

Summary
Background
In many animals, the epidermis is in permanent contact with the environment and represents a first line of defense against pathogens and injury. Infection of the nematode Caenorhabditis elegans by the natural fungal pathogen Drechmeria coniospora induces the expression in the epidermis of antimicrobial peptide (AMP) genes such as nlp-29. Here, we tested the hypothesis that injury might also alter AMP gene expression and sought to characterize the mechanisms that regulate the innate immune response.
Results
Injury induces a wound-healing response in C. elegans that includes induction of nlp-29 in the epidermis. We find that a conserved p38-MAP kinase cascade is required in the epidermis for the response to both infection and wounding. Through a forward genetic screen, we isolated mutants that failed to induce nlp-29 expression after D. coniospora infection. We identify a kinase, NIPI-3, related to human Tribbles homolog 1, that is likely to act upstream of the MAP2K SEK-1. We find NIPI-3 is required only for nlp-29 induction following infection and not following wounding.
Conclusions
Our results show that the C. elegans epidermis actively responds to wounding and infection via distinct pathways that converge on a conserved signaling cassette that controls the expression of the AMP gene nlp-29. A comparison between these results and MAP kinase signaling in yeast gives insights into the possible origin and evolution of innate immunity.
Introduction
The nematode C. elegans responds to bacterial infection by up-regulating the expression of genes encoding antimicrobial proteins [1–5]. Although it is not known how infection is recognized in C. elegans, several conserved signaling pathways are involved in controlling this innate immune response [6]. Among them, a p38 mitogen activated kinase (MAPK) pathway, involving the MAPK PMK-1, the MAPK kinase (MAPKK) SEK-1 and the MAPKK kinase (MAPKKK) NSY-1 is important for the resistance of C. elegans to infection by the Gram-negative bacterium Pseudomonas aeruginosa [7]. This pathway lies downstream of the TIR domain adapter TIR-1, ortholog of the human protein SARM [8]. In addition to a role in host defense, the TIR-1/NSY-1/SEK-1 cassette is also important for neuronal development. Interestingly, while the cassette acts downstream of the calcium/calmodulin-dependent kinase UNC-43 during development [9], unc-43 is not required for resistance to P. aeruginosa [7].
In contrast to our current understanding of the response of C. elegans to bacterial infection, we know little about its anti-fungal innate immune defenses. One model pathogen is the nematode-specific fungus Drechmeria coniospora. Conidia of D. coniospora attach via adhesive knobs to the nematode cuticle and then form appressorial structures from which penetration tubes emerge. These pierce the cuticle and develop into trophic hyphae that traverse the epidermis, eventually colonizing the entire worm [10]. This natural fungal infection leads to the induction of a number of genes that encode conserved glycine- and tyrosine-rich antimicrobial peptides (AMPs) annotated as neuropeptide-like proteins (NLPs). We showed previously that the induction of two of these, nlp-29 and nlp-31 requires tir-1 [11]. How tir-1 is activated by fungal infection is unknown and it is unclear whether up-regulation of nlp gene expression is a consequence of pathogen recognition or part of a response to cellular damage associated with infection.
In the current study, we discovered that the C. elegans epidermis is capable of responding to physical injury. This response includes a wound healing mechanism and induction of nlp-29 and nlp-31 expression. We show that the up-regulation of AMP gene expression upon infection can be genetically separated from that upon injury, but that both responses converge on a common p38 MAPK pathway. Our results give insights into the possible origins and evolution of innate immunity.
Results
An in vivo reporter system for monitoring innate immune responses
To begin dissecting the innate immune response of C. elegans to fungal infection, we generated a strain carrying an integrated transgene (frIs7) with two constructs, the pnlp-29::GFP green fluorescent reporter that is induced upon infection in the epidermis [11], and a pcol-12::dsRed red fluorescent reporter that is expressed constitutively in the epidermis, starting from the late L1 stage. The latter provides an internal control for the functional integrity of the epidermis and non-specific transgene silencing (Figure 1A,B). The analysis of large populations with the COPAS Biosort™ revealed a continuous distribution of fluorescence levels (Figure 1 C,D). While in a typical infection, the mean level of red fluorescence varied by less than 15% between non-infected and infected populations, for green fluorescence there was between a 3- and 8-fold increase (Figure 1C). Within a population, the green/red fluorescence ratio often spanned more than an order of magnitude, with a standard deviation exceeding 150% of the average in some cases. Even if the distribution is not Gaussian, the mean value for the fluorescence ratio is close to the median (Figure 1D) and therefore can be taken as a representative measure of reporter gene expression within the population. As discussed below, the individual variability in reporter gene expression, is no different from that measured for many quantitative phenotypes.
Figure 1. The pnlp-29::GFP reporter is induced by Drechmeria coniospora.

Control wt;frIs7 worms (A) and worms 24 h after infection (B). Red and green fluorescence is visualized simultaneously. C Quantification of fluorescence of infected (yellow) and non-infected (blue) worms. The mean values for green and red fluorescence (in arbitrary, but constant units for each colour) for the uninfected and infected populations were 59 and 287 (green) and 331.5 and 295.0 (red), respectively. D Continuous distribution of fluorescence levels in the population. The analysis of large numbers of individuals revealed a continuous distribution of fluorescence levels, with the median value for the fluorescence ratio (green/red) being close to the mean (triangles). Although there was an extremely broad range of values for individuals (from 4 to 589, and 11 to 700 in arbitrary units for 1369 non-infected and 1232 infected worms, respectively, in this experiment), leading to high standard deviations (157% and 54% of the mean values, respectively; shown as error bars with the mean at the median position), and noise (variance/mean2, 2.4 and 0.3, respectively). Due to the nature of the distribution, standard deviations are not an informative parameter and are not shown on subsequent figures using the Biosort.
Physical injury induces nlp-29 expression
Infection of C. elegans by D. coniospora involves a breach of the cuticle and disrupts epidermal cell integrity [10]. We therefore investigated whether physical wounding of the cuticle and epidermis was sufficient to induce innate immune responses within the epidermis. Microinjection is a routine method to produce transgenic nematodes. Typically, an individual worm, immobilized on an agar pad, is pierced with a fine needle controlled by a micromanipulator. While large breaches in the epidermis cause an eversion of the internal tissues, when correctly performed, this procedure leaves no obvious mark (Suppl. Figure 1A). We found that wounding the epidermis in this way resulted in characteristic autofluorescent scars that formed rapidly and were visible for several days at the wound site. When we pricked the epidermis of adult animals on agar plates, by hand using microinjection needles, often no obvious damage was seen, although sometimes, a small amount of internal material leaked out. In both cases, wounding the epidermis provoked similar autofluorescent scars (Figures 2A–B & 3D–G).
Figure 2. Needle and laser wounding of the epidermis cause scar formation and local cuticle synthesis.
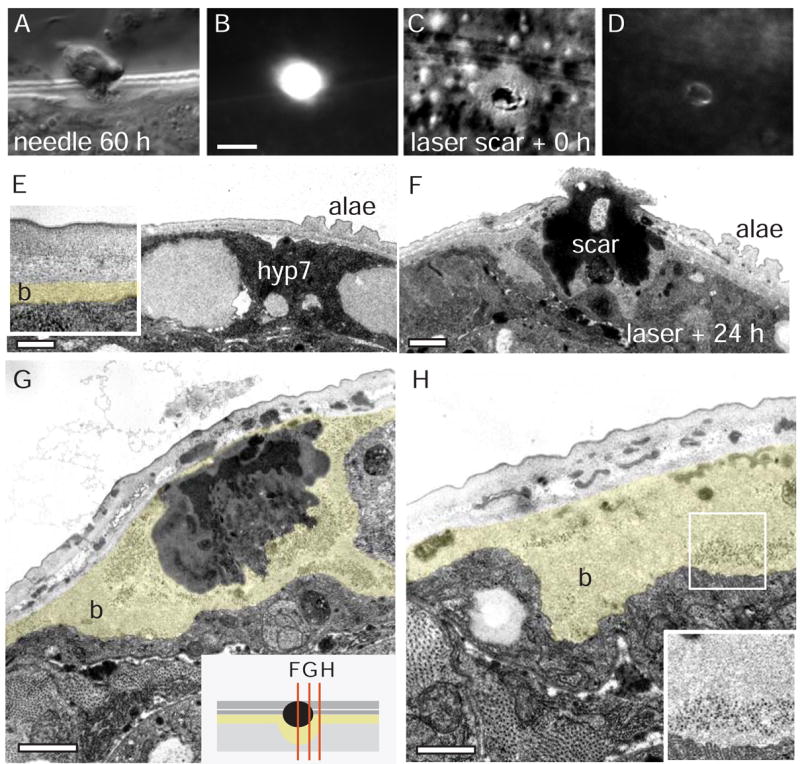
A–D Epidermal scars in wild type (N2) 60 h after needle wound (A,B) and immediately after femtosecond laser wound (C,D); DIC image (panels A, C) and epifluorescence (GFP long pass filter, panels B,D). The autofluorescent material accumulates at the wound immediately (see Suppl. Movies). Scale 10 μm. E–H Ultrastructural analysis of scar and cuticle synthesis at epidermal wounds. The lateral epidermis just ventral to the alae was damaged internally by aiming for the PLM axonal process (in the zdIs5 strain) and cutting it with MHz femtosecond laser. F–H wounded and unwounded (E) sides of the same animal 24 h post wounding. The innermost (basal) layer of cuticle (b) is immediately adjacent to the epidermis hyp7 and is approximately 100 nm thick in unwounded epidermis (b, coloured in inset, E). Surrounding the electron dense material of the scar, the basal layer is up to 20 times thicker than in unwounded cuticle, and contains ribosome-sized electron dense particles (inset, H). Images are all from one animal, sections in F, G, and H are 1.3 μm apart (schematic in inset, G). Scale bar 2 μm.
Figure 3. Epidermal damage induce pnlp-29::GFP.
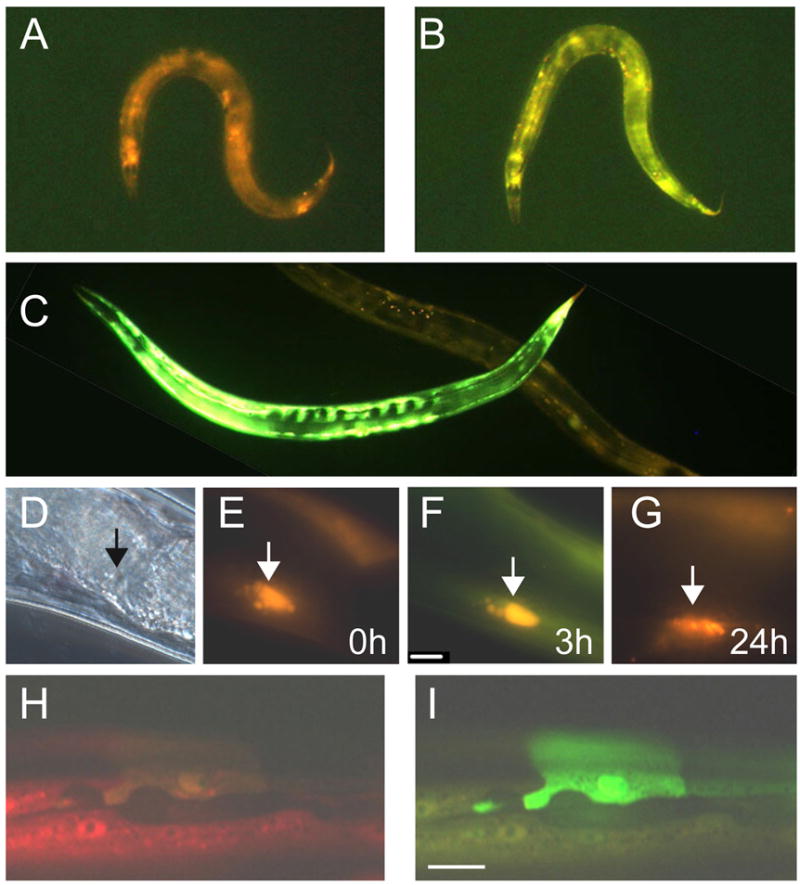
An individual wt;frIs7 worm before (A) and 2 h after needle wounding (B). C An individual that has been wounded with a laser 5 h previously is lying above a mock-wounded worm. D–G An individual worm viewed by DIC (D) or epifluorescence (E) microscopy just after needle wounding. The arrowhead marks the wound site. The same worm 3 hours (F) or 24 hours (G) after wounding. H & I Part of the epidermis of an eff-1(hy21);frIs7 worm. One unfused hyp7 cell shows strong GFP fluorescence (I). With the exception of D (DIC) and H (epifluorescence using a red filter to visualize pcol-12:DsRed only), in all images, red and green fluorescence are visualized simultaneously.
We examined the survival of wounded worms under normal culture conditions. Although some 10% of the wounded worms died within two days after being injured, the remaining worms died at a rate that paralleled that of uninjured worms (Suppl. Figure 1B). The wounded worms also moved normally and showed no other outward signs of trauma. This suggests that wound healing efficiently restores normal physiology.
To cause more precise damage of epidermal cells, we used femtosecond laser pulses focused on the apical surface of the epidermis to create small wounds (Suppl. movie 1). Such laser wounding generally did not cause leakage of internal cytoplasm or fluid, but nevertheless resulted in scars 2–5 μm in diameter (Figure 2C), smaller but otherwise similar to those seen after needle wounding. Laser wound sites accumulated autofluorescent material within seconds of damage (Figure 2D; Suppl. movie 2).
Through ultrastructural analysis of laser wounds, we found that a continuous cuticle layer was re-established within 24 h of damage, suggesting that laser wounding causes a disruption of the epidermis and cuticle that is later repaired. At wound sites, we consistently observed thickening of basal layers of cuticle (Figure 2E–H) suggesting that wounds trigger local secretion of basal cuticle components as part of the barrier repair process.
Both needle and laser wounds induced pnlp-29::GFP expression with a similar time-course. GFP fluorescence was detected within 1 hour of injury, sustained for several hours and then declined (Figure 3A–G). While most experiments were conducted under standard culture conditions in the presence of bacteria, similar results were obtained when we wounded worms on bacteria-free plates (Suppl. Figure 1C). To our knowledge, these are the first indications that C. elegans is able to respond transcriptionally to physical injury.
Defects in the epidermis induce nlp-29 expression
We wondered whether mutants with a defect in the cuticle or epidermis would also show a changed level of pnlp-29::GFP expression. Worms carrying a dominant mutation in the cuticle collagen gene rol-6 exhibited a strong constitutive expression of green fluorescence (results not shown). In C. elegans, almost all of the epidermis is a single multinucleate syncytium called hyp7. When we looked at pnlp-29::GFP expression in eff-1 mutant animals that exhibit abnormal hyp7 cell fusions [12, 13], we also saw a high level of GFP expression, often most intense in non-fused cells (Figure 3H,I). Thus developmental defects and tissue damage lead to the up-regulation of AMP expression.
A conserved p38 MAPK pathway controls AMP gene expression
We have previously shown that infection-induced expression of pnlp-29::GFP is severely compromised upon RNAi of tir-1 [11]. By performing RNAi on the different tir-1 isoforms (Suppl. Figure 2), we established that tir-1b is required for the infection-induced expression of nlp-29, as well as for the response of C. elegans to wounding (Figure 4A). Accordingly, in mutant worms homozygous for tir-1 loss-of-function alleles which affect only the tir-1a, c and e isoforms, but not tir-1b or d, we observed no change in constitutive or induced expression of pnlp-29::GFP, whereas in the tir-1(tm3036) mutant, which affects all isoforms, induction of pnlp-29::GFP was blocked (Figure 4B–C, Suppl. Figure 2). tir-1 acts upstream of the nsy-1/sek-1/pmk-1 p38 pathway to control resistance to the intestinal pathogen P. aeruginosa [8]. We therefore assayed the effect of abrogation of these genes on the expression of pnlp-29::GFP. In nsy-1, sek-1 or pmk-1 mutants there was a marked decrease in pnlp-29::GFP expression following both infection and wounding (Figure 4C–E, results not shown).
Figure 4. nlp-29 induction after infection and injury is dependent on the TIR-1B/p38 pathway.

Quantification with the Biosort of the normalized fluorescence ratio of worms carrying the frIs7 transgene. A RNAi was used to target the different tir-1 isoforms, and fluorescence measured 24 h post-infection (yellow) or 6 h after wounding (green) and compared to control worms (blue). B Mutants affecting the long tir-1 isoforms (ok1052) and (gk264) have an essentially wild type phenotype. C–F Different mutants were analyzed after infection and wounding: sek-1(ag1), sek-1(km4), nsy-1(ky397), pmk-1(km25), mek-1(ks54), jkk-1(km2), unc-16(ju146) and unc-43(n498n1186). G Over-expression of sek-1 in the epidermis, under the control of the col-12 promoter provokes high fluorescence ratios in sek-1 mutants. The number of worms in each sample is given in parentheses.
During neuronal development, the tir-1/nsy-1/sek-1 cascade acts genetically downstream of the CaMK II unc-43 [9, 14]. We found that pnlp-29::GFP expression and induction after infection was unaltered in an unc-43 loss-of-function mutant (Figure 4F). Thus, just as it is not required for the response to P. aeruginosa infection [7], unc-43 appears not to be involved in the response to D. coniospora. Similarly, abrogation of unc-16, which encodes a scaffold protein that interacts with SEK-1 [15], had no effect on constitutive or infection-induced expression of pnlp-29::GFP. Further, jkk-1, kgb-1 or jnk-1 that are elements of JNK MAPK cascades that play an important role in the response of C. elegans to stress [16, 17] were found to be dispensable for pnlp-29::GFP expression (Figure 4F and data not shown). In a mek-1 mutant background, there was a partial reduction in the induction of pnlp-29::GFP (Figure 4F). Therefore, mek-1, which encodes a MKK7 homolog, may have a role in the activation of pmk-1, as previously described in the context of P. aeruginosa infection [16]. The predominant pathway controlling pnlp-29::GFP expression does, however, involve the NSY-1/SEK-1/PMK-1 cassette.
The p38 MAPK pathway acts cell-autonomously in the epidermis
The p38 pathway acts cell-autonomously during neuronal development [18] and in the adult intestine to protect C. elegans from oxidative stress [19]. We tested whether the p38 pathway is required in the epidermis to control AMP gene induction. Expressing sek-1 in the epidermis under the control of the col-12 promoter was more than sufficient to restore pnlp-29::GFP expression in a sek-1 mutant (Figure 4G). When the transgene was transferred to a wild-type background, the transgenic animals had an increased constitutive and induced fluorescence, while in a pmk-1 background, there was essentially no observable fluorescence (Figure 6F). These results strongly suggest that SEK-1 and by extension the entire p38 pathway acts cell-autonomously in the epidermis to control AMP expression.
Figure 6.

A–D Fluorescence images of pnipi-3::GFP transgenic worms showing expression in the intestine, head neurons and motoneurons in a L1 larva (A), in the epidermis in a 3-fold embryo (B). In the adult head (C) and tail (D), expression is seen in the epidermis (arrow) and intestine (arrowhead), as well as a subset of neurons. Scale bar 10 μm. E, F Quantification with the Biosort of the normalized fluorescence ratio of worms carrying the frIs7 transgene following infection (yellow) compared to controls (blue). wt and nipi-3(fr4) mutants, with or without the rescuing transgene containing nipi-3 under its own promoter (E). wt, nipi-3(fr4) and pmk-1 mutants, with or without the transgene containing sek-1 under the control of the col-12 promoter (F). G–N Images of uninfected (G, H, I, J) or infected (K, L, M, N) worms carrying the frIs7 transgene in the wt (G, K) and nipi-3 background (H–L), and in some cases a second transgene driving expression of nipi-3 in the epidermis (I, M) or intestine (J, N). The worms shown in (M) are representative only of the rescued worms (see Suppl. Figure 6).
A genetic screen for genes controlling nlp-29 expression
We next screened for mutants that had a normal level of pcol-12::dsRed expression but that were unable to activate pnlp-29::GFP expression after infection (see Experimental Procedures). From an initial screen of 10,000 haploid genomes, we recovered six Nipi (No Induction of Peptide after Drechmeria Infection) mutants. The six nipi mutations are recessive and were assigned to five complementation groups. Of the five, four affected pnlp-29::GFP expression following both infection and wounding (Figure 5A; NP, unpublished data). The mutants nipi-1(fr1 and fr3) LGIV, nipi-2(fr2) LGX and nipi-3(fr4) LGX showed the most penetrant phenotypes. Complementation tests with available mutants suggested that these 3 nipi mutants do not correspond to known components of the C. elegans p38 MAPK pathway.
Figure 5. nipi-3 encodes a kinase required for the induction of nlp-29 and nlp-31 after infection but not after wounding A.

Quantification with the Biosort of the normalized fluorescence ratio of transgenic worms carrying the frIs7 transgene 24 h post-infection (yellow; left panel) or 6 h after wounding (green; right panels) in wt, nipi-2(fr2) and nipi-3(fr4) mutant worms compared to controls (blue). B Quantification by qRT-PCR of nlp-29 and nlp-31 mRNA in wild-type and nipi-3 mutant worms after infection (yellow and orange, respectively) or wounding (green and lime, respectively). C Quantification with the Biosort of the normalized fluorescence ratio of wt;frIs7 worms 24 h post-infection (yellow) or 6 h after wounding (green) compared to controls (blue), following mock RNAi or RNAi of nipi-3. D Structure of the nipi-3 locus. The location of the fr4 mutation is shown relative to the exon/intron structure of nipi-3; the red shading in 4 exons represents the kinase domain. Grey boxes represent the predicted 5′ and 3′-most exons of the neighbouring upstream and downstream genes, respectively. The first line below the gene indicates the extent of the genomic fragment used to rescue the mutant phenotype; the next line (labelled RNAi), that used for RNAi and the third line, the promoter region used to drive the expression of GFP in the reporter construct. E The nipi-3 gene encodes a protein with similarity to human Tribbles homolog 1 (TRIB1). Part of the predicted amino acid sequence of the NIPI-3 is compared to that of human Tribbles homolog 1 (TRIB1; accession number AAH63292). The N-terminal region outside the kinase domain is underlined; the site corresponding to the nipi-3 mutation is marked by an asterisk. Identical residues are boxed in black, similar residues in grey, using Hofmann and Baron’s Boxshade.
nipi-3 a Tribbles-like kinase is required in the response to infection
We focused on nipi-3(fr4), in which the infection-induced up-regulation of pnlp-29::GFP was essentially abolished, but that still responded strongly to injury (Figure 5A). As expected, in nipi-3 mutants we observed an abrogation of nlp-29 mRNA induction upon infection but not wounding, as measured by qRT-PCR (Figure 5B). We found that the expression of a second AMP gene, nlp-31, which is induced by D. coniospora infection [11], was also up-regulated upon wounding. Only the infection response was affected in the nipi-3 mutant (Figure 5B). These results suggest that nipi-3(fr4) is specifically involved in regulating the transcriptional response to infection.
We identified nipi-3 as K09A9.1, predicted to encode a protein serine/theronine kinase (Figure 5D). When compared to human proteins, NIPI-3 is most similar to Tribbles homolog 1 (TRIB1/hTribbles), especially in the kinase domain (residues 237–461; Figure 5E). Among C. elegans proteins, NIPI-3′s most extended similarity is to the uncharacterized hypothetical protein ZK524.4 and to UNC-43. The region of conservation between NIPI-3 and UNC-43 spans part of the kinase domain and some 100 C-terminal residues (Suppl. Figure 3A). The nipi-3(fr4) mutation is in the kinase domain changing a conserved hydrophobic residue to an asparagine (I307N; Figure 5D&E). The mutated residue is spatially separate from the substrate binding pocket, catalytic site, ATP-binding and activation loops (Suppl. Figure 3B).
nipi-3 mutants also exhibited a temperature-sensitive Dpy phenotype: at 25°C nipi-3 mutants were 20% shorter on average than wild-type worms (Suppl. Figure 4). Reintroduction of a 3.6 kb wild-type genomic DNA fragment comprising the coding and 5′ and 3′ sequences fully rescued the nipi-3(fr4) mutant phenotypes (Figure 5D, 6E; Suppl. Figure 4). Abrogation of nipi-3 function through RNAi recapitulated the effects on pnlp-29::GFP expression seen in a nipi-3(fr4) mutant (Figure 5C) and did not provoke any other obvious phenotype (e.g. embryonic or larval lethality, uncoordinated movement). Taken together these results suggest the fr4 mutation causes a loss of nipi-3 function.
To assay for a direct role of nipi-3 in controlling fungal resistance, we compared the survival of wild-type and mutant worms after D. coniospora infection. There was a significant reduction in the resistance of the nipi-3 mutants, as well as in pmk-1(km25) and tir-1(tm3036) mutants. These mutants also had a reduced lifespan on the E. coli strain OP50 (Suppl. Figure 5). This precludes definitively assigning a role in anti-fungal resistance to nipi-3.
Using a GFP transcriptional reporter construct, we found that nipi-3 is expressed in the epidermis as well as in the pharynx and intestine, in a subset of head neurons and motoneurons, and in the PLM and CAN neurons (Figure 6A–D, results not shown). When nipi-3 was specifically expressed in the epidermis, under the control of the col-12 promoter, the induction of pnlp-29::GFP after infection was restored to wild-type levels in a fraction (ca. 1/5) of the transgenic worms. On the other hand, intestinal expression of nipi-3, under the control of the mlt-2 promoter, completely failed to rescue the mutant phenotype (Figure 6G–N; Suppl. Figure 6). Thus, nipi-3 is a new component of innate immune signaling in C. elegans and acts in the epidermis to control AMP expression after infection.
NIPI-3 bears some similarity to UNC-43, a kinase required for establishing the unique identity of the two AWC chemosensory neurons [14]. Unlike unc-43, nipi-3 is not required for asymmetric gene expression in the AWC neurons (data not shown). UNC-43 forms a complex with TIR-1 and NSY-1 and signals through SEK-1 [9]. In the presence of the pcol-12::sek-1 transgene, the level of constitutive expression of pnlp-29::GFP in a nipi-3 mutant background was elevated, and similar to that in wild-type worms. In contrast, in a pmk-1 mutant background, the level of expression was much lower (Figure 6F). These results imply that nipi-3 does not act downstream of sek-1. After infection, in the nipi-3;pcol-12::sek-1 strain there was a two-fold increase in the expression of pnlp-29::GFP. As discussed below, these results are consistent with nipi-3 acting upstream of sek-1.
Discussion
Continuous Distribution of a Quantitative Signalling Readout
The use of the Biosort allows the level of fluorescent reporter gene expression to be measured in single worms. We observed large differences in pnlp-29::GFP expression between infected individuals. This may in part reflect variation in the precise course of the infection, determined among other things by the density and location of spore attachment, and the initial level of AMP expression. The individual variation recorded for the constitutive expression of pnlp-29::GFP in uninfected worms may be surprising, especially since C. elegans is a self-fertilizing hermaphrodite species, and the individuals in a population are genetically identical. Other studies, measuring the expression of other C. elegans reporter genes with the Biosort have revealed a similar range of expression levels [20, 21]. This type of stochastic variation appears to be a widespread phenomenon. Even in yeast, a wide range of values is seen for the expression of the same gene in different individual cells under identical conditions [22, 23]. In yeast, noise in expression levels generally scales with protein abundance. Exceptions to this rule are the stress response proteins, induced under stressful conditions [24]. It has been proposed that this phenotypic variability might enable cells to adapt better to changing environments [25]. It is possible that the observed variability in pnlp-29::GFP expression also plays an adaptive role. Despite this variability, and regardless of any function, the pnlp-29::GFP transgene constitutes a useful tool for dissecting innate immune signalling pathways.
An epidermal wounding response in C. elegans
We used the pnlp-29::GFP reporter to demonstrate for the first time that C. elegans responds to physical injury of the epidermis by up-regulating AMP gene expression. Since we show elsewhere (Pujol et al. submitted) that increased AMP gene expression renders C. elegans more resistant to infection, this mechanism may help worms counter septic injuries that occur in nature. It has been recently shown that in mammalian skin AMP gene expression is induced by sterile injury [26]. Thus protection of the epidermis from infection in such high-risk situations may be a conserved aspect of animal physiology. In Drosophila, clean wounding causes melanization at the wound site [27]. The C. elegans genome encodes tyrosinases that may be evolutionarily related to the prophenoloxidases responsible for melanin biosynthesis in invertebrates [28]. But when we injured the epidermis of C. elegans, we observed no obvious melanin deposition, but rather the almost immediate accumulation at the wound site of autofluorescent material, followed by the repair of the extracellular matrix of the barrier epithelium. Possible triggers for these wound healing and innate immune responses, include rupture of the epidermal apical plasma membrane, or the disruption of contacts between the epidermis and cuticle.
The response to fungal infection is distinct from the epidermal wounding response
While the repair of the barrier epidermis itself can be considered as an innate immune response in that it protects the organism from opportunistic infection, the isolation of the nipi-3(fr4) mutant shows that in C. elegans separable signalling cascades control gene expression after infection and wounding. Thus this strong loss of function mutant shows a limited induction of the two AMP genes nlp-29 or nlp-31 after infection but a near-normal induction of the same two genes following wounding. In the presence of artificially high levels of sek-1, even in a nipi-3 background, there is sufficient activation of the pathway to lead to high expression of pnlp-29::GFP after infection. Since in the pmk-1;pcol-12::sek-1 strain, there is no such elevated pnlp-29::GFP expression even after infection, it would appear that the p38 MAPK is a key regulator of nlp-29, and consequently that nipi-3 acts through this pathway.
Conservation of MAPK signaling modules
Although the responses to D. coniospora and injury can be genetically distinguished, they do converge on the TIR-domain adapter protein TIR-1B and downstream p38 MAPK pathway components. Taken together with the results of our epistasis experiments, this suggests that nipi-3 acts upstream of tir-1. By analogy with the role of UNC-43 in neurons [9], we speculate that NIPI-3 functions as a direct activator of TIR-1B in the epidermis. This would imply that an infection-associated signal exists upstream of NIPI-3. Whatever the mechanisms involved in triggering the infection-specific epidermal response in C. elegans, it is striking to note that the same signaling cassette (TIR-1/NSY-1/SEK-1) is used in at least three different contexts (Figure 7). This parallels the situation in the yeast Saccharomyces cerevisiae, wherein the NSY-1-like MAP3K Ste11p acts in three different biological processes [29]. Interestingly, Ste11p, and Ste50p, its upstream partner possess SAM domains, like TIR-1 (Figure 7). Indeed, close inspection of the NSY-1 sequence reveals that it contains a divergent SAM domain, within a larger conserved motif, present in the vertebrate proteins, ASK1 and ASK2 (Suppl. Figure 7). Importantly, in mice, upon stimulation by lipopolysaccharide, ASK1 selectively activates the p38 pathway [30]. Moreover, ASK1 regulates the expression of β-defensins and LL37, AMPs produced by human epidermis, in a p38-dependent manner [31]. These findings suggest that SAM domains may have played an ancestral role in innate immune signaling that was complemented by the recruitment of TIR domain-containing proteins. Additionally, our results give strong support to the idea that part of vertebrate innate immunity has its origins in MAPK signaling pathways that arose early in evolution to conserve cellular homeostasis. The finding that NIPI-3, a protein kinase with a catalytic domain that is most similar to the human Tribbles homolog 1 is involved in the response to infection is particularly interesting since this protein has been identified as acting in the p38 pathway in vertebrates [32, 33], thus extending the number of proteins potentially involved in this conserved signaling cascade.
Figure 7.

Multiple signals converge on a p38 MAPK signaling cassette conserved between C. elegans (left panel) and budding yeast (right). NIPI-3 may function analogously to UNC-43 as an activator of TIR-1, specifically in the response to infection.
The activation of nlp-29 and nlp-31 by wounding suggests that in C. elegans, as perhaps in vertebrates [34], tissue damage triggers an innate immune response. The nipi-3 gene, however, defines a genetically distinct response to infection. Our results thus support the existence of a pathogen-specific reaction, in addition to a non-specific protective response. Both share components of a p38-signaling cascade; the former may have evolved more recently. In conclusion, the current work advances our knowledge of host defenses in the nematode C. elegans and contributes to the debate on the origins and evolution of innate immunity.
Experimental Procedures
nipi-3 isolation, cloning and rescue
We mutagenized wt;frIs7 worms with EMS using standard procedures [35]. Synchronized F2 worms were infected at the L4 stage with D. coniospora. After 24 h at 25°C, we screened, either automatically with the Biosort, or visually and manually, for worms that failed to show an elevated level of GFP expression after D. coniospora infection. nipi-3(fr4) was mapped through standard genetic and SNP mapping by analysis of 360 recombinants with the strain CB4856, to a 145 kb region left of lin-15 on LGX, containing 21 predicted genes. Sequencing of the gene K09A9.1 revealed a single point mutation in nipi-3 mutants, a T to A transition (flanking sequences TGAAAACGACGTGATGAGTA and CTACCAAAAGGTTGTGGAGA). A full-length cDNA was generated and sequenced from wild-type worms, leading to a modification of the gene structure predicted in Wormbase (WS180), see Genbank accession number EU043523. The nipi-3(fr4) allele changes an isoleucine to an asparagine at position 307 of the predicted protein sequence. To confirm the identity of nipi-3, the K09A9.1 gene with 1426 bp upstream and 1753 bp downstream was amplified by PCR with the primers JEP963 and JEP964 and microinjected (at 5 ng/μl with 80 ng/μl pBunc-53::GFP [36]) into nipi-3(fr4);frIs7 mutant worms. Two independent lines IG581 nipi-3(fr4);frIs7;frEx195 and IG580 nipi-3(fr4);frIs7;frIs10 were generated; the latter is a spontaneous integrant.
Supplementary Material
Acknowledgments
We thank Y. Duverger and S. Scaglione for worm sorting, M. Fallet for help with image processing, Z. Wu for assistance with the femtosecond laser, H. Agherbi for help with integration of frIs7, O. Zugasti for generating some transgenic strains. Worm sorting analyses were carried out using the facilities of the Marseille-Nice Genopole®. Some nematode strains were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources (NCRR), or by the National Bioresource Project coordinated by S. Mitani. This work was funded by institutional grants from INSERM and the CNRS, the French Ministry of Research Programme de Microbiologie, ACI BDPI and BCMS, the RNG, ANR, the NSF (OISE 0726131) and the French Foreign Ministry (France Berkeley Fund). Y.J. is an Investigator of the Howard Hughes Medical Institute. The Ewbank lab is an FRM équipe labellisée.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mallo GV, Kurz CL, Couillault C, Pujol N, Granjeaud S, Kohara Y, Ewbank JJ. Inducible antibacterial defense system in C. elegans. Curr Biol. 2002;12:1209–1214. doi: 10.1016/s0960-9822(02)00928-4. [DOI] [PubMed] [Google Scholar]
- 2.O’Rourke D, Baban D, Demidova M, Mott R, Hodgkin J. Genomic clusters, putative pathogen recognition molecules, and antimicrobial genes are induced by infection of C. elegans with M. nematophilum. Genome Res. 2006;16:1005–1016. doi: 10.1101/gr.50823006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Troemel ER, Chu SW, Reinke V, Lee SS, Ausubel FM, Kim DH. p38 MAPK Regulates Expression of Immune Response Genes and Contributes to Longevity in C. elegans. PLoS Genetics. 2006;2:e183. doi: 10.1371/journal.pgen.0020183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shapira M, Hamlin BJ, Rong J, Chen K, Ronen M, Tan MW. A conserved role for a GATA transcription factor in regulating epithelial innate immune responses. Proc Natl Acad Sci U S A. 2006;103:14086–14091. doi: 10.1073/pnas.0603424103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong D, Bazopoulou D, Pujol N, Tavernarakis N, Ewbank JJ. Genome-wide investigation reveals pathogen-specific and shared signatures in the response of C. elegans to infection. Genome Biol. 2007;8:R194. doi: 10.1186/gb-2007-8-9-r194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ewbank JJ. Signaling in the Immune Response. WormBook Volume. 2006 doi: 10.1895/wormbook.1.83.1. The C. elegans Research Community, ed. ( http://www.wormbook.org) [DOI] [PMC free article] [PubMed]
- 7.Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H, Tanaka-Hino M, Hisamoto N, Matsumoto K, Tan MW, Ausubel FM. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science. 2002;297:623–626. doi: 10.1126/science.1073759. [DOI] [PubMed] [Google Scholar]
- 8.Liberati NT, Fitzgerald KA, Kim DH, Feinbaum R, Golenbock DT, Ausubel FM. Requirement for a conserved Toll/interleukin-1 resistance domain protein in the Caenorhabditis elegans immune response. Proc Natl Acad Sci U S A. 2004;101:6593–6598. doi: 10.1073/pnas.0308625101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chuang CF, Bargmann CI. A Toll-interleukin 1 repeat protein at the synapse specifies asymmetric odorant receptor expression via ASK1 MAPKKK signaling. Genes Dev. 2005;19:270–281. doi: 10.1101/gad.1276505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dijksterhuis J, Veenhuis M, Harder W. Ultrastructural study of adhesionand initial stages of infection of the nematode by conidia of Drechmeria coniospora. Mycological research. 1990;94:1–8. [Google Scholar]
- 11.Couillault C, Pujol N, Reboul J, Sabatier L, Guichou JF, Kohara Y, Ewbank JJ. TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nat Immunol. 2004;5:488–494. doi: 10.1038/ni1060. [DOI] [PubMed] [Google Scholar]
- 12.Shemer G, Suissa M, Kolotuev I, Nguyen KC, Hall DH, Podbilewicz B. EFF-1 is sufficient to initiate and execute tissue-specific cell fusion in C. elegans. Curr Biol. 2004;14:1587–1591. doi: 10.1016/j.cub.2004.07.059. [DOI] [PubMed] [Google Scholar]
- 13.Mohler WA, Shemer G, del Campo JJ, Valansi C, Opoku-Serebuoh E, Scranton V, Assaf N, White JG, Podbilewicz B. The type I membrane protein EFF-1 is essential for developmental cell fusion. Dev Cell. 2002;2:355–362. doi: 10.1016/s1534-5807(02)00129-6. [DOI] [PubMed] [Google Scholar]
- 14.Sagasti A, Hisamoto N, Hyodo J, Tanaka-Hino M, Matsumoto K, Bargmann CI. The CaMKII UNC-43 activates the MAPKKK NSY-1 to execute a lateral signaling decision required for asymmetric olfactory neuron fates. Cell. 2001;105:221–232. doi: 10.1016/s0092-8674(01)00313-0. [DOI] [PubMed] [Google Scholar]
- 15.Byrd DT, Kawasaki M, Walcoff M, Hisamoto N, Matsumoto K, Jin Y. UNC-16, a JNK-signaling scaffold protein, regulates vesicle transport in C. elegans. Neuron. 2001;32:787–800. doi: 10.1016/s0896-6273(01)00532-3. [DOI] [PubMed] [Google Scholar]
- 16.Kim DH, Liberati NT, Mizuno T, Inoue H, Hisamoto N, Matsumoto K, Ausubel FM. Integration of Caenorhabditis elegans MAPK pathways mediating immunity and stress resistance by MEK-1 MAPK kinase and VHP-1 MAPK phosphatase. Proc Natl Acad Sci U S A. 2004;101:10990–10994. doi: 10.1073/pnas.0403546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizuno T, Hisamoto N, Terada T, Kondo T, Adachi M, Nishida E, Kim DH, Ausubel FM, Matsumoto K. The Caenorhabditis elegans MAPK phosphatase VHP-1 mediates a novel JNK-like signaling pathway in stress response. Embo J. 2004;23:2226–2234. doi: 10.1038/sj.emboj.7600226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanaka-Hino M, Sagasti A, Hisamoto N, Kawasaki M, Nakano S, Ninomiya-Tsuji J, Bargmann CI, Matsumoto K. SEK-1 MAPKK mediates Ca2+ signaling to determine neuronal asymmetric development in Caenorhabditis elegans. EMBO Rep. 2002;3:56–62. doi: 10.1093/embo-reports/kvf001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inoue H, Hisamoto N, An JH, Oliveira RP, Nishida E, Blackwell TK, Matsumoto K. The C. elegans p38 MAPK pathway regulates nuclear localization of the transcription factor SKN-1 in oxidative stress response. Genes Dev. 2005;19:2278–2283. doi: 10.1101/gad.1324805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cardoso C, Couillault C, Mignon-Ravix C, Millet A, Ewbank JJ, Fontes M, Pujol N. XNP-1/ATR-X acts with RB, HP1 and the NuRD complex during larval development in C. elegans. Dev Biol. 2005;278:49–59. doi: 10.1016/j.ydbio.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 21.Rea SL, Wu D, Cypser JR, Vaupel JW, Johnson TE. A stress-sensitive reporter predicts longevity in isogenic populations of Caenorhabditis elegans. Nat Genet. 2005;37:894–898. doi: 10.1038/ng1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Attfield PV, Choi HY, Veal DA, Bell PJ. Heterogeneity of stress gene expression and stress resistance among individual cells of Saccharomyces cerevisiae. Mol Microbiol. 2001;40:1000–1008. doi: 10.1046/j.1365-2958.2001.02444.x. [DOI] [PubMed] [Google Scholar]
- 23.Ryley J, Pereira-Smith OM. Microfluidics device for single cell gene expression analysis in Saccharomyces cerevisiae. Yeast. 2006;23:1065–1073. doi: 10.1002/yea.1412. [DOI] [PubMed] [Google Scholar]
- 24.Bar-Even A, Paulsson J, Maheshri N, Carmi M, O’Shea E, Pilpel Y, Barkai N. Noise in protein expression scales with natural protein abundance. Nat Genet. 2006;38:636–643. doi: 10.1038/ng1807. [DOI] [PubMed] [Google Scholar]
- 25.Barkai N, Shilo BZ. Variability and robustness in biomolecular systems. Mol Cell. 2007;28:755–760. doi: 10.1016/j.molcel.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 26.Sorensen OE, Thapa DR, Roupe KM, Valore EV, Sjobring U, Roberts AA, Schmidtchen A, Ganz T. Injury-induced innate immune response in human skin mediated by transactivation of the epidermal growth factor receptor. J Clin Invest. 2006;116:1878–1885. doi: 10.1172/JCI28422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Gregorio E, Han SJ, Lee WJ, Baek MJ, Osaki T, Kawabata S, Lee BL, Iwanaga S, Lemaitre B, Brey PT. An immune-responsive Serpin regulates the melanization cascade in Drosophila. Dev Cell. 2002;3:581–592. doi: 10.1016/s1534-5807(02)00267-8. [DOI] [PubMed] [Google Scholar]
- 28.Cerenius L, Soderhall K. The prophenoloxidase-activating system in invertebrates. Immunol Rev. 2004;198:116–126. doi: 10.1111/j.0105-2896.2004.00116.x. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz MA, Madhani HD. Principles of MAP kinase signaling specificity in Saccharomyces cerevisiae. Annu Rev Genet. 2004;38:725–748. doi: 10.1146/annurev.genet.39.073003.112634. [DOI] [PubMed] [Google Scholar]
- 30.Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, Koyasu S, Matsumoto K, Takeda K, Ichijo H. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol. 2005;6:587–592. doi: 10.1038/ni1200. [DOI] [PubMed] [Google Scholar]
- 31.Sayama K, Komatsuzawa H, Yamasaki K, Shirakata Y, Hanakawa Y, Ouhara K, Tokumaru S, Dai X, Tohyama M, Ten Dijke P, Sugai M, Ichijo H, Hashimoto K. New mechanisms of skin innate immunity: ASK1-mediated keratinocyte differentiation regulates the expression of beta-defensins, LL37, and TLR2. Eur J Immunol. 2005;35:1886–1895. doi: 10.1002/eji.200425827. [DOI] [PubMed] [Google Scholar]
- 32.Kiss-Toth E, Bagstaff SM, Sung HY, Jozsa V, Dempsey C, Caunt JC, Oxley KM, Wyllie DH, Polgar T, Harte M, O’Neill LA, Qwarnstrom EE, Dower SK. Human tribbles, a protein family controlling mitogen-activated protein kinase cascades. J Biol Chem. 2004;279:42703–42708. doi: 10.1074/jbc.M407732200. [DOI] [PubMed] [Google Scholar]
- 33.Kiss-Toth E, Wyllie DH, Holland K, Marsden L, Jozsa V, Oxley KM, Polgar T, Qwarnstrom EE, Dower SK. Functional mapping and identification of novel regulators for the Toll/Interleukin-1 signalling network by transcription expression cloning. Cell Signal. 2006;18:202–214. doi: 10.1016/j.cellsig.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 34.Matzinger P. Friendly and dangerous signals: is the tissue in control? Nat Immunol. 2007;8:11–13. doi: 10.1038/ni0107-11. [DOI] [PubMed] [Google Scholar]
- 35.Wood WB, editor. The nematode Caenorhabditis elegans. Plainview, N. Y: Cold Spring Harbor Laboratory Press; 1988. [Google Scholar]
- 36.Stringham E, Pujol N, Vandekerckhove J, Bogaert T. unc-53 controls longitudinal migration in C. elegans. Development. 2002;129:3367–3379. doi: 10.1242/dev.129.14.3367. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.