinα-knockout mouse line provides a novel model of cardiac hypertrophy and cardiomyopathy with conduction defects.
Keywords: Xin repeat proteins, N-cadherin, β-catenin, p120-catenin, connexin 43
The intercalated disk contains adherens junctions, desmosomes, and gap junctions that maintain the integrity of the association between cardiomyocytes and enable the myocardium to function in synchrony. The expression and distribution of many of these junctional components are often altered in many types of heart disease (5, 8, 13, 35). However, direct evidence to support a role for these proteins in contributing to cardiomyopathies remains incomplete. The best-characterized example involves the effects of deletion of a key adherens junction component, N-cadherin, on the intercalated disk. N-cadherin functions to mediate Ca2+-dependent homophilic cell-cell adhesion. Conditional deletion of N-cadherin in the adult mouse heart leads to a complete dissolution of the intercalated disk structure and a significant decrease in the expression of the gap junction proteins connexin 43 and connexin 40 as well as the cytoplasmic desmosomal protein plakoglobin. These global perturbations of intercalated disk components subsequently result in dilated cardiomyopathy and in spontaneous ventricular tachycardia and arrhythmic death (12, 15). Although E-cadherin has been shown to restore cardiomyocyte adhesion and cardiac looping in conventional N-cadherin-knockout embryos (18), cardiac-specific expression of E-cadherin results in dilated cardiomyopathy, suggesting that ectopic expression of cadherin in intercalated disks is not sufficient to restore normal cardiac function (7). In addition, mutations in desmosome-associated proteins, including plakoglobin (19), desmoglein-2 (26), and desmoplakin (1), have been identified as the primary cause of arrhythmogenic right ventricular cardiomyopathy, leading to ventricular tachycardia and lethal arrhythmias. Similarly, aberrant desmosome and gap junction organization, with aberrant side-to-side connections, was observed in the hearts of patients with hypertrophic cardiomyopathy (28), providing a first account of the interplay between desmosomal and gap junctional components in human cardiomyopathy. These and other studies demonstrate that the disruption of various intercalated disk components leads to cardiac defects and cardiomyopathies; however, the molecular mechanisms regulating the interplay between these components are complex and remain to be elucidated.
The striated muscle-specific Xin protein was shown to be involved in chicken cardiac morphogenesis by antisense knockdown experiments (33). Subsequent cloning of the mouse homolog identified two Xin genes, mXinα and mXinβ. The Xin protein contains a 16-amino acid repeat region (Xin repeat), a putative DNA binding domain, a putative nuclear localization signal, and a proline-rich region. At the present time, neither nuclear localization nor transcriptional activity of mXinα has been detected. mXinα appears to be restricted to the periphery of cardiomyocytes in early embryos and later specifically localizes to the intercalated disk in near-term embryos through adulthood (29). The Xin repeats from the human homolog (hXinα, Cmya1) of mXinα have been shown to bind actin filaments in vitro (23). However, in vivo mXinα was not found to associate with thin filaments; rather, mXinα colocalizes with N-cadherin and β-catenin at the intercalated disks (29), suggesting that mXinα interacts with one or more of these molecules. Indeed, Xin coimmunoprecipitates with both β-catenin and N-cadherin from chicken embryonic heart lysates, another indication that Xin is a part of the N-cadherin/β-catenin complex (29). In another study, we have shown (9) the direct interaction between mXinα and β-catenin.
To gain insight into mXinα function, mXinα-knockout mice were created. Viable and fertile knockout mice are observed in Mendelian ratios, and an upregulation of the mXinβ gene was detected. mXinα-deficient mouse hearts exhibit cardiac hypertrophy and cardiomyopathy. Ultrastructural examination of mXinα-null mouse hearts reveals myofilament disarray and disrupted intercalated disk structure. The expression levels of N-cadherin, β-catenin, p120-catenin (p120ctn), and connexin 43, but not α-catenin or plakoglobin, are decreased significantly in mXinα-null mice. These data suggest that mXinα plays an important role in regulating the hypertrophic response and maintaining the tightly associated intercalated disk membrane and myofilament organization in normal mice.
MATERIALS AND METHODS
Construction of mXinα targeting vector and generation of mXinα-deficient mice
All animal procedures were performed with the approval of the University of Iowa Institutional Animal Care and Use Committee and were conducted in accordance with the Guide for the Care and Use of Laboratory Animals approved by the National Institutes of Health. To delete the mXinα gene, a targeting vector was constructed (Fig. 1A). The targeting vector, cloned into the NotI/EcoRI sites of pBluescript II SK (Stratagene, La Jolla, CA), contains a mutation to change ATG to ATC and a replacement of 203 bp downstream of ATG with the LacZ-Neor cassette. The resulting construct did not alter the 3' splicing site of intron 1.
Fig. 1.

Targeted deletion of mXinα. A: schematic representation of the targeting strategy. The mXinα locus contains 3 exons (E1, E2 and E3), with the 5′ boundary of E2 only 4 bp upstream of the translation initiation codon (ATG). The location of the 311-bp EcoRI/KpnI probe used for Southern blot analysis is within E3 but outside the targeting vector. T, targeted locus. B: genotyping of mXinα-knockout mouse littermates by Southern blot and PCR analyses. For Southern blot analysis, KpnI-digested genomic DNA was hybridized with a 311-bp probe, which detected 10-kb and 15-kb KpnI fragments from endogenous and targeted loci, respectively. For PCR genotyping, genomic DNAs were analyzed with primers designed to amplify the endogenous (E) and targeted (T) loci. The locations of the PCR amplified products for E (573 bp) and T (630 bp) are shown in A. C: Northern and Western blot analyses. For Northern blot analysis, total RNAs were hybridized with the labeled mXinα cDNA. A 5.8-kb mXinα message was detected in both wild-type and heterozygous RNA lanes, but not in the homozygous RNA lane. The same membrane was reprobed with labeled GAPDH cDNA to show RNA loading. For Western blot analysis, total proteins extracted from age-matched wild-type (lane 1), heterozygous (lane 2), and homozygous (lane 3) hearts were immunoblotted with anti-mXinα and anti-GAPDH antibodies. A 150-kDa mXinα protein was detected in the wild-type and heterozygous mouse hearts, but not in the homozygous mouse heart. The mXinα-null hearts appear to have more GAPDH protein than the wild-type heart (left). Although most protein bands from these heart extracts show equal intensity by Coomassie blue stain, the myosin heavy chains (MHCs; indicated by 194 kDa) are significantly decreased in the mXinα-deficient hearts (right). Western blot with anti-α-tubulin antibody shows that wild-type and mXinα-deficient mouse hearts contained equal amounts of α-tubulin (bottom left).
The linearized targeting vector was electroporated into embryonic stem (ES) cells at the University of Iowa Gene Targeting Facility. After selection, ES cells were screened for the presence of the targeted locus by Southern blot analysis. Germ line transmission of the targeted locus was determined by Southern blot analysis (33) and PCR genotyping. PCR genotyping was carried out, using tail DNA to amplify a 630-bp fragment spanning the Neor-mXinα adjoining region of the targeted locus (T in Fig. 1A) or a 573-bp fragment spanning the intron 1–E2 region of the endogenous mXinα locus (E in Fig. 1A). Primer pairs for amplification of Neor-mXinα or intron 1–E2 are 5′-GCGATGCCTGCTTGCCGAAT-3′ and 5′-GCATCTAGCCGCCAGTTCTCA-3′ or 5′-GGAATTCAAACATTGGCTCTGCC-3′ and 5′-CTCATTTGGATGGTTGGTGT-3′, respectively.
Cloning of mXinβ cDNA and RT-PCR analyses of mXinα isoforms and hypertrophy response genes
For mXinβ cDNA cloning, a custom-made cDNA library was prepared from adult mXinα-null mouse hearts and used for low-stringency screening with cXin probe (nt 1977–2649) and mXinα probe (nt 140–1744) as described previously (33). The composite sequences of the inserts from six positive, overlapping clones contain 4,382 bp, represented by pBKX-2, 3, and 4 (GenBank accession nos. AY775570, AY775571, and AY775572).
Total RNA isolated from mouse hearts was used for cDNA synthesis with random hexamers and SuperScript II reverse transcriptase (Invitrogen, San Diego, CA) as described previously (33). For cloning mXinα isoforms, the primer pairs either flanking (Pa-a: nt 3140–3162 and Pa-b: nt 3974–3993) or within intron 2 (Pa-d: nt 3139–3161 and Pa-e: nt 3886–3909, as well as Pa-c: nt 3562–3583 and Pa-b) were designed to specifically amplify mXinα isoforms with standard PCR conditions. The resulting PCR products were cloned into pCRII-TOPO vector (Invitrogen) and sequenced. For the analysis of hypertrophy response genes, previously published primer pairs and PCR conditions for atrial natriuretic factor (ANF), α-myosin heavy chain (MHC), β-MHC, skeletal α-actin, cardiac α-actin, and GAPDH were adapted and carried out (36, 38). Quantitative real-time RT-PCR was performed with the SYBR Green method, with primer pairs designed by ABI software (Applied Biosystems), on the ABI7000 Sequence Detection System (Center for Comparative Genomics, University of Iowa).
Northern and Western blot analyses
Total RNA isolation and Northern blot analysis were performed as previously described (33). The labeled probes included 311-bp mXinα, pBKX-2 mXinβ (nt 1–3319), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Whole hearts dissected from ∼6- to 9-mo-old wild-type, heterozygous, and homozygous mXinα-knockout littermates were homogenized and used for Western blot analysis as described previously (29, 37). The antibodies used included rabbit anti-mXinα (U1013) (29) and mouse monoclonal anti-GAPDH (Research Diagnostics, Flanders, NJ) and anti-α-tubulin (DM1A, a generous gift from Dr. Steven Blose, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). Monoclonal anti-β1-integrin, anti-α-catenin, and anti-plakoglobin antibodies were purchased from BD Biosciences Pharmingen (San Diego, CA). Rabbit anti-connexin 43 and monoclonal anti-N-cadherin, anti-β-catenin, and anti-p120ctn antibodies were obtained from Zymed Laboratories (South San Francisco, CA). Rabbit anti-desmoplakin was purchased from Serotec (Oxford, UK). FA3 monoclonal antibody specifically recognizes α-MHC, as characterized previously (10).
Generation of peptide antibodies specific to mXinα or mXinβ
After amino acid sequence comparison between mXinα and mXinβ, a region of specific peptide sequence with high antigenic index was identified by Lasergene Protean software (DNAstar, Madison, WI). Synthetic peptides to these regions [for mXinα, amino acid (aa) 564 EMIHQQEQQKPEEEEGKGPG 583; for mXinβ, aa 181 TSSSYTSTRQRKETSTSSYS 200] were made, and specific polyclonal antibodies were prepared in rabbits (U1697 for α-peptide-specific and U1741 for β peptide-specific) by Genemed Synthesis (South San Francisco, CA). The specificity of these antibodies was examined by immunofluorescence microscopy on both cultured rat neonatal cardiomyocytes (34) and frozen sections of mouse hearts and by Western blot analysis of mouse heart extracts.
Immunofluorescence and image analysis
Frozen sections from wild-type, heterozygous, and mXinα-null mouse hearts were used in immunofluorescence microscopy as previously described (29). The primary antibodies used included rabbit anti-mXinα and anti-connexin 43 as well as monoclonal anti-N-cadherin, anti-α-catenin, anti-plakoglobin, and anti-p120ctn. Quantification of connexin 43 immunofluorescence results was performed with a protocol adapted from previously described methods (25, 27). Frozen sections from three hearts each of adult wild-type and mXinα-null mice were immunostained with anti-connexin 43 antibody. All sections were processed in the same manner. For each specimen, five randomly selected fields (×63 objective lens) of longitudinally oriented myocytes were analyzed by ImageJ software (http://rsb.info.nih.gov/ij) to quantify the number and size/area of connexin 43-containing fluorescence spots. The level of background fluorescence was relatively low and was subtracted with the ImageJ program under the same conditions for each test field. The immunoreactive label representing connexin 43 antigen was concentrated in discrete areas between cell abutments, representing intercalated disk-associated connexin 43. Some signal was also observed between the myocytes but along the long axis of the myocytes, representing laterally localized connexin 43. Phase-contrast images were used to further confirm localizations of the fluorescence spots/bands. With the “analyze particles” function of ImageJ software and an arbitrary threshold level of 100 pixels, total number and size/area of label were automatically calculated for each image. Subsequently, the total number and size of fluorescent label that was localized laterally along the myocytes were calculated separately. The number and size/area of laterally localized connexin 43 were expressed as a percentage of total connexin 43 staining. A one-way ANOVA was used to test for statistical significance.
In situ hybridization
For section in situ hybridization with digoxigenin-labeled riboprobes, mouse hearts were dissected, fixed, and embedded in Paraplast Plus as described previously (29). Fifteen-micrometer sections were cut and hybridized with labeled riboprobe prepared from pBKX-2 as described previously (33).
Heart weight-to-body weight ratio analysis
Adult wild-type, heterozygous, and homozygous mXinα-knockout mice at an age of ∼5–11 mo were weighed in grams. The whole hearts from these mice were dissected, rinsed with phosphate-buffered saline to remove blood, blotted dry, and weighed in milligrams.
Histological staining
Adult mouse hearts dissected from wild-type, heterozygous, and homozygous littermates were fixed overnight in 10% formalin-10% DMSO, dehydrated, cleared, and embedded in Paraplast Plus. Serial sections were mounted onto Superfrost Plus slides, rinsed in xylene, rehydrated, and stained with either hematoxylin and eosin (H&E) or Masson's trichrome stain, following standard procedures. Quantification of fibrosis was determined from 30–50 randomly selected microscopic fields of sections prepared from 3 different hearts of each genotype with Image-Pro Express analysis software (Media Cybernetics, Silver Spring, MD).
Transmission electron microscopy
Three 12-wk-old male mice of each genotype were anesthetized, and the hearts were perfused with Locke's solution containing 0.5% heparin-5% lidocaine to arrest the heart in diastole and avoid clotting. The hearts were then perfusion fixed in Karnovsky solution and dissected. The left ventricle of each heart sample was dissected into 1-mm thin slices and fixed in Karnovsky solution at 4°C overnight. The samples were treated sequentially with 1% OsO4 and 2.5% uranyl acetate, dehydrated, and embedded in epon resin. Ultrathin sections were cut, mounted on Formvar-coated grids, poststained, and examined under a Hitachi H-7000 transmission electron microscope (Central Microscopy Research Facility, University of Iowa).
Electrocardiographic recordings of isolated, perfused hearts
The intact heart was isolated from ∼3- to 4-mo-old wild-type and mXinα-knockout mice and mounted on Langendorff apparatus via aortic cannula. Immediately, the heart was retrogradely perfused through the aortic cannula from a heated storage cylinder with 37°C oxygenated Krebs-Henseleit solution to produce an isolated, perfused Langendorff preparation (20, 21). The electrocardiogram (ECG) was recorded by a Hugo Sachs Elektronik-Harvard apparatus. ECG parameters such as P wave, PQ interval, QRS interval, QT interval, and R-R interval (heart rate) were calculated from ECG tracings as described previously by Kirchhoff et al. (11).
RESULTS
Generation and characterization of mXinα-knockout mice
After electroporation of the targeting vector into ES cells and selection in G418, 189 resistant clones were obtained and analyzed by Southern blot. We obtained 20 positive ES clones containing a 15-kb band of targeted locus and a 10-kb band of mXinα locus (Fig. 1B). Two independent clones were used to generate chimeric founders. Heterozygous mice were crossed to generate mXinα-null mice. The resulting heterozygous crosses showed a ratio of 1.1:1.8:1 for wild-type:heterozygote: homozygote from 189 mice genotyped, providing evidence that mXinα-null mice are viable.
Loss of the mXinα message in homozygous mice was confirmed by Northern blot analysis on total RNAs isolated from hearts of each genotype (Fig. 1C). In the heterozygous heart, decreased mXinα message was also seen. The membrane was reprobed with GAPDH to reveal equal RNA loading. Western blot analysis of total proteins extracted from hearts of each genotype further verified loss of mXinα protein in homozygotes (Fig. 1C) and reduced levels of mXinα protein in the heterozygote compared with the wild-type mouse (Fig. 1C, Western blot, top left). When this membrane was probed with anti-GAPDH, a significant increase in GAPDH protein was found in the mXinα-null heart. Moreover, the Coomassie blue-stained gel of total extracts revealed a decrease in the amount of MHC band (indicated by 194 kDa) in heterozygous and homozygous hearts (Fig. 1C, Western blot, right). In contrast, similar Western blot analysis showed that there was no significant change in the amount of α-tubulin in the mXinα-knockout mouse hearts (Fig. 1C, Western blot, bottom left).
Insertion of the LacZ-Neo cassette at the beginning of exon 2 within the targeted gene allowed the endogenous mXinα promoter to control the expression of the LacZ gene. To determine the expression pattern of the knockin LacZ gene, whole-mount embryos at embryonic day (E)8.0, E8.25, E9.0, and E13.5 were dissected and stained for β-galactosidase (β-gal) expression. β-gal was observed throughout the heart tube at E8.0 (Supplemental Fig. 1A) but not in the E7.5 embryo (data not shown), consistent with the first detectable level of mXinα protein expression (29).1 Paraffin sectioning of E8.0–8.25 embryos revealed β-gal expression exclusively in the myocardial layer (Supplemental Fig. 1, B and C). β-gal staining was detected throughout both the looped (E9.0) and the septated (E13.5) hearts in addition to expression within the skeletal muscle of the E13.5 embryo (Supplemental Fig. 1, E–G). A wild-type E9.0 embryo was used as a control and demonstrated no background staining with the β-gal assay (Supplemental Fig. 1D). β-gal expression was continuous within the heart from E9.0 through adulthood (data not shown). Therefore, the β-gal expression in mXinα-deficient mice recapitulated endogenous mXinα expression and did not interfere with normal cardiac development.
Upregulation of mXinβ in mXinα-knockout mice
Immunofluorescence microscopy of wild-type hearts with U1013 anti-mXinα antibody revealed that mXin colocalizes with N-cadherin at the intercalated disk (Supplemental Fig. 2A, a and b). In mXinα-null hearts, some fluorescent signal coinciding with N-cadherin was still detected (Supplemental Fig. 2A, c and d). This suggested the possibility of a second mXin gene, whose protein product is recognized by the U1013 anti-mXinα antibody. Indeed, with low-stringency Southern blot a second band in mouse genomic DNA was detected, demonstrating the presence of a Xin family within the mouse genome (data not shown). Cloning of the second gene was carried out, and six independent overlapping clones were obtained. The total cDNA length derived from these clones is 4,382 bp (GenBank accession nos. AY775570–AY775572), including an ATG initiation codon at bp 100. This second mXin gene is referred to as mXinβ.
mXinβ RNA, detected by in situ hybridization, was specifically expressed at discrete regions in the heart that highly resemble intercalated disks (Supplemental Fig. 2B). Hybridization with control sense probe did not result in any signal (Supplemental Fig. 2Bb). The signal detected by the mXinβ probe was significantly increased in both heterozygous and mXinα-null mice compared with the wild-type mouse (Supplemental Fig. 2B, c and d, respectively). This upregulation of mXinβ in mXinα-deficient mice was further confirmed by Northern blot analysis (Supplemental Fig. 2C).
Prolonged exposure of Western blots reacted with U1013 anti-mXinα antibody revealed two additional high-molecular-mass bands (labeled “mXinα-a” and “mXinβ” in Fig. 2I). Using antibodies generated against an α-specific peptide (U1697) and a β-specific peptide (U1741), we have further identified the highest-molecular-mass band as mXinβ, and the other band near the 250-kDa marker is an additional mXinα isoform called mXinα-a. In cultured neonatal cardiomyocytes, mXinβ, like mXinα, is enriched in cell-cell contacts (arrows in Fig. 2, D and H), at adhesion sites, and along stress fibers. Immunofluorescent staining on frozen sections of adult mouse heart with these antibodies showed the same intercalated disk localization for both mXinα and mXinβ (Fig. 2, J and K). RT-PCR was used to confirm the presence of a large mRNA for mXinα isoform mXinα-a in the heart (Supplemental Fig. 3). When the Pa-d and Pa-e primer pair was used to amplify an RT product prepared from total heart RNA, a 771-bp product was detected. Similarly, a 431-bp fragment was amplified when primer Pa-c and Pa-b were used. These results suggest the presence of at least one additional mXinα mRNA species in the heart, called mXinα-a, which contains the intron sequence between exon 2 and exon 3. When primer pairs (Pa-a and Pa-b) flanking this intron were used in PCR, two minor bands (853 and 675 bp) and a major band (462 bp) were detected (Supplemental Fig. 3). The 853-bp and 462-bp products are consistent with the presence of at least two mXinα isoforms, a minor form of mXinα-a and a major form of mXinα. The nucleotide sequences of these PCR fragments (except 675 bp) further confirmed two isoforms of mXinα in the heart. The 675-bp amplified product was a PCR artifact due to mismatch priming and amplified a fragment from glucose phosphate isomerase 1.
Fig. 2.

Characterization of mXinα and mXinβ peptide antibodies. A–H: double-label immunofluorescence microscopy was performed on rat neonatal cardiomyocytes with rabbit polyclonal α-peptide-specific antibody (U1697), β-peptide-specific antibody (U1741), or their respective preimmune serum and with mouse monoclonal anti-cardiac troponin T antibody (CT3). Rhodamine-conjugated goat anti-rabbit IgG and fluorescein-conjugated goat anti-mouse IgG were used for the secondary antibody in the indirect immunofluorescence. Arrows point to the cell-cell contacts, in which cardiac troponin T is absent but both mXinα and mXinβ are enriched. Bar, 10 μm. I: Western blot analysis of cardiac protein extract with U1013 and peptide-specific (U1697 and U1741) antibodies. The α-peptide-specific antibody (U1697) recognizes both mXinα (150 kDa) and mXinα-a (∼260 kDa) isoform in 7.5% gel blot, whereas the β-peptide-specific antibody reacts with mXinβ band (∼320 kDa) in 6.0% gel blot. The U1013 antibody against amino acids 1–532 of mXinα recognizes both mXinα and mXinα-a bands and cross-reacts with mXinβ. J–L: immunofluorescence microscopy was performed on adult mouse frozen heart sections with rabbit polyclonal U1013, U1697, and U1741 and rhodamine-conjugated goat anti-rabbit secondary antibody. Similar to the U1013 staining, both α-peptide-specific (U1697) and β-peptide-specific (U1741) antibodies recognize the antigens localized to the intercalated disks, and their respective preimmune antibodies do not show this staining pattern (data not shown). Bar, 10 μm.
Like mXinα, mXinα-a was significantly decreased in the heterozygous sample and completely absent in the homozygous sample, as detected by Western blot analysis with U1013 (Fig. 3B), consistent with the idea of mXinα-a as an alternatively spliced isoform of the same mXinα gene. In contrast, the mXinβ protein in the same blot was proportionally increased in the mXinα-deficient mouse hearts (Fig. 3B). Therefore, it appears that the upregulation of mXinβ in the mXinα-knockout heart is at both the message and the protein levels. This, together with the same localization of mXinα and mXinβ, suggests that the increased expression of mXinβ likely plays a compensatory role. It should also be noted that the Coomassie blue-stained gel analysis of total protein extracts from each genotype also revealed decreased levels of MHC in the mXinα-deficient hearts (Fig. 3A). However, there was no significant change in α-tubulin levels in the mXinα-deficient mouse heart (Fig. 3C), demonstrating equal sample loading.
Fig. 3.
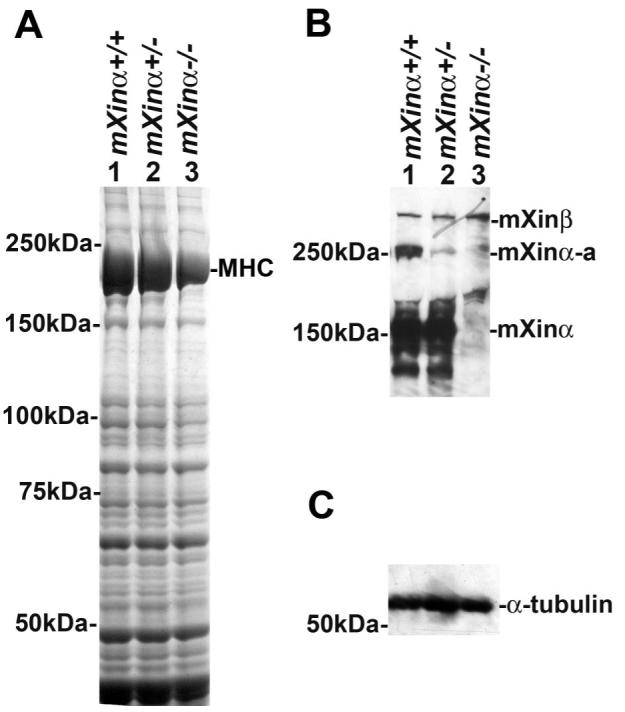
Upregulation of mXinβ protein in mXinα-deficient mouse hearts. A: Coomassie blue-stained gel analysis of total heart extracts from each genotype demonstrates decreased levels of MHC in mXinα-deficient hearts. B: longer exposure of the Western blot with U1013 anti-mXinα antibody detects mXinα, mXinα-a, and mXinβ bands in wild-type and heterozygous samples. Both mXinα and its isoform, mXinα-a, are absent in the mXinα-null sample. In contrast, mXinβ appears to be proportionally increased in the heterozygous and homozygous knockout samples. C: Western blot with anti-α-tubulin antibody shows that there is no significant change in the amount of α-tubulin, demonstrating equal loading.
Cardiac hypertrophy in mXinα-deficient mice
Heterozygous and mXinα-null hearts are modestly enlarged compared with wild-type littermates at 9 mo of age (Fig. 4A, a-c). Additionally, H&E-stained frontal heart sections exhibit thickening of the ventricular myocardial wall and increased trabeculation, suggestive of hypertrophic cardiomyopathy (Fig. 4A, d-f).
Fig. 4.
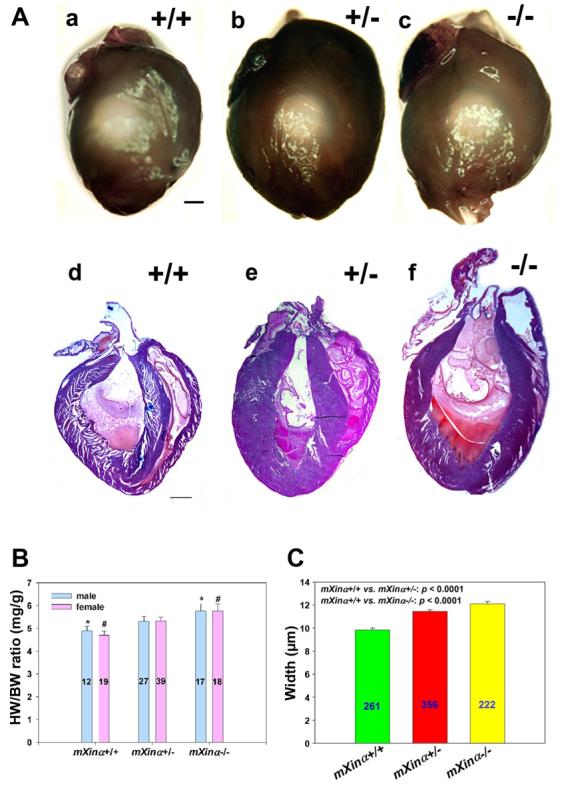
mXinα-deficient mice exhibit cardiac hypertrophy. A: representative set of hearts dissected from 9-mo-old wild-type (+/+) and mXinα-deficient (+/− and −/−) mouse littermates is shown. Whole hearts were isolated (a–c), sectioned, and stained with hematoxylin and eosin (d–f). Both heterozygous and homozygous hearts appear to be modestly enlarged compared with wild-type hearts. Additionally, the ventricles of the mXinα-deficient hearts appear to be thickened with increased trabeculation. Bar, 1 mm. B: while there is no significant difference between males and females of the same genotype, mXinα−/− mice have a statistically significant increase in heart weight-to-body weight ratio (HW/BW) compared with wild-type mice (*P = 0.024 male, #P = 0.004 female; ANOVA). Nos. of animals measured are indicated within the bars. C: mXinα-deficient mice have hypertrophied cardiomyocytes. Average widths of myofibers in micrometers were determined from longitudinal sections of hearts for at least 3 mice of each genotype at 9–11 mo of age. Means ± SE for each genotype are displayed graphically. The average width differences between mXinα+/+ and mXinα-deficient myofibers were statistically significant by 1-way ANOVA. Nos. of myofibers measured for each genotype are indicated within the bars.
To assess the extent of hypertrophy, heart weight-to-body weight ratios (HW/BW) were determined from wild-type and mXinα-deficient mice at 3–9 mo of age. The HW/BW (mean ± SE) for wild-type males was 4.89 ± 0.20, compared with 5.31 ± 0.22 and 5.76 ± 0.32 for heterozygous and homozygous males, respectively. The HW/BW for wild-type females was 4.69 ± 0.20, compared with 5.32 ± 0.17 and 5.77 ± 0.30 for heterozygous and homozygous females, respectively (Fig. 4B). These ratios are statistically significant by one-way ANOVA between wild-type and homozygous hearts. Although there was no significant difference between wild-type and heterozygous hearts, a trend of enlarged heterozygous hearts was observed. No significant difference was observed between males and females of the same genotype.
To investigate whether cardiomyocytes from mXinα-deficient mice are hypertrophic, we employed differential interference contrast microscopy to compare myofiber width in heart sections from at least three mice of each genotype at 9–11 mo of age. With Leica Openlab software, the widths of longitudinal myofibers of similar orientation from frontal sections of hearts of each genotype were measured. The myofiber width of the wild-type heart averaged 9.83 ± 0.17 μm, whereas the heterozygous and homozygous myofiber widths averaged 11.45 ± 0.27 and 12.11 ± 0.22 μm, respectively (Fig. 4C). These differences are statistically significant by one-way ANOVA.
To further characterize the hypertrophic phenotype observed in the mXinα-deficient mice, RT-PCR analysis was used to evaluate the expression of known hypertrophy response genes, including ANF, β-MHC, α-MHC, skeletal α-actin, and cardiac α-actin. The results from this analysis demonstrate that ANF expression is modestly increased in mXinα-deficient mice (Fig. 5). In addition, β-MHC transcript levels are increased specifically in mXinα-null mice, with no change in α-MHC expression. There is also a slight decrease in cardiac α-actin in mXinα-null mice, whereas no difference in skeletal α-actin expression was observed between wild-type and mXinα-deficient mice (Fig. 5). The observed increase in ANF expression levels was further confirmed with quantitative real-time RT-PCR analysis of four sets of age-matched littermates. Compared with wild-type ANF levels (set to a relative fold value of 1) 1.648 ± 0.109-fold and 1.674 ± 0.266-fold increases in expression were observed for heterozygotes and homozygotes, respectively, after normalization with GAPDH levels. Quantitative real-time RT-PCR analysis with primers for other hypertrophy genes studied was also performed but did not result in specific amplification of these genes.
Fig. 5.

RT-PCR analysis of hypertrophy response gene transcript levels. RT-PCR results demonstrate that atrial natriuretic factor (ANF) expression is modestly increased in mXinα-deficient mice. In addition, β-MHC transcript levels are increased in mXinα-null mice, with no change in α-MHC expression. A slight decrease in cardiac α-actin was also observed in mXinα-null mice, whereas there was no difference in skeletal α-actin expression between wild-type and mXinα-deficient mice. As expected, mXinα transcript levels were not detected in the mXinα-null sample, and the levels of control GAPDH expression were not different among samples.
Masson's trichrome stain was used to detect evidence of interstitial fibrosis in the mXinα-deficient mice. Several areas dispersed throughout the mXinα-deficient heart sections stained positive for collagen, indicative of fibrosis (Fig. 6A). In contrast, very little, if any, fibrosis was present in the wild-type heart sections (Fig. 6A, a, d, g). The amount of fibrosis detected in mXinα-deficient mice increased with age from 3 to 14 mo, while very little fibrosis was observed in older wild-type mice (Fig. 6A). Additionally, the tissue morphology appeared disrupted in the older mXinα-deficient mice compared with the older wild-type mice and younger mice of all genotypes. Quantification of fibrosis demonstrated that these differences are statistically significant by one-way ANOVA between wild-type and mXinα−/− mouse hearts at 3, 9, and 14 mo of age (Fig. 6B). A significant difference in fibrosis was also observed between wild-type and mXinα+/− mouse hearts at 14 mo. These results suggest that mXinα-deficient mice are predisposed to hypertrophic cardiomyopathy.
Fig. 6.

mXinα-deficient mouse hearts show increased interstitial fibrosis. A: Masson's trichrome-stained myocardial sections demonstrate a significant increase in interstitial fibrosis in heterozygous (b, e, h) and homozygous (c, f, i) hearts compared with wild-type (a, d, g) hearts. B: the amount of interstitial fibrosis increases with increasing age, as shown in the graphical representation of the quantified data. This increased fibrosis in mXinα-null hearts compared with wild-type hearts was statistically significant at 3 mo (*P = 0.012), 9 mo (#P < 0.001), and 14 mo (+P < 0.001) by ANOVA. The increase in fibrosis in the heterozygote compared with wild-type at 14 mo was also statistically significant (&P < 0.001).
Changes in expression and localization of known intercalated disk proteins in mXinα-deficient hearts
To determine whether any changes in the expression and/or distribution of known intercalated disk proteins occur in mXinα-deficient hearts, Western blot analysis and immunofluorescence microscopy were carried out. Representative Western blot results are shown in Fig. 7A. Quantitation of the Western blots in various exposures from four sets of samples after normalization with α-tubulin indicated that there was no statistically significant change in the expression of α-catenin, plakoglobin, and β1-integrin (Fig. 7B). However, the expression levels of N-cadherin, β-catenin, p120ctn, connexin 43 (both phosphorylated and nonphosphorylated forms, see Fig. 7A), and desmoplakin in mXinα-null hearts were significantly lower than the levels in wild-type hearts by ANOVA. The levels of α-MHC were also significantly decreased in mXinα-deficient hearts. In contrast, GAPDH protein levels in homozygous hearts were 1.5-fold higher than in wild-type hearts. Immunofluorescence microscopy revealed that most known intercalated disk proteins do not significantly change their localization in mXinα-deficient hearts, except p120ctn and connexin 43 (Figs. 8 and 9). The localization of p120ctn changed from more diffuse distribution in the wild-type heart (Fig. 8, G and J) to discrete band/puncta staining in the mXinα-null heart (Fig. 8, I and L). In the wild-type heart, connexin 43 was mainly localized to the intercalated disks at cell termini (arrowheads in Fig. 9D), with few lateral cell-cell connections. In contrast, connexin 43 distributions in the mXinα-null heart showed a significant increase in lateral cell-cell connections (arrows in Fig. 9F). Quantification of this lateral connexin 43 localization demonstrated a significant increase in the number of incidences of laterally associated connexin 43 antigen, as the percentage of lateral connexin 43 increased from 12.3% in the wild-type heart to 17.8% in the mXinα-null heart (P = 0.041). In addition, the relative size/area representing this lateral localization was significantly increased from 11.03% in the wild-type heart to 30.01% in the mXinα-null heart (P = 0.017).
Fig. 7.

Changes in expression of various known intercalated disk proteins in mXinα-deficient mouse hearts. A: representative Western blot analysis comparing protein expression levels between wild-type, heterozygote, and mXinα-null littermates is shown. The connexin 43 blot shown here only represents phosphorylated forms; as shown in Fig. 10, the anti-connexin 43 antibody detected both nonphosphorylated and phosphorylated forms (4 bands in the SDS gel) of connexin 43 from wild-type and mXinα-null hearts. B: quantification of Western blot data from 4 sets of heart samples demonstrates that mXinα, N-cadherin, connexin 43 (both nonphosphorylated and phosphorylated forms), and α-MHC proteins are significantly decreased in mXinα-deficient hearts (*P < 0.05) by ANOVA. Additionally, p120-catenin (p120ctn), β-catenin, and desmoplakin protein levels are significantly decreased in mXinα-null mouse hearts (P < 0.05) by ANOVA analysis (*) or t-test (#). In contrast, GAPDH levels are significantly increased in the mXinα-null (*P = 0.002) samples compared with the wild-type samples.
Fig. 8.

Localization of α-catenin, plakoglobin, and p120ctn in wild-type and mXinα-deficient mouse hearts. Indirect immunofluorescence microscopy was performed on frozen sections of hearts from each genotype with anti-α-catenin (A–C), anti-plakoglobin (D–F), or anti-p120ctn (G–L). The phase-contrast images in J–L correspond to the fluorescent images shown in G–I, respectively. The localization of α-catenin or plakoglobin was not altered in mXinα-deficient hearts, whereas the distribution of p120ctn in mXinα-deficient hearts appears changed from a more diffuse stained pattern to a discrete banding pattern. Bar, 10 μm.
Fig. 9.

Expression and localization of connexin 43 in wild type (mXinα+/+) and mXinα-null (mXinα−/−) mouse hearts. A: Western blot analysis of total protein extract from hearts of 2 sets of wild-type (mXinα +/+) and mXinα-null (−/−) mice exhibited 4 bands, representing the expression of phosphorylated and nonphosphorylated states of connexin 43 antigen (indicated by arrowheads). Connexin 43 expression appears to be decreased for these various phosphorylated states in mXinα-null mice compared with wild-type littermates. B–G: indirect immunofluorescence with anti-connexin 43 antibody performed on wild-type (mXinα+/+) and mXinα-null (mXinα−/−) mouse hearts demonstrated an increase in laterally distributed connexin 43 in the mXinα-null hearts. While the wild-type hearts exhibited the typical connexin 43 localization pattern predominantly at the cell-cell junctions, perpendicular to the longitudinal myocytes (arrowheads in D–G), the mXinα-null hearts showed increased connexin 43 aberrantly localized laterally, parallel to the myocytes (arrows in F and G). Quantification of this observation with ImageJ software demonstrated a significant increase in the number of incidences of lateral connexin 43 localization (B) as well as % of laterally localized connexin 43-stained areas (C) in the mXinα-null heart compared with the wild-type heart (P = 0.041 and 0.017, respectively). Phase-contrast images were used to identify lateral localization (E and G) of connexin 43 in the immunostained sections (D and F). Scale bar, 50 μm.
Abnormal intercalated disk ultrastructure and myofiber disarray in mXinα-null mice
The specific localization of mXinα at the intercalated disk and changes in the expression levels of intercalated disk components prompted a qualitative ultrastructural examination of the intercalated disk region of mXinα-deficient mice compared with wild-type littermates. A clear boundary between the A and I bands of sarcomeres near the intercalated disk is observed in wild-type and heterozygous hearts (Fig. 10, A and B). This boundary is absent in the mXinα-null heart (Fig. 10C); some thick filaments aberrantly extend into the I band at the intercalated disk of the mXinα-null heart (Fig. 10C, arrows). The gap junctions (Fig. 10, A and B) near the adherens junctions are easily identified in wild-type and heterozygous hearts but, while still present, are observed less frequently in mXinα-null hearts. The filamentous web underlying the adherens junction is less dense in mXinα-deficient hearts than in wild-type hearts. While the adjacent faces of the intercalated disks of the wild-type heart are closely and densely associated, the mXinα-deficient intercalated disks are more detached, with greater separation (Fig. 10, A-C).
Fig. 10.

A–C: transmission electron microscopy of intercalated disk structures. Ventricular muscle from wild-type mice (A) at 3 mo of age shows a well-defined boundary between A and I bands of myofibrils near the intercalated disk. Densely stained filamentous webs underneath the adherens junctions and gap junction (GJ) are readily observed. Ventricular muscle from the heterozygous littermate (B) appears to have a similar ultrastructure at the intercalated disk except for a less densely stained web. In contrast, disorganized myofibrils at the adherens junctions were frequently detected in ventricular muscle from the homozygous littermate (C). The thick filaments (indicated by white arrows) often extended to the I band region near the intercalated disk. In addition to less densely stained web, regions of the 2 membranes from adjacent cardiomyocytes seem to be more separated. Bar, 1 μm. D–F: transmission electron microscopy of myofibrils. Cardiac myofibrils from the wild-type mice (D) exhibit well-defined sarcomeres. The Z lines are tight and dense, located at the middle of the I bands. In contrast, cardiac myofibrils at the affected areas from the homozygous littermates (E) show diffused and widened Z lines. There is no clear boundary between A and I bands of sarcomeres. However, at other unaffected areas (F), cardiac myofibrils from the homozygous littermates appear to have organization similar to that seen in the wild-type mice. Bar, 1 μm.
The ultrastructure of the wild-type sarcomeres is highly organized, with distinct I and A bands and a definitive H zone and M lines (Fig. 10D). The Z disks are well aligned, and of a consistent length throughout the heart. In contrast, the I and A bands are not well defined in the severely affected areas of the mXinα-null heart (Fig. 10E). The sarcomeres appear distorted and compacted, as the length from Z line to Z line is decreased compared with the wild-type sarcomeres. The Z lines in mXinα-null hearts are also diffuse and thickened. However, some regions of mXinα-null hearts appear to have a more organized sarcomere structure (Fig. 10F).
ECG analysis reveals prolonged QT intervals in mXinα-deficient hearts
Previous patch-clamp studies revealed that mXinα-deficient mouse cardiomyocytes had abnormal electrophysiological characteristics, including an increase in Na+ inward currents and decreases in L-type Ca2+ currents, transient outward K+ currents, and Ba2+-sensitive inward rectifier K+ currents (3). These together with the observed changes in connexin 43 expression and localization in the mXinα-deficient mouse heart suggest that knockout mouse hearts may also have conduction defects. Therefore, we performed ECG recordings on isolated, perfused hearts (Langendorff preparations) of ∼3- to 4-mo-old mice of each genotype. A representative ECG tracing for each genotype is shown in Fig. 11, and Table 1 summarizes the ECG parameters calculated from tracings of multiple samples of each genotype. The P wave represents depolarization of the atrium, the PQ interval represents atrioventricular conduction, the QRS interval represents ventricular depolarization, and the QT interval represents depolarization and repolarization of the ventricle. mXinα-null mice displayed an increased P wave duration compared with wild-type and heterozygous mice (P < 0.05, ANOVA). Furthermore, mXinα-deficient mice showed significantly prolonged QT intervals but no difference in QRS intervals (Table 1). These data on the prolonged P wave and QT interval suggest a defect in atrial depolarization and ventricular repolarization.
Fig. 11.

ECG tracings of isolated, perfused hearts from wild-type (mXinα+/+) and mXinα-deficient (mXinα+/− and mXinα−/−) mice at 3–4 mo of age. The definition for P wave as well as PQ, QRS, and QT intervals is indicated in the wild-type trace. mXinα-null mouse heart exhibits prolonged P wave and QT interval, with normal QRS interval, suggesting a defect in the conduction of atrium and ventricle, respectively.
Table 1.
Conduction parameters determined by ECG recording of isolated, perfused hearts (Langendorff preparations) from wild-type and mXinα-deficient mice
| mXinα+/+ | mXinα+/− | mXinα−/− | |
|---|---|---|---|
| No. of mice | 11 | 10 | 7 |
| P wave | 15.3±0.4 | 15.5±0.6 | 19.1±0.4* |
| PQ interval | 38.6±1.9 | 40.0±2.7 | 40.6±3.8 |
| QRS interval | 9.0±0.4 | 10.2±0.6 | 9.0±0.4 |
| QT interval | 41.7±1.1 | 54.8±3.2* | 73.2±6.6* |
| R-R interval | 193.6±14.8 | 176.0±10.3 | 235.1±17 |
Values (in ms) are means ± SE. ECG traces were recorded at 37°C.
P < 0.05, significant difference between wild-type (mXinα+/+) and mXinα-deficient (mXinα+/− or mXinα−/−) preparations.
DISCUSSION
mXinα and mXinβ evolve from a single gene, and both have overlapping and unique functions
Although embryonic lethality was expected based on previous antisense knockdown experiments in chicken embryos (33), viable and fertile mXinα-null mice were obtained. The presence of mXinβ in the intercalated disk of the mouse heart, coupled with the upregulation of mXinβ at both the message and protein levels in mXinα-knockout mice, provides a reasonable explanation for viable mXinα-null mice. Previous antisense results did not completely rule out the possibility of a Xin gene family in the chicken (33). However, database homology searches against the complete chicken genome did not support the existence of such a Xin gene family in the chicken. Moreover, amino acid sequence comparisons revealed that cXin has 42.8% and 33.4% identity to mXinα and mXinβ, respectively. Similar percent identity (42.7% and 33.3%) is also obtained when comparing cXin to the human homologs, hXinα and hXinβ. The percent identity between mXinα and mXinβ, or between hXinα and hXinβ, is only 28%. These results are consistent with the notion that mXinα and mXinβ have evolved from a single Xin gene, and that cXin may be evolutionarily closer to mXinα. On the other hand, mXinα, mXinβ, and cXin all contain the Xin repeat, which is now known as a novel class of actin-binding domain (4, 16, 23). Interestingly, there are 27 Xin repeats in cXin, which is closer to the 28 repeats present in mXinβ than to the 15 repeats in mXinα. Together, these results support a single Xin gene in the chicken and further imply that mXinα and mXinβ may have evolved to perform distinct functions in cardiomyocytes. However, the fact that both mXinα and mXinβ have similar domain structures, similar tissue expression pattern, and similar intracellular localization (16, 17) suggests that they may also play overlapping roles in cardiac development and function. The upregulation of mXinβ in mXinα-null mice further supports this overlapping role, and argues for compensation by mXinβ in the absence of mXinα.
mXin messages and proteins preferentially localize to intercalated disks
We demonstrated that the mXinβ messages were preferentially localized to what appeared to be the intercalated disks of wild-type and mXinα-deficient hearts. The majority of mXinα messages were also found at this specific structure (data not shown). In contrast, intercalated disk localization was not observed for cardiac troponin T messages (data not shown). The localization of mXin mRNAs to the intercalated disk, where the protein is utilized, may provide an efficient way for mXin localization, given the fact that mXin is also capable of binding to actin filaments. In another study, we have shown (9) that mXinα directly binds to β-catenin, and that aa 535–636 of mXinα, located within the Xin repeats, are both necessary and sufficient for this interaction. The overlap of the β-catenin-binding domain with the actin-binding domain on mXinα may provide another way for ensuring mXinα localization exclusively to the intercalated disk and not to the thin filaments.
mXinα-deficient mice represent a novel model of cardiomyopathy with conduction defect
In this study, we have shown that a lack of mXinα in the heart results in abnormal intercalated disk ultrastructure, eventually leading to myofibril disarray, fibrosis, cardiac hypertrophy, and cardiomyopathy. Despite these findings, the intercalated disk structure appears relatively intact at the light microscopy level. Thus this animal model of cardiomyopathy differs from that derived from the N-cadherin condition knockout mouse (12), in which a complete dissolution of the intercalated disk structure was observed. In mXinα-knockout mouse hearts, decreased expression levels of p120ctn, β-catenin, N-cadherin, and desmoplakin were detected, suggesting that perturbations of the adherens junctions and desmosomes are present. This is in contrast to other mouse models for dilated cardiomyopathy such as the muscle LIM protein (MLP)-knockout mouse (2) and the tropomodulin-overexpressing transgenic (TOT) mouse (31). Both MLP−/− and TOT mice show significant upregulation of adherens junction components but not desmosomal components (6, 24). In addition to decreases in the expression of N-cadherin, β-catenin, p120ctn, and desmoplakin, mXinα-knockout mouse hearts consistently show not only a significant decrease in the expression of connexin 43 but also an alteration in its localization. These data are similar to previous results first demonstrating abnormal localization of desmosomal and gap junctional components in human hypertrophic cardiomyopathy hearts, where the authors suggest that this remodeled gap junction localization may contribute to the cardiac arrhythmias associated with hypertrophic cardiomyopathy (28). Indeed, ECG analyses of isolated, perfused hearts show that mXinα-deficient mice have a significantly prolonged QT interval but no change in QRS interval, providing evidence for a conduction defect. In a patch-clamp study of ventricular myocytes prepared from wild-type and mXinα-deficient mice, we have observed significant reductions in transient outward K+ currents (Ito) and Ba2+-sensitive inward rectifier K+ currents (Ik1) in mXinα−/− myocytes (3), which are important components for repolarizing cardiomyocytes. An optical mapping technique has been used to compare the conduction velocity on the front surface of ventricles of wild-type and mXinα-null hearts with the MED64 recording system (MED-P515A, Panasonic). The results revealed that the conduction velocity in mXinα-null ventricles was significantly slower than that in wild-type ventricles (14). This slower conduction velocity together with the depressed Ito and Ik1 detected in ventricular myocytes is consistent with a prolonged QT period in mXinα-null ECG. Although the mechanism causing a wider P wave in mXinα-null ECG remains unclear, MED64 recording studies on left atrial-pulmonary vein myocardium preparations and connexin 40 studies on its expression and localization may provide evidence to support a wider P wave in the mXinα-null heart. Together, these data demonstrate that mXinα-knockout mice represent a novel model for the study of cardiomyopathy with conduction defects.
Molecular mechanisms underling cardiomyopathy in mXinα-null mice
In response to abnormal external or internal load/agonists, the adult heart can undergo hypertrophic growth, which often progresses to dilated cardiomyopathy, or can alternatively undergo dilation directly. This pathological hypertrophy reactivates many fetal cardiac genes and represses their corresponding adult counterparts. For example, in the adult mouse heart, cardiomyopathy and pressure overload increase embryonic β-MHC and decrease adult α-MHC (32). This isoform transition correlates well with the observed decrease in cardiac contractility. Consistent with such general gene expression changes associated with cardiac hypertrophy, mXinα-deficient hearts exhibit a significant reduction in α-MHC protein as well as cardiac α-actin but not skeletal α-actin message levels. However, the degree of increased α-skeletal actin expression has been shown to be directly related to the severity of the cardiac hypertrophy phenotype, with definite changes only observed when the cardiac mass was increased by >20% (30). Additionally, increased ANF and β-MHC messages were observed, consistent with the hypertrophic phenotype. In contrast, GAPDH, a major enzyme in the glycolytic pathway, was found to be increased in mXinα-deficient hearts compared with wild-type hearts, although the GAPDH message level did not change significantly. This is also consistent with the fact that hypertrophic hearts generally exhibit a transition from an oxidative to a more anaerobic metabolism, such as glycolysis, which involves GAPDH (22).
In the present study, we have shown that cardiac hypertrophy in mXinα−/− mice begins with abnormal intercalated disk ultrastructure as early as 3 mo of age. This structural alteration is accompanied by a significant decrease in the expression of N-cadherin, β-catenin, and p120ctn, suggesting that the hypertrophy that results in mice around 9–11 mo may be due to the impaired organization of the intercalated disk and instability of adhesions. In concert with this suggestion, mXinα has been shown to interact directly with β-catenin and actin filaments (9, 16). Through these interactions, mXinα may orchestrate the structural integrity of the N-cadherin cellular interface and its connection to the actin cytoskeleton.
Supplementary Material
ACKNOWLEDGMENTS
We thank Qinchuan Wang and Shaun Grosskurth for providing RT product and advising on RT-PCR experiments.
GRANTS
This work has been supported by National Heart, Lung, and Blood Institute Grant HL-075015 to J. J.-C. Lin and Grant NSC95-2320-B016-013 to C.-I. Lin from the National Science Council, Taipei, Taiwan, Republic of China. H. W. Sinn was supported by a predoctoral fellowship from the American Heart Association, Heartland Affiliate.
Footnotes
The online version of this article contains supplemental material.
REFERENCES
- 1.Alcalai R, Metzger S, Rosenheck S, Meiner V, Chajek-Shaul T. A recessive mutation in desmoplakin causes arrhythmogenic right ventricular dysplasia, skin disorder, and woolly hair. J Am Coll Cardiol. 2003;42:319–327. doi: 10.1016/s0735-1097(03)00628-4. [DOI] [PubMed] [Google Scholar]
- 2.Arber S, Hunter JJ, Ross JJ, Hongo M, Sansig G, Borg J, Perriard JC, Chien KR, Caroni P. MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy and heart failure. Cell. 1997;88:393–403. doi: 10.1016/s0092-8674(00)81878-4. [DOI] [PubMed] [Google Scholar]
- 3.Cheng CP, Loh YX, Lin CI, Lai YJ, Chen YC, Sytwu HK, Gustafson-Wagner EA, Lin JJC. Electrophysiological characteristics of ventricular myocytes of Xinα-deficient mice. In: Bologna Kimchi A., editor. International Proceedings: Advances in Heart Disease. MEDIMOND; Italy: 2005. pp. 25–29. [Google Scholar]
- 4.Cherepanova O, Orlova A, Galkin VE, van der Ven PF, Furst DO, Jin JP, Egelman EH. Xin-repeats and nebulin-like repeats bind to F-actin in a similar manner. J Mol Biol. 2006;356:714–723. doi: 10.1016/j.jmb.2005.11.082. [DOI] [PubMed] [Google Scholar]
- 5.Dupont E, Matsushita T, Kaba R, Vozzi C, Coppen SR, Khan N, Kaprielian R, Yacoub MH, Severs NJ. Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol. 2001;33:359–371. doi: 10.1006/jmcc.2000.1308. [DOI] [PubMed] [Google Scholar]
- 6.Ehler E, Horowits R, Zuppinger C, Price RL, Perriard E, Leu M, Caroni P, Sussman M, Eppenberger HM, Perriard JC. Alterations at the intercalated disk associated with the absence of muscle LIM protein. J Cell Biol. 2001;153:763–772. doi: 10.1083/jcb.153.4.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferreira-Cornwell MC, Luo Y, Narula N, Lenox JM, Lieberman M, Radice GL. Remodeling the intercalated disc leads to cardiomyopathy in mice misexpressing cadherins in the heart. J Cell Sci. 2002;115:1623–1634. doi: 10.1242/jcs.115.8.1623. [DOI] [PubMed] [Google Scholar]
- 8.Fujio Y, Yamada-Honda F, Sato N, Funai H, Wada A, Awata N, Shibata N. Disruption of cell-cell adhesion in an inbred strain of hereditary cardiomyopathic hamster (Bio 14.6) Cardiovasc Res. 1995;30:899–904. [PubMed] [Google Scholar]
- 9.Gustafson-Wagner E, Lin JLC, Sinn H, Wang DZ, Reiter RS, Yang B, Williamson R, Chen J, Jaacks S, Lin JJC. Targeted deletion of mXinα gene, encoding an intercalated disc protein, leads to cardiac hypertrophy (Abstract) Circulation. 2006;114:II-54. [Google Scholar]
- 10.Jin JP, Malik ML, Lin JJC. Monoclonal antibodies against cardiac myosin heavy chain. Hybridoma. 1990;9:597–608. doi: 10.1089/hyb.1990.9.597. [DOI] [PubMed] [Google Scholar]
- 11.Kirchhoff S, Nelles E, Hagendorff A, Kruger O, Traub O, Willecke K. Reduced cardiac conduction velocity and predisposition to arrhythmias in connexin 40-deficient mice. Curr Biol. 1998;8:299–302. doi: 10.1016/s0960-9822(98)70114-9. [DOI] [PubMed] [Google Scholar]
- 12.Kostetskii I, Li J, Xiong Y, Zhou R, Ferrari VA, Patel VV, Molkentin JD, Radice GL. Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ Res. 2005;96:346–354. doi: 10.1161/01.RES.0000156274.72390.2c. [DOI] [PubMed] [Google Scholar]
- 13.Kostin S, Klein G, Szalay Z, Hein S, Bauer EP, Schaper J. Structural correlate of atrial fibrillation in human patients. Cardiovasc Res. 2002;54:361–379. doi: 10.1016/s0008-6363(02)00273-0. [DOI] [PubMed] [Google Scholar]
- 14.Lai YJ, Chen YY, Cheng CP, Lin JJC, Chudorodova SL, Roshchevskaya IM, Roshchevsky MP, Chen YC, Lin CI. Changes in ionic currents and reduced conduction velocity in hypertrophied ventricular myocardium of Xinα-deficient mice. Anadolu Kardiyol Derg. 2007;7(Suppl 1):90–92. [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Patel VV, Kostetskii I, Xiong Y, Chu AF, Jacobson JT, Yu C, Morley GE, Molkentin JD, Radice GL. Cardiac-specific loss of N-cadherin leads to alteration in connexins with conduction slowing and arrhythmogenesis. Circ Res. 2005;97:474–481. doi: 10.1161/01.RES.0000181132.11393.18. [DOI] [PubMed] [Google Scholar]
- 16.Lin JJC, Gustafson-Wagner EA, Sinn HW, Choi S, Jaacks SM, Wang DZ, Evans SM, Lin JLC. Structure, expression, and function of a novel intercalated disc protein, Xin. J Med Sci. 2005;25:215–222. doi: 10.1901/jaba.2005.25-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin JJC, Wang DZ, Reiter RS, Wang Q, Lin JLC, Williams HS. Differentially expressed genes and cardiac morphogenesis. In: Tomanek RJ, Runyan R, editors. Formation of the Heart and Its Regulation. Birkhauser; Boston, MA: 2001. pp. 75–96. [Google Scholar]
- 18.Luo Y, Ferreira-Cornwell M, Baldwin H, Kostetskii I, Lenox JM, Lieberman M, Radice G. Rescuing the N-cadherin knockout by cardiac-specific expression of N- or E-cadherin. Development. 2001;128:459–469. doi: 10.1242/dev.128.4.459. [DOI] [PubMed] [Google Scholar]
- 19.McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–2124. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- 20.Neely JR, Liebermeister H, Battersby EJ, Morgan HE. Effect of pressure development on oxygen consumption by isolated rat heart. Am J Physiol. 1967;212:804–814. doi: 10.1152/ajplegacy.1967.212.4.804. [DOI] [PubMed] [Google Scholar]
- 21.Ng WA, Grupp IL, Subramaniam A, Robbins J. Cardiac myosin heavy chain mRNA expression and myocardial function in the mouse heart. Circ Res. 1991;69:1742–1750. doi: 10.1161/01.res.68.6.1742. [DOI] [PubMed] [Google Scholar]
- 22.Olson EN, Schneider MD. Sizing up the heart: development redux in disease. Genes Dev. 2003;17:1937–1956. doi: 10.1101/gad.1110103. [DOI] [PubMed] [Google Scholar]
- 23.Pacholsky D, Vakeel P, Himmel M, Lowe T, Stradal T, Rottner K, Furst DO, van der Ven PFM. Xin repeats define a novel actin-binding motif. J Cell Sci. 2004;117:5257–5268. doi: 10.1242/jcs.01406. [DOI] [PubMed] [Google Scholar]
- 24.Perriard JC, Hirschy A, Ehler E. Dilated cardiomyopathy: a disease of the intercalated disc? Trends Cardiovasc Med. 2003;13:30–38. doi: 10.1016/s1050-1738(02)00209-8. [DOI] [PubMed] [Google Scholar]
- 25.Peters NS, Severs NJ, Rothery SM, Lincoln C, Yacoub MH, Green CR. Spatiotemporal relation between gap junctions and fascia adherens junctions during postnatal development of human ventricular myocardium. Circulation. 1994;90:713–725. doi: 10.1161/01.cir.90.2.713. [DOI] [PubMed] [Google Scholar]
- 26.Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, Frigo G, Vettori A, Valente M, Towbin J, Thiene G, Danieli GA, Rampazzo A. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:1171–1179. doi: 10.1161/CIRCULATIONAHA.105.583674. [DOI] [PubMed] [Google Scholar]
- 27.Saffitz JE, Green KG, Kraft WJ, Schechtman KB, Yamada KA. Effects of diminished expression of connexin43 on gap junction number and size in ventricular myocardium. Am J Physiol Heart Circ Physiol. 2000;278:H1662–H1670. doi: 10.1152/ajpheart.2000.278.5.H1662. [DOI] [PubMed] [Google Scholar]
- 28.Sepp R, Severs NJ, Gourdie RG. Altered patterns of cardiac intercellular junction distribution in hypertrophic cardiomyopathy. Heart. 1996;76:412–417. doi: 10.1136/hrt.76.5.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sinn HW, Balsamo J, Lilien J, Lin JJC. Localization of the novel Xin protein to the adherens junction complex in cardiac and skeletal muscle during development. Dev Dyn. 2002;225:1–13. doi: 10.1002/dvdy.10131. [DOI] [PubMed] [Google Scholar]
- 30.Stilli D, Bocchi L, Berni R, Zaniboni M, Cacciani F, Chaponnier C, Musso E, Gabbiani G, Clement S. Correlation of alpha-skeletal actin expression, ventricular fibrosis and heart function with the degree of pressure overload cardiac hypertrophy in rats. Exp Physiol. 2006;91:571–580. doi: 10.1113/expphysiol.2005.032607. [DOI] [PubMed] [Google Scholar]
- 31.Sussman MA, Baque S, Uhm CS, Daniels MP, Price RL, Simpson DG, Terracio L, Kedes L. Altered expression of tropomodulin in cardiomyocytes disrupts the sarcomeric structure of myofibrils. Circ Res. 1998;82:94–105. doi: 10.1161/01.res.82.1.94. [DOI] [PubMed] [Google Scholar]
- 32.Swynghedauw B. Developmental and functional adaptation of contractile proteins in cardiac and skeletal muscles. Physiol Rev. 1986;66:710–730. doi: 10.1152/physrev.1986.66.3.710. [DOI] [PubMed] [Google Scholar]
- 33.Wang DZ, Reiter RS, Lin JLC, Wang Q, Williams HS, Krob SL, Schultheiss TM, Evans S, Lin JJC. Requirement of a novel gene, Xin, in cardiac morphogenesis. Development. 1999;126:1281–1294. doi: 10.1242/dev.126.6.1281. [DOI] [PubMed] [Google Scholar]
- 34.Wang Q, Sigmund CD, Lin JJC. Identification of cis elements in the cardiac troponin T gene conferring specific expression in cardiac muscle of transgenic mice. Circ Res. 2000;86:478–484. doi: 10.1161/01.res.86.4.478. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Gerdes AM. Chronic pressure overload cardiac hypertrophy and failure in guinea pigs. III. Intercalated disc remodeling. J Mol Cell Cardiol. 1999;31:333–343. doi: 10.1006/jmcc.1998.0886. [DOI] [PubMed] [Google Scholar]
- 36.Wang Z, Wang DZ, Pipes GC, Olson EN. Myocardin is a master regulator of smooth muscle gene expression. Proc Natl Acad Sci USA. 2003;100:7129–7134. doi: 10.1073/pnas.1232341100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Warren KS, Lin JLC, McDermott JP, Lin JJC. Forced expression of chimeric human fibroblast tropomyosin mutants affects cytokinesis. J Cell Biol. 1995;129:697–708. doi: 10.1083/jcb.129.3.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xing W, Zhang TC, Cao D, Wang Z, Antos CL, Li S, Wang Y, Olson EN, Wang DZ. Myocardin induces cardiomyocyte hypertrophy. Circ Res. 2006;98:1089–1097. doi: 10.1161/01.RES.0000218781.23144.3e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.