

SUMMARY
Glycoprotein B (gB) is one of the essential components for infection by herpes simplex virus-1 (HSV-1). Although several cellular receptors that associate with glycoprotein D (gD), such as herpes virus entry mediator (HVEM) and Nectin-1, have been identified, specific molecules that mediate HSV-1 infection by associating with gB have not been elucidated. Here, we found that paired immunoglobulin-like type 2 receptor (PILR) α associates with gB, and cells transduced with PILRα become susceptible to HSV-1 infection. Furthermore, HSV-1 infection of human primary cells expressing both HVEM and PILRα was blocked by either anti-PILRα or anti-HVEM antibody. Our results demonstrate that cellular receptors for both gB and gD are required for HSV-1 infection and that PILRα plays an important role in HSV-1 infection as a co-receptor that associates with gB. These findings uncover a crucial aspect of the mechanism underlying HSV-1 infection.
INTRODUCTION
Herpes simplex virus type 1 (HSV-1) is the prototype of the diverse α-herpesvirus family, which generally causes mucocutaneous lesions but also is involved in lethal encephalitis. HSV-1 establishes latent infections in neurons of peripheral ganglia and may reactivate to cause recurrent lesions (Whitley and Roizman, 2001). Entry of HSV-1 into cells depends upon interactions between cell surface receptors and viral proteins on the virion. Among the surface glycoproteins of HSV-1, gB, gD, gH, and gL are involved in the entry of HSV-1 and mutant viruses that lack one of these four glycoproteins cannot infect cells (Spear, 2004). gD interacts with herpesvirus entry mediator (HVEM), Nectin-1, and specific sites on heparan sulfate generated by certain 3-O-sulfotransferases (Montgomery et al., 1996; Geraghty et al., 1998; Warner et al., 1998; Shukla et al., 1999). Because gD is an essential glycoprotein for HSV-1 to infect a cell, interactions between gD and these cellular receptors play an important role in HSV-1 entry into cells (Spear et al., 2000). Although gD itself is probably not a membrane fusogen, binding of gD with cellular receptors induces membrane fusion mediated by the gH / gL heterodimer and gB (Krummenacher et al., 2005).
Both gB and gH have been reported to play an essential role in membrane fusion during HSV-1 infection (Cai et al., 1988; Forrester et al., 1992; Turner et al., 1998). Although neither gB nor gH / gL has an obvious hydrophobic fusion peptide sequence, recent analyses suggested that gB and gH possess putative fusion peptides that may be involved in membrane fusion during HSV-1 infection (Gianni et al., 2005; Heldwein et al., 2006). On the other hand, gB has been suggested to associate with cell surface heparan sulfate (Herold et al., 1994); however, cells deficient in heparan sulfate synthesis are still permissive to HSV-1 infection (Banfield et al., 1995), and mutant HSV-1, from which the poly-lysine sequence in gB that is responsible for heparan sulfate binding is deleted, is still infectious (Laquerre et al., 1998). More recently, it has been reported that a soluble form of gB binds to heparan sulfate-deficient cells and blocks HSV-1 infection of some cell lines (Bender et al., 2005). These findings suggest that molecules other than heparan sulfate mediate HSV-1 infection by associating with gB. It has been reported that gC also binds to heparan sulfate and is involved in the binding of HSV-1 to the cell surface, although gC-deficient virus is still infectious (Herold et al., 1991). Therefore, binding of gC to heparan sulfate is not essential for viral entry, similar to the binding of gB to heparan sulfate (Herold et al., 1994; Laquerre et al., 1998).
Immune cells express an abundance of cell surface receptors that regulate their activation. In some cases, highly related receptors have evolved where one receptor in the family has inhibitory functions, whereas another receptor in the family mediates activating functions. Paired immunoglobulin like-type 2 receptor (PILR) is one of these “paired” receptor families (Fournier et al., 2000; Mousseau et al., 2000; Shiratori et al., 2004). The inhibitory PILRα has an immunoreceptor tyrosine-based inhibition motif (ITIM) in its cytoplasmic domain and delivers inhibitory signals (Fournier et al., 2000). By contrast, the activating PILRβ associates with the immunoreceptor tyrosine-based activation motif (ITAM)-bearing DAP12 adapter molecule and delivers activating signals (Shiratori et al., 2004). Because most mammals possess PILR genes, PILR likely plays an important role in the regulation of immune cells (Wilson et al., 2006). Previously, we have shown that the mouse inhibitory PILRα and activating PILRβ specifically recognize mouse CD99 and these receptors are involved in the regulation of immune responses (Shiratori et al., 2004).
Here, we found that HSV-1-infected cells express a ligand for inhibitory PILRα and that gB of HSV-1 is a ligand for PILRα. Furthermore, we determined that interactions between gB and PILRα mediate HSV-1 infection. Analyses using PILRα-transfectants, HSV-1 mutants, and primary cells expressing both HVEM and PILR indicated that cellular receptors for both gB and gD are required for HSV-1 infection.
RESULTS
Cloning of a PILRα ligand expressed on HSV-1-infected cells
When we analyzed HSV-1-infected cells for ligands that bind human PILRα, we found that HSV-1-infected human 293T cells were specifically stained with human PILRα-Ig fusion protein (Figure 1A). PILRα-Ig bound equally to cells infected with several HSV-1 strains. Similarly, NIH3T3 cells or Vero cells infected with HSV-1 were also recognized by human PILRα-Ig fusion protein (data not shown). This suggested that HSV-1 encodes a ligand for PILRα or that HSV-1 induces a cellular ligand for PILRα in infected cells. In order to identify the ligand for human PILRα present on HSV-1-infected cells, infected cells were lysed and the ligand for PILRα was immunoprecipitated with PILRα-Ig, followed by SDS-PAGE analysis. PILRα-Ig, but not control Ig, specifically precipitated 110 kDa molecules from HSV-1-infected cells (Figure 1B). We extracted the 110 kDa protein, and then analyzed the protein by using LC/MS/MS mass spectrometry. Surprisingly, 29 peptide sequences that were identical to gB were obtained by the analysis (Figure S1). In addition, 2 peptide sequences that were identical to gH were also identified. This suggested that the ligand detected on HSV-1-infected cells is likely to be gB and not a cellular ligand induced by HSV-1 infection.
Figure 1. PILRα ligand expressed on HSV-1-infected cells is gB.

(A) Expression of a ligand for PILRα on HSV-1-infected cells. 293T cells or HSV-1 (strain F, VR3, SC16 or KOS)-infected 293T cells were stained with human PILRα-Ig (solid line) or a control Ig fusion protein (CD200-Ig, dotted line).
(B) Immunoprecipitation of PILRα ligand from HSV-1-infected cells. Lysates of HSV-1-infected or non-infected 293T cells were immunoprecipitated with PILRα-Ig and immunoprecipitates were separated by SDS-PAGE, followed by silver staining.
(C) Western blot analysis of the HSV-1 PILRα ligand. Lysate from HSV-1-infected cells was immunoprecipitated with PILRα-Ig, Nectin-1-Ig, or CD200-Ig (control). Immunoprecipitates were separated by SDS-PAGE and were blotted with anti-gB or anti-gD Ab. Ig fusion proteins used for immunoprecipitation were detected by anti-human IgG Ab.
Specific binding of PILRα to gB
In order to confirm that the ligand for PILRα is gB, lysates of HSV-1-infected cells were immunoprecipitated with PILRα-Ig, followed by Western blot analysis. The 110 kDa protein precipitated from HSV-1-infected cells by PILRα-Ig was confirmed to react with an anti-gB mAb but not anti-gD mAb (Figure 1C). On the other hand, Nectin1-Ig clearly precipitated gD, as well as small amounts of gB. Control Ig did not precipitate either gB or gD. This indicated that the 110 kDa protein precipitated by PILRα-Ig is mainly gB.
Next, we analyzed the specificity of binding of PILRα to gB. 293T cells transfected with a full-length gB cDNA did not express significant amounts of gB on the cell surface (data not shown). Therefore, we generated a mutant gB that lacks the C-terminal 40 amino acids in the cytoplasmic tail (gBΔC) because the cytoplasmic domain of gB contains an intracellular retention signal that impairs cell surface expression of wild-type gB (Beitia Ortiz de Zarate et al., 2004). Unlike wild-type gB, gBΔC was expressed well on the cell surface of 293T cells upon transient transfection. We co-transfected gBΔC, gD, or gH and gL together with GFP into 293T cells, and analyzed binding of PILRα-Ig and Nectin-1-Ig to GFP-positive transfectants by flow cytometry (Figure 2A). Cell surface expression of these glycoproteins was confirmed by staining with mAbs against these glycoproteins. PILRα-Ig clearly bound cells transfected with gBΔC but not gD or gH and gL. Mouse PILRα-Ig also recognized cells transfected with gBΔC, but not with gD or gH and gL (data not shown). On the other hand, Nectin-1-Ig fusion protein bound cells expressing gD, but not gB or gH and gL. These data indicated that PILRα specifically binds to gB, but not gD or gH and gL.
Figure 2. Specific interaction between PILRα and gB.

(A) Specific binding of human PILRα to gB. Mutant gB that lacks the C-terminus 40 amino acids, gD, or gH and gL was co-transfected with GFP into 293T cells. Transfected cells were stained with PILRα-Ig and Nectin-1-Ig fusion proteins, and anti-gB, anti-gD, and anti-gH mAbs (solid line). Cells were also stained with control Ig fusion protein or control mAb (dotted line). The staining patterns of GFP-positive cells are shown.
(B) PILRα-Ig does not recognize cells infected with gB-deficient HSV-1. Non-infected 293T cells and 293T cells infected with wild-type HSV-1, gB-deficient HSV-1, or revertant HSV-1 were stained with PILR-Ig, Nectin-1-Ig, anti-gB mAb, and anti-gD mAb (solid line). Cells were also stained with control Ig fusion protein or control mAb (dotted line).
To further confirm the specificity of the binding of PILRα to gB, we analyzed cells infected with gB-deficient HSV-1. Because gB-deficient virus does not infect cells, gB-deficient virus that possesses gB protein on the virion was produced by using gB-transfected complementing cells (Cai et al., 1988). Similar to the data presented in Figure 1, cells infected with wild-type HSV-1 were stained with anti-gB and anti-gD mAb, as well as PILRα-Ig (Figure 2B). Cells infected with gB-deficient HSV-1 were stained with anti-gD mAb, but not with anti-gB mAb or PILRα-Ig fusion protein. Cells infected with a revertant virus that expresses wild-type gB were recognized by both anti-gB mAb and PILRα-Ig fusion protein. These data indicated that PILRα specifically recognizes gB and suggest that the small amounts of gH detected by mass spectrometry analysis were likely a result of co-precipitation with gB.
HSV-1 infection of PILR transfectants
gB is a viral protein conserved in all herpesviruses and plays an important role in HSV-1 infection (Cai et al., 1988; Heldwein et al., 2006). HSV-1 that lacks gB cannot infect cells (Cai et al., 1988). Although receptors for gB have been suggested to be expressed on the cell surface (Bender et al., 2005), specific receptors that associate with gB and mediate HSV-1 infection have not been identified. We analyzed whether the interaction between gB and PILRα is involved in HSV-1 infection. Because CHO-K1 cells express an unknown endogenous cellular ligand for PILRα, we isolated PILRα-ligand-negative CHO-K1 cells by flow cytometry and transfected human PILRα into these PILRα-ligand-negative CHO-K1 cells in order to avoid potential interactions between human PILRα and the endogenous hamster PILRα ligand. When the PILRα-ligand-negative CHO-K1 cells were transfected with human PILRα, these cells were effectively infected by HSV-1 expressing a GFP marker, whereas mock-transfected CHO-K1 cells were not infected by GFP-HSV-1 (Figures 3A and 3B). Similar results were obtained when HSV-1 infection was analyzed by fluorescence microscopy (Figure 3C). These data indicated that human PILRα is involved in HSV-1 infection by associating with gB.
Figure 3. HSV-1 infection of PILRα-transfected CHO-K1 cells.

(A) PILRα-ligand-negative CHO-K1 cells were transiently transfected with the pMx-IRES-DsRed expression vector containing human PILRα. Transfected CHO-K1 cells were infected with HSV-1-GFP, and cells expressing GFP within the DsRed-positive population were analyzed by flow cytometry.
(B) Proportions of cells expressing GFP, gated on human PILRα (DsRed)-positive cells, are shown. Mean ± SD of triplicate analyses are shown.
(C) CHO-K1 cells were transiently transfected with PILRα-IRES-DsRed or mock-IRES-DsRed expression vectors and cells expressing DsRed were purified by using a cell sorter. The transfected cells were infected with HSV-1-GFP and expression of GFP was analyzed by fluorescence microscopy.
Generation of neutralizing anti-human PILRα monoclonal antibody
We addressed whether a direct interaction between human PILRα and gB is involved in HSV-1 infection. For this purpose, we generated a mAb that specifically blocks the interaction between human PILRα and gB. We identified an anti-PILRα mAb (M4) that completely blocked the binding of PILRα-Ig to gB-transfected cells. As shown in Figure S2A, anti-PILRα mAb M4 specifically recognized mouse Ba/F3 cells transfected with human PILRα but not parental Ba/F3 cells. Anti-PILRα mAb M4 did not bind to HVEM- or Nectin-expressing cells (data not shown). Furthermore, when human PILRα-Ig fusion protein was pre-incubated with the anti-PILRα M4 mAb, the PILRα-Ig fusion protein did not bind the cells transfected with the gBΔC (Figure S2B). This indicated that the anti-PILRα M4 mAb completely blocks the interaction between human PILRα and gB.
Inhibition of HSV-1 infection of PILRα transfectants by anti-PILRα mAb or soluble PILR
We analyzed whether the anti-PILRα mAb blocks HSV-1 infection of PILRα-transfected cells. When human PILRα transfectants were infected with HSV-1 in the presence of anti-PILRα mAb, infection was completely blocked by the anti-human PILRα mAb in a dose-dependent manner (Figure 4A). By contrast, an isotype-matched mAb did not affect HSV-1 infection of PILRα transfectants. In addition, other anti-PILRα mAbs that did not block the binding of PILRα to gB also did not block HSV-1 infection of PILRα transfectants (data not shown).
Figure 4. Inhibition of HSV-1 infection by anti-PILRα mAb or by PILRα-Ig.

(A) Inhibition of HSV-1 infection by anti-PILRα mAb. CHO-K1 cells were transiently transfected with human PILRα in the pMx-IRES-DsRed expression vector, and cells were infected with HSV-1-GFP in the presence of various concentrations of anti-PILRα or control mAb. Proportions of infected cells were determined by flow cytometry.
(B) Inhibition of HSV-1 infection by PILRα-Ig fusion protein. CHO-K1 cells transiently transfected with human PILRα were infected with HSV-1-GFP in the presence of various concentrations of PILRα-Ig or control Ig fusion protein. The proportion of infected cells was determined by flow cytometry. Mean ± SD of triplicate analyses are shown.
Because PILRα-Ig directly binds to gB of HSV-1, soluble PILRα-Ig fusion protein might block the interaction between gB and human PILRα. To test this possibility, we analyzed HSV-1 infection of PILRα-expressing cells in the presence or absence of the human PILRα-Ig fusion protein. As shown in Figure 4B, HSV-1 infection of human PILRα-transfected CHO-K1 cells was significantly blocked by PILRα-Ig fusion protein, but not by a control Ig fusion protein. These data indicated that a direct interaction between PILRα and gB mediates HSV-1 infection.
Role of gD in infection of PILRα-expressing cells
gD, as well as gB, is an essential viral protein for HSV-1 infection because gD-deficient HSV-1 is non-infectious (Ligas and Johnson, 1988). We addressed whether gD is involved in PILRα-mediated HSV-1 infection. PILRα-transfected CHO cells were infected with a gD-deficient virus produced by normal Vero cells (gD (−)-HSV) or gD-expressing complementing Vero cells (gD (+)-HSV). gD itself has been reported to be dispensable for the production of HSV-1 virions (Ligas and Johnson, 1988). Indeed, equal amounts of gB were detected in these viruses by Western blot analysis, suggesting that the amounts of virions were similar (Figure S3). When PILRα-transfected CHO cells were inoculated with these gD-deficient HSV-1, gD (+)-HSV was able to infect the PILRα-transfected CHO cells but not gD (−)-HSV, at least not to any significant extent (Figure 5A). This indicated that gD is required for PILRα-mediated HSV-1-infection. We then addressed whether binding of gD to certain cellular receptors are required for HSV-1 infection of PILRα-transfected CHO cells. We generated a gD-Ig fusion protein and tested the ability of this fusion protein to block HSV-1 infection of PILRα-transfected CHO. As shown in Figure 5B, HSV-1 infection of PILRα-transfected CHO cells was inhibited by the gD-Ig fusion protein in a dose-dependent manner (Figure 5B). These data suggested that association of gD with certain cell surface molecules is required for PILRα-mediated HSV1 infection.
Figure 5. Requirement of gD in PILRα-mediated HSV-1 infection.

(A) Requirement of gD in HSV-1 infection of PILRα-expressing cells. PILRα- or mock-transfected CHO cells were infected with gD-deficient virus produced by normal Vero cells (gD (−) HSV-1, closed circle) or gD-transfected Vero cells (gD (+) HSV-1, open circle). Proportions of ICP4 (a viral protein produced immediately after infection)-positive cells, detected by flow cytometry, are shown as mean ± SD of triplicate analyses.
(B) Inhibition of HSV1 infection by soluble gD. PILRα- or mock-transfected CHO cells were infected with HSV-GFP in the presence of gD-Ig (closed circle) or control Ig (open circle) fusion protein. Proportions of GFP-positive cells are shown as mean ± SD of triplicate analyses.
In order to further examine the mechanism of PILRα-mediated HSV-1 infection, we employed a cell fusion assay. gB, gD, gH, and gL were co-transfected with GFP into CHO cells and PILRα was co-transfected with RFP into CHO cells. Purified GFP- or RFP-positive cells were co-cultured for 8 h and cell fusion was analyzed by fluorescence microscopy. When CHO cells co-transfected with gB, gD, gH, and gL were co-cultured with PILRα-transfected CHO cells, significant numbers of CHO cells expressing both GFP and RFP were observed (Figure 6). Furthermore, cells expressing both GFP and RFP were multinucleated. CHO cells co-transfected with gB, gD, gH and gL did not show cell fusion with mock-transfected CHO cells. CHO cells transfected with gD, gH, and gL also did not show cell fusion with PILRα-transfected CHO cells. When CHO cells transfected with gB, gH, and gL were analyzed, most cells did not show cell fusion with PILRα-transfected CHO cells. However, a very low frequency of CHO cells transfected with gB, gH, and gL, but not gD, showed cell fusion with PILRα-transfected CHO cells (Figures 6 and S4). The size of fused CHO cells transfected with gB, gH, and gL were significantly smaller compared to CHO cells transfected with gB, gD, gH, and gL. Thus, the efficiency of cell fusion induced by CHO cells transfected with only gB, gH, and gL was much lower than that by CHO cells transfected with gB, gD, gH, and gL, just as viral entry in the absence of gD was also very much reduced in efficiency (Figure 5A).
Figure 6. Cell fusion mediated by interaction between gB and PILRα.
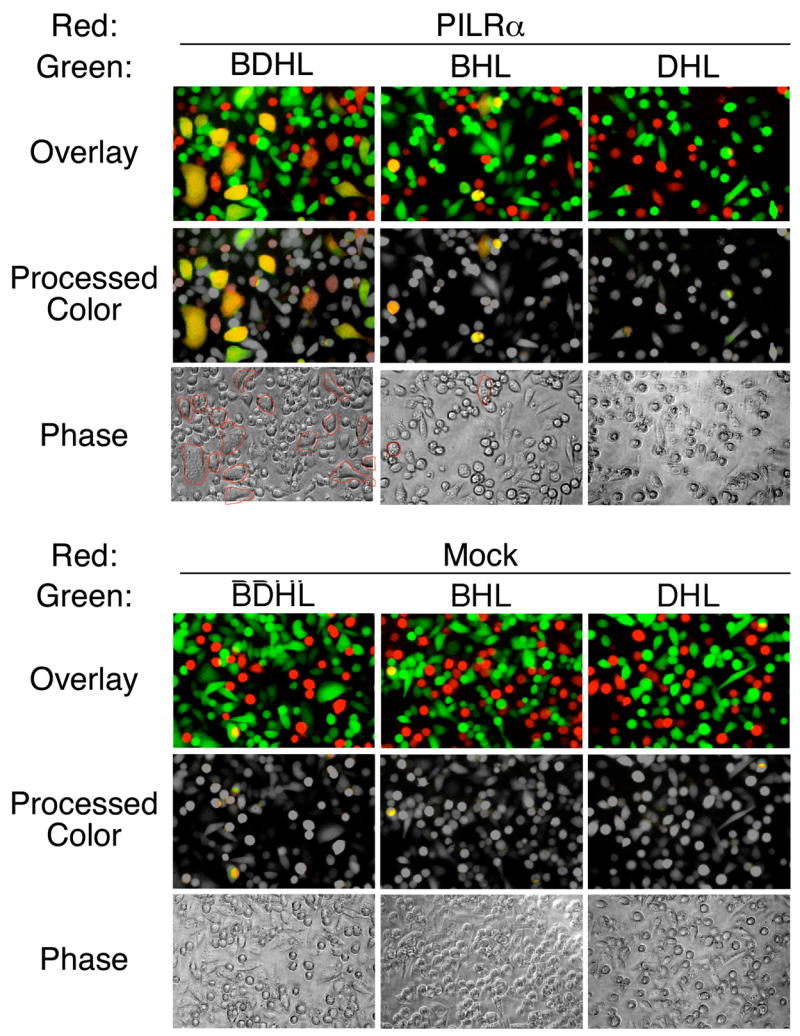
Cell fusion assay between CHO cells transfected with HSV-1 glycoproteins and PILRα. gB, gD, gH, and gL (BDHL), gB, gH, and gL (BHL), or gD, gH, and gL (DHL) were co-transfected into CHO-K1 cells with GFP. PILRα or control plasmid (Mock) was co-transfected into CHO-K1 cells with RFP. GFP- and RFP-expressing cells were purified by using flow cytometry and were co-cultured. After 8 h, cells were analyzed by fluorescence microscopy. Photographs taken by green and red filters were overlaid (Overlay). Green and red colors of non-fused cells were converted to gray color, and yellow colors of fused cells were left unchanged (Processed color). Photographs taken using phase contrast are also shown (Phase). Multinuclear cells are circled in red.
These data indicated that interactions between gB and PILRα are involved in membrane fusion during HSV-1 infection. gD-deficient virus infected PILRα-transfected cells very inefficiently, if at all, and minimal cell fusion was observed by interactions between gB and PILRα in the absence of gD. Therefore, interaction of gD with certain cellular molecules is required for optimal membrane fusion even in the presence of gB and PILRα.
PILRα-mediated HSV-1 infection of primary cells
We addressed whether PILRα is involved in the infection of primary human cells. Previously, PILRα has been reported to be expressed mainly on monocytes, granulocytes, and dendritic cells (Fournier et al., 2000). When amounts of PILRα transcript in various human tissues were analyzed by quantative real-time RT-PCR, significant amounts of PILRα transcripts were detected not only in PBMC but also in various tissues including the nervous system (Figure S5A). In particular, brain and cerebellum expressed relatively high amounts of PILRα transcripts. Nucleic acid sequences of PCR products amplified from whole brain and cerebellum were identical to PILRα and similar results were obtained by using different primer sets (data not shown). When tissue sections from several tissues were analyzed by immunohistochemistry, various types of cells including neurons were stained with anti-PILRα mAb (Figure S5B). These data suggested that PILRα is broadly expressed in various tissues and expression of PILRα is not limited to the myeloid lineage.
When expression of PILRα, HVEM, and Nectin-1 on human PBMC was analyzed by flow cytometry, all of the CD14-positive monocyte population expressed both HVEM and PILRα, but not Nectin-1 (Figure 7A). On the other hand, a CD14-negative population, which predominantly consisted of lymphocytes, expressed HVEM, but not significant amounts of PILRα or Nectin-1. Expression levels of HVEM on the CD14-negative and -positive populations were similar. Therefore, we separated PBMC into CD14-positive and -negative populations and infected these cells with HSV-1 in the presence or absence of a neutralizing anti-PILRα mAb (Figure 7B).
Figure 7. PILRα-mediated HSV-1 infection in primary cells.

(A) Expression of HVEM, Nectin-1, and PILRα on human PBMC. PBMC were stained with anti-HVEM, anti-Nectin-1, or anti-PILRα mAbs, along with anti-CD14 mAb.
(B) PILRα-mediated HSV-1 infection of CD14-positive PBMC. Freshly isolated human CD14-positive or -negative PBMC were infected with various amounts of HSV-1-GFP in the presence or absence of anti-human PILRα mAb or control mAb (10 μg/ml) and the proportion of infected cells was determined by flow cytometry.
(C) Role of HVEM in HSV-1 infection of monocytes. Freshly isolated human CD14-positive monocytes were infected with HSV-1 GFP in the presence of anti-HVEM serum or control serum at the indicated concentrations.
(D) Role of gD in HSV-1 infection of primary monocytes. Freshly isolated human CD14-positive monocytes were infected with gD-deficient virus produced by normal Vero cells (gD(−) HSV-1) or gD-transfected Vero cells (gD(+) HSV-1). Proportions of ICP4-positive (infected) cells were determined by flow cytometry. All the data are shown as mean ± SD of triplicate analyses.
CD14-positive monocytes, which express both PILRα and HVEM, were susceptible to HSV-1 infection. In contrast, HSV-1 did not infect the CD14-negative population, although the CD14-negative population expresses HVEM at the same level as the CD14-positive monocytes. gD-Ig fusion protein bound CD14-negative population (Figure S6A) and binding of gD-Ig fusion protein to CD14-negative population was blocked by anti-HVEM Ab (Figure S6B). This suggested that HVEM expressed on the CD14-negative population is accessible to gD, although the CD14-negative population was resistant to HSV-1 infection. Furthermore, HSV-1 infection of monocytes, which express both PILRα and HVEM, was efficiently blocked by anti-human PILRα mAb, but not by a control mAb (Figure 7B). In addition, HSV-1 infection of monocytes was also efficiently blocked by antiserum against HVEM but not by control serum (Figure 7C). Similar to PILRα-transfected CHO cells, the gD-deficient virus did not infect monocytes (Figure 7D). These data demonstrated that interactions of both gB and gD with their specific cellular receptors are required for HSV-1 infection of primary cells and that PILR functions as a co-receptor for HSV-1 infection. This also indicated that cellular receptors for either gB or gD alone are not enough to mediate HSV-1 infection. Resistance of CD14-negative PBMC that express HVEM, but not PILRα, to HSV-1 infection also suggests that cellular receptors for gD alone are not sufficient to mediate HSV-1 infection.
DISCUSSION
Here, we have shown that PILRα mediates HSV-1 infection by associating with gB. We have also demonstrated that anti-PILRα or anti-HVEM Ab blocks HSV-1 infection of monocytes, which constitutively express both PILRα and HVEM. Furthermore, we have found that cellular receptors for gD are required for HSV-1 infection of PILRα-expressing cells by use of gD-deficient HSV-1 and anti-HVEM serum. These data indicated that interactions of both gB and gD with specific cellular receptors are required for HSV-1 infection. These findings also established that cellular receptors for either gB or gD alone are not enough to mediate HSV-1 infection. Resistance of CD14-negative PBMC that express HVEM, but not PILRα, to HSV-1 infection also revealed that cellular receptors for gD alone are not sufficient to mediate HSV-1 infection of primary cells. Taken together, PILRα functions as an entry co-receptor for HSV-1 infection.
CHO cells transfected with cellular receptors for gD, such as HVEM or Nectin, become susceptible to HSV-1 infection (Montgomery et al., 1996; Geraghty et al., 1998; Warner et al., 1998; Shukla et al., 1999). CHO cells transfected with PILRα also become susceptible to HSV-1 infection. Interestingly, soluble gD blocked the HSV-1 infection of PILRα-transfected CHO. In addition, gD-deficient HSV-1 did not infect PILRα-transfected CHO cells. These data indicated that association of gB with PILRα alone does not mediate HSV-1 infection. CHO cells might express certain receptors for gD that alone are not enough to mediate HSV-1 infection. Indeed, CHO cells can be infected, albeit very inefficiently, by some strains of HSV, indicating the presence of weak entry receptor activity (Shieh et al., 1992). Significant amounts of transcripts for D-glucosaminyl 3-O-sulfotransferase-3, which generates the specifically modified heparan sulfate recognized by gD (Shukla et al., 1999), were detected in CHO cells by RT-PCR (data not shown). Therefore, low levels of gD ligands expressed on CHO cells, which do not mediate HSV-1 infection by themselves, seem to be required for HSV-1 infection even when a specific cellular receptor for gB, PILRα, is expressed. Furthermore, it has recently been reported that soluble gB blocks HSV-1 infection of several cell lines (Bender et al., 2005). Their findings also support the important role of cellular receptors for both gB and gD in HSV-1 infection. On the other hand, gB, as well as gC, binds cell surface heparan sulfate (Herold et al., 1991; Herold et al., 1994). However, the interaction of these envelop glycoproteins with heparan sulfate is involved in the binding of virus to the cell surface but not in viral entry (Herold et al., 1991; Laquerre et al., 1998). Therefore, PILRα plays an important role in HSV-1 infection as a cellular entry receptor for gB in collaboration with cellular receptors for gD.
The association of gD with a cellular receptor is thought to trigger membrane fusion mediated by other viral envelope proteins, gH/gL and gB (Krummenacher et al., 2005). gB has been proposed as a viral fusion protein, together with the gH / gL heterodimer (Turner et al., 1998). Recently, it has been reported that gD and gH / gL are sufficient for hemi-fusion of membranes but that gB is required to form a fusion pore (Subramanian and Geraghty, 2007). Indeed, gH possesses a putative fusion sequence and mutations in this sequence cause loss of infectivity (Gianni et al., 2005). Recent analysis of the crystal structure of gB suggested that gB exhibits a structure similar to that of vesicular stomatitis virus (VSV) G protein (Heldwein et al., 2006; Roche et al., 2006), which is fully competent by itself to mediate viral entry and viral fusion. The regions of gB homologous to the VSV G fusion loops are considered to be putative gB fusion loops even though their hydrophobicity is not representative for general fusion peptides (Heldwein et al., 2006). Considering that cellular receptors for both gB and gD are required for HSV-1 infection, interaction between gB and PILRα might function, along with gD-receptor interactions, to trigger putative gB fusion loops to contact cell membranes at viral fusion.
PILRα possesses an ITIM in its cytoplasmic domain and crosslinking of PILRα with Ab downregulated the activation of PILRα-expressing cells (Fournier et al., 2000). Therefore, PILRα might be involved in immune evasion by associating with gB. On the other hand, the ITIM sequence shares a motif with the tyrosine-based sorting signals that are involved in the internalization of cell surface molecules (Ohno et al., 1995). However, mutant PILRα that lacks the cytoplasmic domain still mediates HSV-1 infection (our unpublished observation). Therefore, the ITIM sequence of PILRα is not required for HSV-1 entry.
When we analyzed PILRα expression in various tissues, we found that the expression of PILRα is not restricted to the myeloid linage. Significant amounts of PILRα were detected in various tissues, including the nervous system. This suggested that PILRα might be involved in HSV-1 infection in various tissues. However, because expression levels of PILRα in most of these tissues are low compared to that in PBMC, there might be other receptors that mediate HSV-1 infection by associating with gB. In the present study, we have identified for the first time a specific cell surface receptor that mediates HSV-1 infection by associating with gB. Cellular receptors for both gB and gD have been shown to play an important role in HSV-1 infection of human primary cells and other cells. Thus, it seems likely that an understanding of HSV-1 entry and membrane fusion will require defining the sequence and consequences of the binding of gD and gB to two different sets of entry receptors.
EXPERIMENTAL PROCEDURES
Cell lines
293T and COS-7 were purchased from Riken Cell Bank, Japan. CHO-K1 was purchased from Health Science Research Resources Bank, Japan. CHO-K1 cells were stained with PILRα-Ig, and cells that did not stain with PILR-Ig were isolated by flow cytometry using a FACSAria (Becton Dickinson). A single cell clone obtained from the sorted cells was used as the PILR-ligand-negative CHO-K1 cell line for introduction of human PILRα.
Plasmids
A cDNA fragment of human PILRα (GenBank accession number: AF161080) was amplified by RT-PCR from human PBMC cDNA and cloned into pMXs-IRES-DsRed vector. Mouse MHC class I H2-Kb cloned into pMXs-IRES-DsRed was used for control transfections. cDNA fragments corresponding to the extracellular domain of human PILRα, HVEM, and Nectin-1 were amplified by PCR and these fragments were inserted into the Xho I cloning site of a modified pME18S expression vector that contained a mouse CD150 leader segment at the N-terminus and the Fc segment of human IgG1 at the C-terminus (GenBank accession number: AAH14667; positions: 249–479), in which the leucines at position 266 and 267 of the Fc were mutated to alanine and glutamine, respectively, in order to reduce the affinity of binding to cellular Fc receptors (Shiratori et al., 2004). Furthermore, histidine at position 467 was mutated to arginine in order to reduce the affinity of binding to HSV-1 Fc receptor (gE) (Chapman et al., 1999). cDNA fragments corresponding to the HSV-1 gB that lacks cytoplasmic 40 amino acids (GenBank accession number: M14164), full length gD (GenBank accession number: X14112), gH, and gL were amplified by PCR and were cloned into pcDNA3.1 expression vector (Invitrogen). Primers used for PCR are described in supplementary experimental procedures.
gB-expressing cell line
Because gB-deficient virus is non-infectious, we generated gB transfectants to produce infective gB-deficient virus. A 3.3 kb Xho I – Bam HI fragment of the cosmid pBC1014 (Kawaguchi et al., 1997), encoding the entire gB open-reading frame and its flanking sequences, was cloned into pBluescript KS+ (Stratagene) to yield pBS-gBL. pBC1014 was kindly provided by Dr. Bernard Roizman. A DNA fragment encoding HSV-1(F) nucleotides 55667 to 55803 amplified by PCR was digested with Xho I and Not I and substituted with the Xho I –Not I fragment of pBS-gBL. The resultant plasmid pBS-gB contains HSV-1(F) nucleotides 55803 to 52589. A DNA fragment containing the promoter region of α27 gene (HSV-1(F) nucleotide 112701 to 113673) amplified by PCR and a Xho I - Kpn I fragment of pBS-gB, containing the entire open-reading frame of gB and its polyadenylation signal, were sequentially cloned into pBluescript KS+ to yield pα27-gB. To construct pcDNA5/FRT/α27-gB, the Sph I – Kpn I fragment of pα27-gB was cloned into the Sph I and Kpn I sites of pcDNA5/FRT (Invitrogen). Flp-In-CV-1/α27-gB cells, which express gB proteins under control of the α27 promoter, were generated by cotransfection of pcDNA5/FRT/α27-gB with pOG44 (Invitrogen) into Flp-In-CV-1 (Invitrogen) according to the manufacturer’s instruction.
Viruses
Wild-type HSV-1 (F, VR3, SC16 and KOS strains) and recombinant HSV-1 (strain F) carrying GFP (YK333) (Tanaka et al., 2004) were used in this study. The recombinant HSV-1 expresses GFP driven by the Egr-1 promoter; the virus particle itself does not contain GFP. Therefore, only the cells infected with HSV-1 express GFP. Indeed, GFP expression in HSV-1-GFP infected cells was well correlated with ICP0 expression (Tanaka et al., 2004), indicating that cells expressing GFP were infected with HSV-1. Virus titers were determined by using Vero cells as previously described (Tanaka et al., 2004). gD-deficient virus (Warner et al., 1998) and gD-transfected Vero cells (VD60) (Ligas and Johnson, 1988) were previously described. gD-deficient virus produced in gD-transfected Vero cells and wild-type virus produced in normal Vero cells were used to infect normal Vero cells and viruses were collected 2 d later. Because gD-deficient virus does not infect cells, the amount of virions was determined by Western blot analysis of gB. The viral stocks containing the same amount of gB were sequentially diluted and used for infection of PILRα-expressing cells.
gB-deficient virus HSV-1 was constructed basically as described previously (Tanaka et al., 2004). A mutated HSV-1 genome carrying the substitution of a part of the gB sequence (HSV-1(F) nucleotides 53617–55793), which encodes the signal peptides and the endoplasmic region of gB, with the kanamycin-resistance gene was generated in E. coli YEbac202 harboring the full length HSV-1 genome cloned into a bacmid and pGETrec (Narayanan et al., 1999), as described previously (Tanaka et al., 2005). The gB deletion virus (YK701) was reconstituted by transfection of the mutated HSV-1 genome into Flp-In-CV-1/α27-gB, as described previously (Tanaka et al., 2003). Deletion of the gB sequence was confirmed by Southern blotting.
Revertant virus that carries gB was constructed from the gB-deficient bacmid. A DNA fragment containing HSV-1(F) nucleotides 55793 to 56586 amplified by PCR, and the Xho I - Bam HI fragment of pBC-gBL was sequentially cloned into pCR2.1 (Invitrogen) to yield pCRxgB. pCRxgB contains HSV-1(F) nucleotides 56586 to 52589. In recombinant virus YK702, the gB sequence deleted from YK701 was restored by cotransfection of YK701 DNA with pCRxgB into Flp-In-CV-1/a27-gB cells. Plaques were isolated and purified on Vero cells. Restoration of the original sequence was confirmed by Southern blotting.
Ig-fusion protein
Plasmids for Ig fusions proteins with a mutant Fc portion that has low affinity to cellular Fc receptor and HSV-1 Fc receptor (glycoprotein E) were constructed as described above. COS-7 cells were transiently transfected with pME18S-human PILRα-IgG Fc, pME18S-human HVEM-IgG Fc, pME18S-human Nectin-1-IgG Fc, or pME18S-gD-IgG Fc vector and culture supernatants were collected. As a control, purified human CD200-Ig Fc fusion protein was used (Shiratori et al., 2005). Ig fusion proteins were purified by protein A affinity chromatography.
Establishment of monoclonal antibodies
BALB/c mice were immunized with human PILRα-Ig fusion protein and TiterMax Gold adjuvant. Two weeks after immunization, lymph node cells were fused with SP2/0 and hybridomas that recognized human PILRα-transfected Ba/F3 cells (mouse pro-B cell line) were obtained. Among the hybridomas that recognize human PILRα, specific mAbs that blocked the binding of PILRα-Ig to cells expressing the extracellular domain of gB were selected. One hybridoma, M4, (IgG1 isotype) which blocked the binding of PILRα-Ig to the extracellular domain of gB, was used in this study. Antibody produced in culture supernatant was purified by protein A affinity chromatography. As a control, anti-Flag mAb M2 (mouse IgG1, Sigma) was used. Anti-HVEM mAb (clone 122) and anti-Nectin-1 mAb (clone CK8) were purchased from MBL and Zymed, respectively.
Transfection and infection
CHO-K1 cells were transiently transfected with Lipofectamine 2000 (Invitrogen) or GeneJuice (Novagen) in F-12 medium containing 10% FCS. The day after transfection, the medium was replaced with F-12 containing 1% FCS. Two days after transfection, transfectants were mixed with HSV-1-GFP, followed by centrifuge at 32o C at 1100 x g for 2 h. PBMC were obtained by using Ficoll-Paque PLUS (Amersham Biosciences) and CD14-positive and -negative cells were separated by using a MACS purification system (Miltenyi Biotec). PBMC were cultured for 2 h in Advanced RPMI-1640 (Invitrogen) containing 1% FCS and were mixed with HSV1-GFP, followed by centrifuge at 32oC at 1100 x g for 2 h. Twelve hours after infection, cells were fixed with 4% paraformaldehyde dissolved in phosphate buffered saline and expression of GFP or DsRed was analyzed by using a FACSCalibur (Becton Dickinson). In the experiments in which HSV-1 that has no fluorescence marker was used, HSV-1-infected cells were fixed with paraformaldehyde, were permeabilized by Triton-X and stained with anti-ICP4 mAb (clone 10F1, Virusys Corp.), and the proportions of stained cells were determined by flow cytometry. In the blocking experiments, cells were infected with HSV-1 in the presence of control or anti-HVEM serum (Montgomery et al., 1996).
Flow cytometry
Cells were incubated with human Ig fusion proteins or primary mouse mAbs, followed by PE-conjugated anti-human IgG or anti-mouse IgG Ab (Jackson Immunoresearch). Expression of viral glycoproteins was analyzed by using anti-gB (clone 1105, Rumbauhg-Goodwin Institute), anti-gD (clone DL6), and anti-gH (clone 53-S, ATCC) mAbs. Stained cells were analyzed by using a FACSCalibur. Data were analyzed by CellQuest Pro software (Becton Dickinson) or FlowJo software (Tree Star, Inc.).
Immunoprecipitation and immunoblotting
Cells were disrupted in lysis buffer (20 mM Tris, 150 mM NaCl, pH 7.5) containing 1% Brij 98 (Sigma). Lysates were immunoprecipitated with PILRα-Ig or Nectin-1-Ig. The immunoprecipitates were eluted by boiling with SDS-PAGE sample buffer and separated on 5–20% polyacrylamide gels. Gels were silver stained (Bio-Rad) or proteins were transferred onto PVDF membranes (Millipore). The membranes were blotted with anti-gB mAb (clone 1105), anti-gD (clone DL6) mAb, or rabbit anti-HSV Ab (ab20535, Abcam).
In-gel digestion and mass spectrometry analysis
Gel pieces from SDS-PAGE were washed and treated with destaining solution (15 mM potassium ferricyanide, 50 mM sodium thiosulfate). After reduction with 10 mM DTT, proteins were alkylated with 55 mM iodoacetamide and then digested for 16 h at 37o C with sequence-grade trypsin (Promega). The resulting peptides were extracted from the gel with 0.1% TFA in 2% ACN. The extracts were evaporated, and the residue was dissolved with 0.1% TFA in 2% ACN. Samples were then analyzed by using a nano-LC (Ultimate, LC packing) and ESI-Q-Tof MS/MS (Q-Tof Ultima API, Micromass). Mass spectrometry data were analyzed with the MASCOT program (Matrix Science Ltd.).
Cell fusion assay
PILRα and RFP (TurboRFP, Evrogen) were co-transfected to PILR-ligand negative CHO cells. gBΔC, gD, gH, gL, and GFP were co-transfected into normal CHO cells. Mouse MHC class I Kb was transfected as a control. 2 d after transfection, GFP- or RFP-positive cells were purified by flow cytometry. 1.5 x 104 CHO cells transfected with HSV-1 glycoproteins and PILRα were co-cultured for 8 h in 384-well tissue culture plates (Greiner). Cells were analyzed by fluorescence microscopy (Zeiss) and photographs were taken with a D50 digital camera (Nikon). Images were processed by Canvas software (ACD Systems).
Quantative analysis of PILRα transcript in human tissues
Total RNAs from human various tissues were purchased from Clontech. Total RNA of human PBMC was isolated by using a RNeasy mini kit (QIAGEN). The first-strand cDNA was synthesized with random hexamers by using SuperScript III reverse transcriptase (Invitrogen). Primers used for amplification of human β-actin and human PILRα were as follows: β-actin, sense primer (5’-GTG ATG GTG GGA ATG GGT CAG -3’), anti-sense primer (5’-TTT GAT GTC ACG CAC GAT TTC C -3’); PILRα, sense, (5’-AAG GTC AGC AGC GGA CTA AA-3’), anti-sense, (5’-CAG TCT TGA GAG GGC TGT CC-3’). Quantitative real-time PCR was accomplished with SYBR Green PCR Master Mix reagents (Applied Biosystems) and 7900HT First real-time PCR system (Applied Biosystems).
Immunohistochemical analysis
Formalin-fixed paraffin-embedded tissues were sectioned by standard protocols. Immunohistochemistry was performed using routine methods. Briefly, 4 mm-thick tissue sections were deparaffinized in xylene and dehydrated with ethanol. The sections were rehydrated with phosphate buffered saline containing 0.01% Tween-20 (PBST) and incubated with 0.3% hydrogen peroxidase to quench endogenous peroxidase activity. The sections were incubated with anti-PILRα or anti-keratin mAbs overnight at 4°C, and after washing with PBST, they were incubated with the Envision Dual Link solution (Dako, Glostrup, Denmark) for 30 min at room temperature. The reaction products were visualized with diaminobenzidine (Dako), and the nuclei were counterstained with hematoxylin for 90 s. Finally, the slides were mounted with the Entellan Neu reagent (Merck, Whitehouse Station, NJ) and coverslips for subsequent analysis.
Supplementary Material
Acknowledgments
We thank Dr. Mark Orr and Dr. Katsumi Maenaka for helpful discussion, Dr. Tatsuo Suzutani for providing HSV strains and Ms. R. Hirohata and M. Matsumoto for technical assistance. We also thank Dr. K. Saito of DNA-chip Development Center for Infectious Diseases (RIMD, Osaka University) for mass spectrometry analysis. This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science and Culture, Japan (Y.K. and H.A.). P.G.S. is supported by National Institutes of Health grant AI36293. L.L.L. is an American Cancer Society Research Professor and is supported by National Institutes of Health grant AI64520.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Banfield BW, Leduc Y, Esford L, Schubert K, Tufaro F. Sequential isolation of proteoglycan synthesis mutants by using herpes simplex virus as a selective agent: evidence for a proteoglycan-independent virus entry pathway. J Virol. 1995;69:3290–3298. doi: 10.1128/jvi.69.6.3290-3298.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beitia Ortiz de Zarate I, Kaelin K, Rozenberg F. Effects of mutations in the cytoplasmic domain of herpes simplex virus type 1 glycoprotein B on intracellular transport and infectivity. J Virol. 2004;78:1540–1551. doi: 10.1128/JVI.78.3.1540-1551.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender FC, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J Virol. 2005;79:11588–11597. doi: 10.1128/JVI.79.18.11588-11597.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai WH, Gu B, Person S. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J Virol. 1988;62:2596–2604. doi: 10.1128/jvi.62.8.2596-2604.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman TL, You I, Joseph IM, Bjorkman PJ, Morrison SL, Raghavan M. Characterization of the interaction between the herpes simplex virus type I Fc receptor and immunoglobulin G. J Biol Chem. 1999;274:6911–6919. doi: 10.1074/jbc.274.11.6911. [DOI] [PubMed] [Google Scholar]
- Forrester A, Farrell H, Wilkinson G, Kaye J, Davis-Poynter N, Minson T. Construction and properties of a mutant of herpes simplex virus type 1 with glycoprotein H coding sequences deleted. J Virol. 1992;66:341–348. doi: 10.1128/jvi.66.1.341-348.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier N, Chalus L, Durand I, Garcia E, Pin JJ, Churakova T, Patel S, Zlot C, Gorman D, Zurawski S, et al. FDF03, a novel inhibitory receptor of the immunoglobulin superfamily, is expressed by human dendritic and myeloid cells. J Immunol. 2000;165:1197–1209. doi: 10.4049/jimmunol.165.3.1197. [DOI] [PubMed] [Google Scholar]
- Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science. 1998;280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- Gianni T, Martelli PL, Casadio R, Campadelli-Fiume G. The ectodomain of herpes simplex virus glycoprotein H contains a membrane alpha-helix with attributes of an internal fusion peptide, positionally conserved in the herpesviridae family. J Virol. 2005;79:2931–2940. doi: 10.1128/JVI.79.5.2931-2940.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. Crystal structure of glycoprotein B from herpes simplex virus 1. Science. 2006;313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- Herold BC, Visalli RJ, Susmarski N, Brandt CR, Spear PG. Glycoprotein C-independent binding of herpes simplex virus to cells requires cell surface heparan sulphate and glycoprotein B. J Gen Virol. 1994;75:1211–1222. doi: 10.1099/0022-1317-75-6-1211. [DOI] [PubMed] [Google Scholar]
- Herold BC, WuDunn D, Soltys N, Spear PG. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J Virol. 1991;65:1090–1098. doi: 10.1128/jvi.65.3.1090-1098.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Van Sant C, Roizman B. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J Virol. 1997;71:7328–7336. doi: 10.1128/jvi.71.10.7328-7336.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Supekar VM, Whitbeck JC, Lazear E, Connolly SA, Eisenberg RJ, Cohen GH, Wiley DC, Carfi A. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 2005;24:4144–4153. doi: 10.1038/sj.emboj.7600875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laquerre S, Argnani R, Anderson DB, Zucchini S, Manservigi R, Glorioso JC. Heparan sulfate proteoglycan binding by herpes simplex virus type 1 glycoproteins B and C, which differ in their contributions to virus attachment, penetration, and cell-to-cell spread. J Virol. 1998;72:6119–6130. doi: 10.1128/jvi.72.7.6119-6130.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligas MW, Johnson DC. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by β-galactosidase sequences binds to but is unable to penetrate into cells. J Virol. 1988;62:1486–1494. doi: 10.1128/jvi.62.5.1486-1494.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RI, Warner MS, Lum BJ, Spear PG. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87:427–436. doi: 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
- Mousseau DD, Banville D, L'Abbe D, Bouchard P, Shen SH. PILRα, a novel immunoreceptor tyrosine-based inhibitory motif-bearing protein, recruits SHP-1 upon tyrosine phosphorylation and is paired with the truncated counterpart PILRβ. J Biol Chem. 2000;275:4467–4474. doi: 10.1074/jbc.275.6.4467. [DOI] [PubMed] [Google Scholar]
- Narayanan K, Williamson R, Zhang Y, Stewart AF, Ioannou PA. Efficient and precise engineering of a 200 kb β-globin human/bacterial artificial chromosome in E. coli DH10B using an inducible homologous recombination system. Gene Ther. 1999;6:442–447. doi: 10.1038/sj.gt.3300901. [DOI] [PubMed] [Google Scholar]
- Ohno H, Stewart J, Fournier MC, Bosshart H, Rhee I, Miyatake S, Saito T, Gallusser A, Kirchhausen T, Bonifacino JS. Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science. 1995;269:1872–1875. doi: 10.1126/science.7569928. [DOI] [PubMed] [Google Scholar]
- Roche S, Bressanelli S, Rey FA, Gaudin Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science. 2006;313:187–191. doi: 10.1126/science.1127683. [DOI] [PubMed] [Google Scholar]
- Shieh MT, WuDunn D, Montgomery RI, Esko JD, Spear PG. Cell surface receptors for herpes simplex virus are heparan sulfate proteoglycans. J Cell Biol. 1992;116:1273–1281. doi: 10.1083/jcb.116.5.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiratori I, Ogasawara K, Saito T, Lanier LL, Arase H. Activation of natural killer cells and dendritic cells upon recognition of a novel CD99-like ligand by paired immunoglobulin-like type 2 receptor. J Exp Med. 2004;199:525–533. doi: 10.1084/jem.20031885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiratori I, Yamaguchi M, Suzukawa M, Yamamoto K, Lanier LL, Saito T, Arase H. Down-regulation of basophil function by human CD200 and human herpesvirus-8 CD200. J Immunol. 2005;175:4441–4449. doi: 10.4049/jimmunol.175.7.4441. [DOI] [PubMed] [Google Scholar]
- Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell. 1999;99:13–22. doi: 10.1016/s0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- Spear PG. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol. 2004;6:401–410. doi: 10.1111/j.1462-5822.2004.00389.x. [DOI] [PubMed] [Google Scholar]
- Spear PG, Eisenberg RJ, Cohen GH. Three classes of cell surface receptors for alphaherpesvirus entry. Virology. 2000;275:1–8. doi: 10.1006/viro.2000.0529. [DOI] [PubMed] [Google Scholar]
- Subramanian RP, Geraghty RJ. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc Natl Acad Sci USA. 2007;104:2903–2908. doi: 10.1073/pnas.0608374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J Virol. 2003;77:1382–1391. doi: 10.1128/JVI.77.2.1382-1391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Kodaira H, Nishiyama Y, Sata T, Kawaguchi Y. Construction of recombinant herpes simplex virus type I expressing green fluorescent protein without loss of any viral genes. Microbes Infect. 2004;6:485–493. doi: 10.1016/j.micinf.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Nishiyama Y, Sata T, Kawaguchi Y. The role of protein kinase activity expressed by the UL13 gene of herpes simplex virus 1: the activity is not essential for optimal expression of UL41 and ICP0. Virology. 2005;341:301–312. doi: 10.1016/j.virol.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Turner A, Bruun B, Minson T, Browne H. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J Virol. 1998;72:873–875. doi: 10.1128/jvi.72.1.873-875.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner MS, Geraghty RJ, Martinez WM, Montgomery RI, Whitbeck JC, Xu R, Eisenberg RJ, Cohen GH, Spear PG. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology. 1998;246:179–189. doi: 10.1006/viro.1998.9218. [DOI] [PubMed] [Google Scholar]
- Whitley RJ, Roizman B. Herpes simplex virus infections. Lancet. 2001;357:1513–1518. doi: 10.1016/S0140-6736(00)04638-9. [DOI] [PubMed] [Google Scholar]
- Wilson MD, Cheung J, Martindale DW, Scherer SW, Koop BF. Comparative analysis of the paired immunoglobulin-like receptor (PILR) locus in six mammalian genomes: duplication, conversion, and the birth of new genes. Physiol Genomics. 2006;27:201–218. doi: 10.1152/physiolgenomics.00284.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.