

Abstract
Nitric oxide (NO) causes S-glutathiolation of the reactive cysteine-674 in the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA), thus increasing SERCA activity, and inhibiting Ca2+ influx and migration of vascular smooth muscle cells (VSMC). Because increased VSMC migration contributes to accelerated neointimal growth and atherosclerosis in diabetes, the effect of culture of VSMC in high glucose (HG) was determined. Rat aortic VSMC were exposed to normal (5.5 mmol/L) or high (25 mmol/L) glucose for 3d, and serum-induced cell migration during 6 h into a wounded cell monolayer was measured 5 min after adding the NO donor S-nitroso-N-acetylpenicillamine (SNAP) or 24 h after interleukin-1 β (IL-1β) to express inducible nitric oxide synthase (iNOS). In normal glucose, SNAP or IL-1β significantly inhibited migration in cells infected with adenovirus to express GFP or SERCA wild type (WT), but not with a C674S SERCA mutant. After HG, NO failed to inhibit migration, nor did it decrease calcium-dependent association of calmodulin with calcineurin, indicating that NO failed to decrease intracellular calcium levels via SERCA. In contrast, overexpression of SERCA WT, but not the SERCA C674S mutant, preserved the ability for NO to inhibit migration despite exposing the cells to HG. The antioxidant, Tempol, or overexpression of superoxide dismutase also prevented the effects of HG. Further studies showed that both biotinylated-iodoacetamide and NO-induced biotinylated glutathione labeling of SERCA C674 were decreased by HG, and a sequence-specific sulfonic acid antibody detected oxidation of the C674 SERCA thiol. These results indicate that failure of NO to inhibit migration in VSMC exposed to HG is due to oxidation of the SERCA reactive cysteine-674.
Keywords: Nitric oxide, High glucose, Migration, Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase
Introduction
Vascular dysfunction is among the most common complications associated with diabetes, and chronic hyperglycemia appears to be an important contributor in this process [1]. Vascular smooth muscle cell (VSMC) migration is an important pathological process in neointimal hyperplasia and atherosclerosis, and acceleration of these pathological processes underlie the higher rates of cardiovascular disease in diabetes. A role of oxidative stress in vascular disease associated with diabetes has been advanced by the recent demonstration that hyperglycemia enhances the proliferation of VSMC, which is blocked by antioxidants [2].
Migration of VSMC depends upon growth factor-induced increases in cytosolic Ca2+ [3], and may be inhibited by nitric oxide (NO) by preventing the increase in Ca2+ [4,5]. Following artery injury in vivo, NO produced primarily by inducible NO synthase (iNOS) inhibits smooth muscle cell migration and proliferation as evidenced by the fact that genetic deletion of iNOS [6] exacerbates experimental neointimal lesions and iNOS gene transfer decreases neointimal formation [7]. The sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) plays an important role in maintaining intracellular Ca2+ homeostasis through its ability to pump cytosolic Ca2+ into the SR/ER stores, which in turn regulates store-dependent Ca2+ influx [4]. NO stimulates SERCA activity, increases intracellular Ca2+ stores, inhibits store-dependent Ca2+ influx, and lowers cytosolic Ca2+, thereby inhibiting Ca2+-dependent cell processes including smooth muscle contraction [4] and migration [8]. Importantly, overexpression of SERCA in injured carotid arteries inhibits neotinimal growth [9].
In our previous studies, we proposed a molecular mechanism by which NO causes cyclic GMP-independent vascular relaxation by increasing SERCA activity by a thiol redox-dependent mechanism. Briefly, NO increases SERCA activity by inducing the reversible S-glutathiolation of specific cysteines in SERCA, predominantly on the most reactive thiol on cysteine-674 [10]. Formation of this adduct on normal SERCA is associated with increased Ca2+-uptake into intracellular stores. In a pathological model of atherosclerosis, cysteine-674 was irreversibly oxidized due to prolonged oxidative stress, thereby accounting for impaired NO-induced S-glutathiolation and activation of SERCA. The potential importance of redox regulation of SERCA cysteine-674 was recently demonstrated by the finding that expression of a SERCA C674S mutant prevented the inhibition by NO of serum-induced migration of human erythroid leukemia cells and rat aortic VSMC in culture [8]. The NO-induced glutathiolation of the stably overexpressed SERCA C674S mutant was dramatically decreased compared to WT SERCA protein, and the inhibition of calcium influx by NO was prevented [10]. In the current study, we hypothesized that during oxidant stress, such as that produced by hyperglycemia in diabetes, redox-dependent SERCA stimulation might be inhibited by oxidation of cysteine-674. Therefore, we tested the effect of high glucose on smooth muscle cell migration and determined the role of SERCA cysteine-674. We found that although it did not affect VSMC migration directly, HG decreased NO-induced S-glutathiolation of SERCA and prevented the inhibition of migration by an NO donor or cytokine-induced iNOS expression. Overexpression of wild type (WT) SERCA, or antioxidants, but not the C674S SERCA mutant preserved the effect of NO, indicating that HG exerted an oxidant effect that was dependent on cysteine-674. Indeed, HG significantly increased oxidation of SERCA cysteine-674 as indicated by thiol labeling and immunochemical methods. Therefore, these studies indicate not only a novel mechanism by which oxidants associated with HG may enhance cell migration by oxidation of a specific SERCA cysteine that normally mediates redox-dependent stimulation of the enzyme, but also shows that the beneficial effects of antioxidants to maintain the action of NO may be ascribed to maintaining the redox active state of a specific amino acid residue on SERCA.
Methods
Construction of adenoviral expression vectors for SERCA 2b WT and SERCA 2b C674S
Full-length human SERCA 2b constructed in pcDNA 3.1 was a gift from Dr. Jonathan Lytton. The mutagenesis of cysteine-674 was performed as previously described using the Stratagene mutagenesis kit [10]. Briefly, the primers for C674S were: 5′-CTGCCTGAACGCCCGCTCTTTTGCTGAGTTGAAC-3′ (boldface type indicates codon in which mutation was made, underline indicates mutated nucleotide). The inserts of SERCA 2b WT and SERCA 2b C674S were excised from pcDNA3.1 using PME-1 and ligated into the EcoRV site of pShuttle-CMV. The correct direction was confirmed by enzyme restriction analysis as well as sequencing. pShuttle-SERCA 2b WT and C674S were linearized and then recombined with pAdEasy in BJ5183 cells and colonies were screened for the appropriate constructs using restriction enzyme analysis. The appropriate cosmids were cut by PAC-1 digestion and infected into HEK-293 cells. Adenoviral colonies were chosen and amplified as previously described [10].
Rat aorta vascular smooth muscle cell (VSMC) culture
VSMCs from passage 8 to 12 were used and cultured in DMEM medium with 10% FBS as described [11].
Biotinylated-iodoacetamide labeling of SERCA Cysteine-674
The method for biotinylated-iodoacetamide (b-IAM) labeling of the reactive thiol on cysteine-674 in SERCA followed those previously reported [10] with modifications. Briefly, cells were lysed in buffer A (Tris-HCL 50 mmol/L pH8.5, NaCl 150 mmol/L, MgCl2 5mmol/L, DETA-PAC 50 μmol/L, PMSF 2mmol/L, Triton 0.5% and 100 μmol/L NEM) on ice for 50 min. Pretreatment with this low concentration of NEM was used to minimize incorporation of b-IAM label into protein that was partially denatured during cell lysis according to previous studies [12, 13]. The excess NEM was removed by gel filtration using Biospin 6 columns (Biorad). The cell lysate was then incubated with b-IAM (1 mmol/L) in buffer B (MES 50 mmol/L pH 6.5, NaCl 150 mmol/L, MgCl2 5 mmol/L, DETA-PAC 50 μmol/L, PMSF 2 mmol/L and Triton X-100 1%) in the dark at 25°C for 30 minutes. The labeling reaction was terminated by adding β-mercaptoethanol (β-ME) to a final concentration of 50 mmol/L. The excess reagent was removed by Biospin 6 columns. Finally, 500 μg cell lysate protein was incubated with 50 μL streptavidin-Sepharose beads overnight at 4°C. The beads were rinsed 3 times using buffer C (Tris-HCl l25 mmol/L pH 7.4, NaCl 500 mmol/L, MgCl2 5 mmol/L and 2% SDS), the b-IAM labeled proteins were released by Laemmli buffer with 5 mol/L urea and 5% β-ME at 55°C for at least 30 min. Proteins were separated by SDS-PAGE, and SERCA was detected by immunoblot. IID8 monoclonal anti-SERCA antibody, which does not detect rat SERCA in VSMC, was used to detect overexpressed human SERCA (Affinity Bioreagent, IID8 910, 1:2,000). In addition a custom anti-peptide rabbit polyclonal antibody raised against the SERCA sequence K30-A43 which is common to human and rat, was used to detect both human and rat SERCA (see supplemental figure 3 in reference 8 and supplemental figure 1A).
Detection of S-glutathiolation of SERCA
The cells were preincubated in physiological salt solution (PSS, KCl 4.7 mmol/L, CaCl2 2.5 mmol/L, NaCl 118.3 mmol/L, KH2PO4 1.2mmol/L, MgSO4 0.6 mmol/L, NaHCO3 25 mmol/L, and dextrose 5.5 mmol/L) containing 250 μmol/L biotinylated GSH ester for 1 h. The biotinylated GSH ester was prepared as previously described [14]. Then NO gas (10 μmol/L) prepared as previously described was added to a dish of cells while swirling for 1 min. Cells were lysed immediately in a cell lysis buffer (Cell Signaling) with NEM (10 mmol/L) added to block further thiol reactions. Excess reagents were removed with Biospin 6 columns. Cell lysate protein (1000 μg) was incubated with 50 μL streptavidin-Sepharose beads overnight at 4°C. The beads were rinsed 3 times using buffer C, and the S-glutathiolated proteins were released by elution buffer (buffer A with 10 mmol/L DTT) at 55°C for at least 30 min. Each sample was diluted in Laemmli buffer containing 5% β-ME, separated by SDS-PAGE, and SERCA was detected by immunoblotting.
Wounded monolayer migration assay in adenoviral infected VSMC
VSMC were seeded into 6-well cell culture plates in DMEM with 10% FBS. When 80% confluent, cells were infected with GFP, or human SERCA 2b WT or SERCA 2b C674S mutant adenovirus in DMEM with 0.1% FBS. Two days later, cells were switched to medium (0.1% FBS) containing 5.5 mmol/L glucose, 5.5 mmol/L glucose plus 19.5 mmol/L mannose, or 25 mmol/L glucose for an additional 3 days. In pilot studies, we were able to select concentrations of adenovirus to overexpress similar protein levels of SERCA WT and SERCA C674S in VSMC 2 days following infection, and infection had no notable effect on cell morphology. These concentrations were used in all experiments. For cell migration studies, the NO donor, S-nitroso-N-acetylpenicillamine (SNAP), was added 5 min before the scratch injury by replacing DMEM with the same medium containing SNAP (200 μmol/L). SNAP was chosen because its half-life in aqueous solution at 37°C is approximately 360 min, and the concentration used provided a nearly constant NO concentration of 1–1.5 μmol/L over the 6 h period for the migration measurements as calculated previously [15]. Similar results in these studies were obtained with DETA-NONOate [8]. The cell monolayer was wounded by making a scratch with a plastic 200 μL pipette tip. After the injury, cells were gently rinsed with PBS to remove unattached cells and incubated in the dark with DMEM with 10% FBS with or without SNAP. The scratch was marked at three sites that were photographed (at 40× magnification) using a Nikon Diaphot 300 microscope. The width of the scratch line was measured at each of the three sites and averaged at each time point using SPOT Advanced software. Tempol (100 μmol/L) was added throughout the 3-day exposure to HG as well as during the 6 h migration assay. IL-1β (5 ng/mL) was used to induce iNOS expression, which was detected by anti-iNOS antibody (1:1,000, Transduction Laboratories) 1 day after IL-1β application. IL-1β or IL-1β together with the iNOS inhibitor, N6-(1-iminoethyl)-L-lysine (L-NIL, 10 μmol/L) was added to DMEM 24 h after VSMC were infected with adenoviral SERCA for 3 days before the migration assay. Adenovirus CuZnSOD and MnSOD (10 pfu/cell) were used to infect VSMC in HG for 2 days and IL-1β was added for the final 24 h before the migration assay.
Cell membrane preparation
VSMC were cultured in DMEM containing normal glucose and 10% FBS. After cells reached subconfluence, they were switched to DMEM with 0.1% FBS containing normal glucose or HG for 3 days. Cells were washed with cold PBS and lysis buffer was added (10 mM Tris-HCl, 0.3 M sucrose, 1% Triton, pH 7.0, containing protease inhibitors and PMSF). All the following steps were done at 4ºC. Cells were scraped and collected in Eppendorf tubes and underwent sonication three times. Supernatant was collected after centrifuging at 14,000 rpm for 10 min, which then was centrifuged at 43,000 rpm for 1 h. The pellet was dissolved in lysis buffer stored at −80 ºC.
Immunoblotting for SERCA C674-SO3H
A polyclonal antibody was made against the human SERCA peptide, 669CLNARC*FARV678, in which cysteine-674 (*) was substituted with cysteric acid, the SO3H thiol oxidation product of cysteine. Immunochemical staining with the antibody was verified to be specific by showing that the immunogenic peptide blocked the staining, but neither the scrambled peptide nor the peptide with reduced cysteine had any effect (Ying et al., unpublished).
Calcium-dependent association of calmodulin with calcineurin
To assess chronic changes in intracellular calcium caused by NO during the migration assay period, calcium-dependent association of calmodulin (CaM) with calcineurin (PP2B) and was detected after CaM immuno-precipitation. After 3 d in normal glucose or HG, VSMC were treated similarly to cell migration studies, except that the NO donor used was DETA-NONOate (300 μmol/L) and cell lysates were collected after 1 h. 10 μg rabbit anti-CaM antibodies (ZYMED laboratories) were added to 1 mg cell lysate in each group, and rotated overnight at 4ºC. Protein A Sepharose beads were used to pull down the antibody bound proteins. The bound proteins were eluted with Laemmli buffer containing 5% β-ME, separated by SDS-PAGE, and CaM and PP2B (BD Biosciences) were detected by immuno-blot.
Data analysis
All experiments were repeated at least 3 times unless otherwise indicated. Data are expressed as means ± SEM. The bands on immunoblots were quantified by densitometry (Molecular analyzer, Biorad). Statistics were analyzed with SPSS 13.0 as indicated for each experiment, and statistical significance was accepted for a P value less than 0.05. Paired comparisons within one cell group treated with or without SNAP, or IL-1β were analyzed with paired Student t-test. Unpaired Student t-test was used for comparisons made between cells overexpressing WT SERCA and C674S SERCA. When comparisons were made among multiple groups, an ANOVA followed by a post hoc S-N-K test was used.
Results
The effect of exogenous NO released from NO donor, SNAP, on VSMC migration in normal glucose, high mannose and high glucose
Using a polyclonal anti-SERCA antibody K30/A43 that detects both human and rat SERCA there was about 3-fold increase of SERCA expression levels after adenovirus infection (supplemental figure 1A). There was no significant difference in SERCA protein expression after infection with SERCA WT or SERCA C674S mutant of VSMC exposed to normal glucose, high mannose, or high glucose (Supplemental figure 1B). Six hours after wounding the cell monolayer in normal glucose, SNAP significantly inhibited the migration of cells infected with either Ad-GFP or Ad-SERCA WT, but had no significant effect in cells infected with Ad-SERCA C674S (Figure 1A and B). In contrast, SNAP failed to inhibit migration in Ad-GFP infected cells exposed to high glucose. SNAP inhibited migration in cells exposed to a high concentration of mannose, a non-metabolized glucose analog, similarly to cells exposed to normal glucose, indicating that the osmolarity of the high glucose was not a factor. Interestingly, overexpression of SERCA WT, but not the SERCA C674S mutant, maintained the ability of SNAP to inhibit migration despite exposing the cells to HG (Figure 1C). These results indicate that high glucose prevents the inhibition of migration by SNAP, and that SERCA WT, by a mechanism involving cysteine-674 can overcome the effect of high glucose.
Figure 1.
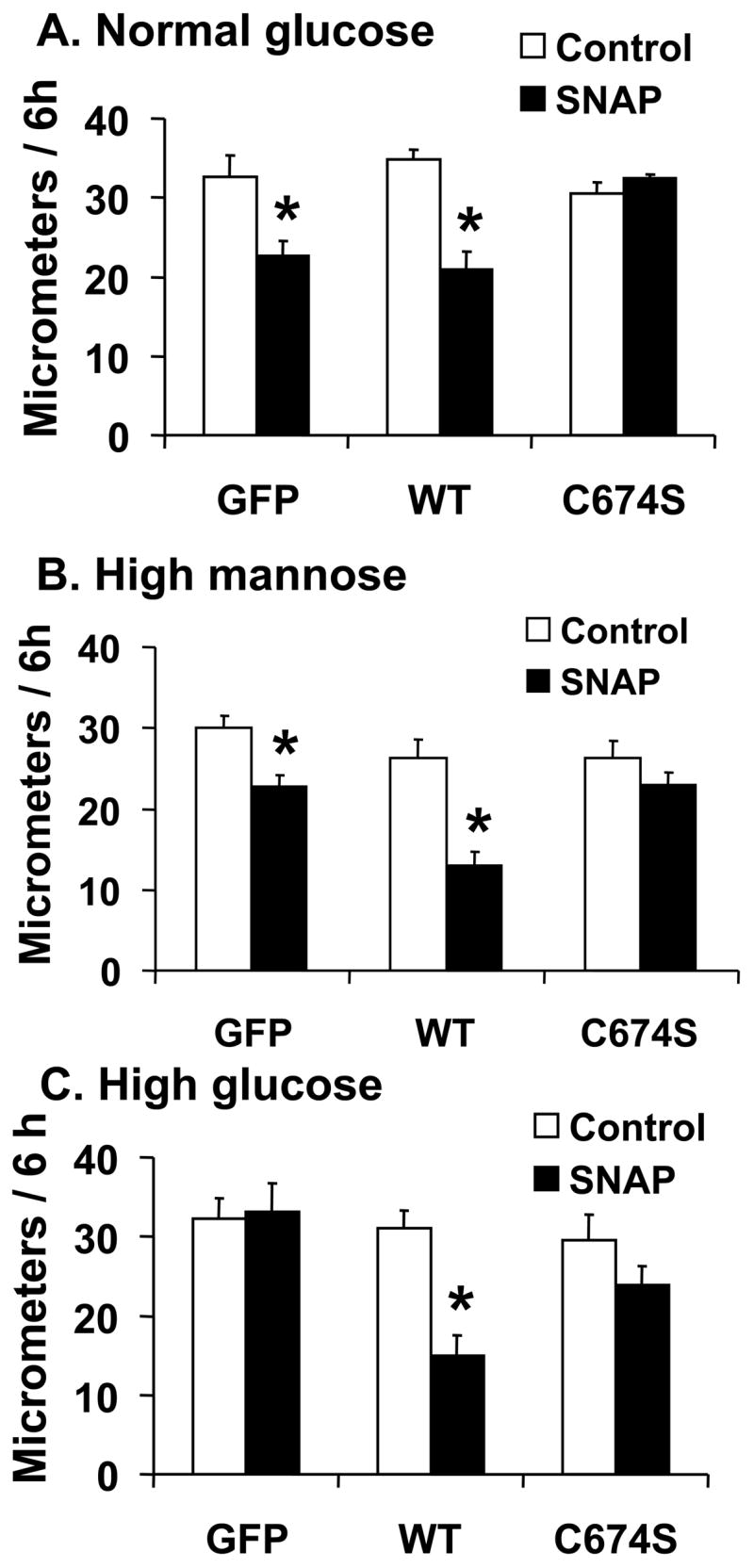
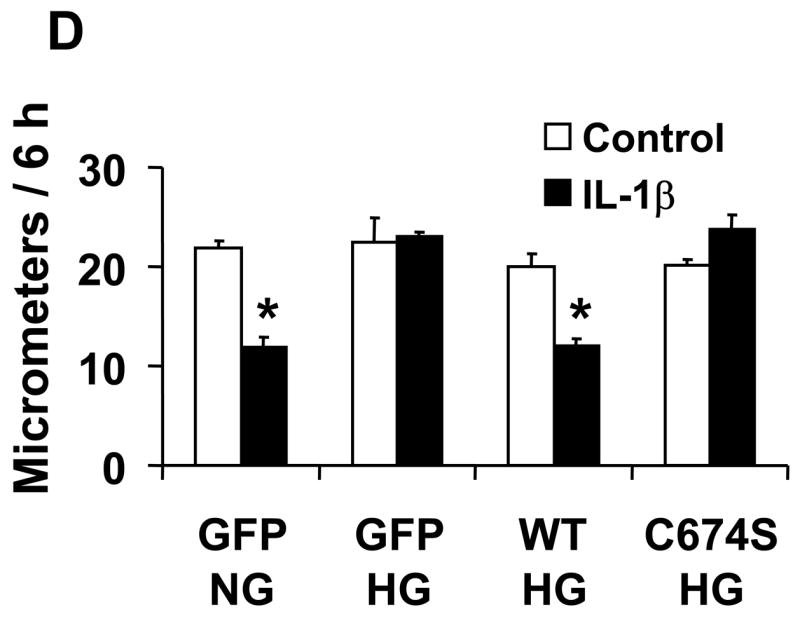
The effect of NO on the migration of rat aortic vascular smooth muscle cells (VSMC). A and B: NO donor SNAP significantly inhibited the migration of cells infected with either Ad-GFP or Ad-SERCA WT, but had no effect in cells infected with Ad-SERCA C674S in normal glucose (5.5 mmol/L) or high mannose (19.5 mmol/L plus glucose 5.5 mmol/L). C: In cells exposed to high glucose (25 mmol/L), SNAP did not inhibit migration. Overexpression of SERCA WT, but not SERCA C674S, preserved the ability of SNAP to inhibit migration. The results (A, B and C) are n=6 (mean ± SEM). *P<0.05, paired t-testbetween cells treated or not with SNAP. D: Cells were infected with Ad-WT or Ad-C674S SERCA for 2 d and then switched to medium containing NG or HG for an additional 3 days. Interleukin-1β (IL-1β, 5 ng/mL) was added 24 h before the migration assay to induce iNOS expression which releases NO. Ad-GFP served as a control. The results are n=5 (mean ± SEM). *P<0.05, paired t-test between cells treated or not with IL-1β.
The effect of endogenous NO released by iNOS on the migration of VSMC in normal and high glucose
In cultured VSMC exposed to normal or high glucose for 3 days, iNOS was expressed similarly following 24 h induction by IL-1β (Supplemental figure 1C). The overexpression of SERCA WT and SERCA C674S mutant was significantly greater in VSMC exposed to IL-1β; nevertheless, SERCA expression was similar in cells exposed to normal and high glucose (Supplemental figure 1C).
In VSMC exposed to normal glucose, IL-1β significantly inhibited migration in cells infected with GFP (Figure 1D), but the inhibitory effect of IL-1β on migration was absent in cells exposed to high glucose. Similar to their effect on the response to SNAP, overexpression of SERCA WT, but not SERCA C674S mutant, preserved the ability of IL-1β to inhibit migration in high glucose. To confirm that the effect of IL-1β to inhibit migration in VSMC exposed to high glucose is through endogenous NO released by iNOS, the iNOS inhibitor, L-N6-(1-iminoethyl) lysine hydrochloride (L-NIL, 10 μmol/L), was used to block the synthesis of NO by iNOS. In cells infected with WT SERCA and exposed to high glucose, L-NIL blocked the inhibition of migration caused by IL-1β in VSMC (Supplemental figure 2). These results indicate that high glucose prevents inhibition of VSMC migration caused by NO produced endogenously from iNOS, and that overexpression of SERCA WT restores the ability of endogenous NO to inhibit migration of cells exposed to high glucose by a mechanism involving cysteine-674.
NO-induced biotinylated glutathione and biotinylated iodoacetamide labeling of SERCA is decreased, and sulfonic acid SERCA is increased by high glucose, indicating oxidation of cysteine-674
We hypothesized that oxidants arising during exposure to high glucose could oxidize the reactive thiol on cysteine 674. To test this hypothesis, biotinylated GSH ester was used to label SERCA in uninfected cells exposed to normal or high glucose. NO gas (10 μmol/L) was added to induce S-glutathiolation of endogenous SERCA, and S-glutathiolated SERCA was pulled down with streptavidin-Sepharose beads, separated by SDS PAGE, and immuno-blotted with anti-SERCA antibody. As shown in Figure 2A with similar expression of total SERCA protein, b-GSS-SERCA was significantly decreased in cells exposed to high glucose. To specifically probe the redox status of the reactive cysteine-674 in uninfected VSMC exposed to high glucose, b-IAM was employed, which primarily labels reduced cysteine-674 [8]. High glucose decreased b-IAM labeling of SERCA by approximately 75%, indicating significant oxidation of cysteine-674 (Figure 2B). Thus, high glucose oxidizes cysteine-674 explaining the decreased NO-induced S-glutathiolation of SERCA.
Figure 2.


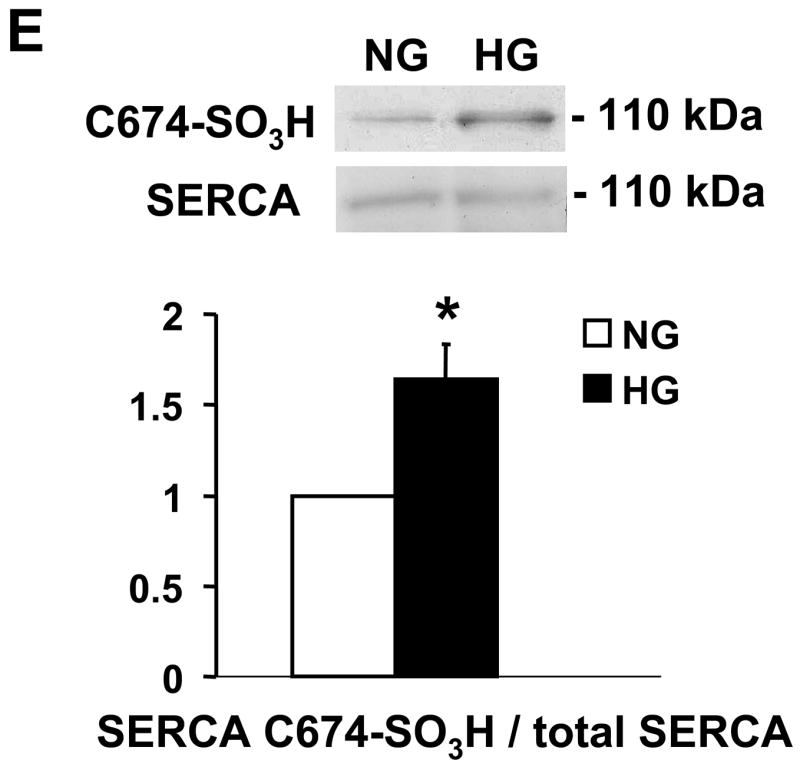
NO-induced biotinylated glutathione (b-GSH) and biotinylated iodoacetamide (b-IAM) labeling of SERCA and cysteine-674 sulfonic acid oxidation. A: b-GSH and b-IAM labeling of SERCA in uninfected VSMC exposed for 3 days to normal glucose (NG) or high glucose (HG). SERCA was detected with K30/A43 antibody. B: Bar graph shows summary of densitometry performed in three experiments indicating that both b-IAM labeling of SERCA and b-GSH binding to SERCA in uninfected VSMC exposed to HG were significantly less than that in NG. *P<0.05, paired t-test between cells treated with NG and HG. C. NO-induced S-glutathiolation of SERCA in VSMC infected with WT or C674S mutant SERCA exposed to NG or HG. SERCA was detected with IID8 910 antibody. D. Bar graph shows summary of 3 independent experiments indicating that NO-induced S-glutathiolation of SERCA was decreased in cells infected with SERCA WT exposed to HG compared to those exposed to NG (*P<0.05). Despite the fact that S-glutathiolation of SERCA was significantly decreased in cells infected with SERCA WT exposed to HG, the level remained significantly above that in cells infected with SERCA C674S mutant (# P<0.05). E: In membranes prepared from VSMC, a sequence-specific antibody against the SERCA cysteine-674 sulfonic acid shows increased detection in cells exposed to HG compared to cells exposed to NG. Total SERCA expression was not different. The bar graph in the lower panel summarizes 3 independent experiments showing that high glucose significantly increases the amount of the irreversible oxidation of SERCA expressed as the ratio of the densitometric values for detection with the SERCA C674-SO3H antibody with the total SERCA detected by the K30 antibody. *P<0.05, paired t-test.
In order to understand why overexpression of SERCA WT overcame the inhibition caused by high glucose, NO-induced b-GSS-SERCA labeling was assessed after VSMC infected with either SERCA WT or C674S mutant were exposed to normal or high glucose (Figure 2C and D). As expected, NO-induced S-glutathiolation of SERCA was greater in cells infected with SERCA WT compared with those infected with SERCA C674S mutant regardless of exposure to high or low glucose. Similar to that observed in uninfected cells (Figure 3A), the NO-induced S-glutathiolation of overexpressed SERCA WT was significantly decreased in cells exposed to high glucose (Figure 2C and D). However, the residual NO-induced S-glutathiolation of SERCA in VSMC infected with SERCA WT and exposed to high glucose was significantly greater than that observed in cells infected with the SERCA C674 mutant (Figure 2D). These data indicate that NO-induced S-glutathiolation of SERCA cysteine-674 is decreased by high glucose exposure despite overexpressing SERCA WT, but that the amount of SERCA that NO can S-glutathiolate after exposure to high glucose remains significantly above that in cells infected with the SERCA C674S mutant.
Figure 3.


Tempol and overexpression of Cu/Zn SOD or MnSOD preserve NO-induced inhibition of migration in VSMC exposed to HG. A: Summary of cell migration in uninfected VSMC exposed to high glucose showing that treatment with Tempol has no effect on migration of cells in the absence of SNAP, but protects the ability of SNAP to inhibit migration of cells exposed to HG. N=3, *P<0.05, two-way ANOVA. B: Infection of cells with adenoviral vectors to overexpress Cu/Zn SOD and MnSOD preserved the ability of IL-1β induced iNOS to inhibit migration in VSMC exposed to HG. N=3, *P<0.05, two-way ANOVA. C: Tempol prevents the decrease in b-IAM labeling of SERCA in uninfected VSMC exposed to high glucose. The bar graph summarizes densitometry data from 3 independent experiments. *P<0.05, one-way ANOVA.
To further assess the oxidation of SERCA cysteine-674, a specific antibody was developed that targets the irreversible oxidized sulfonic acid at SERCA cysteine-674 (C674-SO3H). As shown in Figure 2E, the amount of the irreversible oxidation of SERCA in membranes prepared from VSMC exposed to high glucose was significantly increased compared to that in cells exposed to normal glucose.
Tempol and overexpression of Cu/ZnSOD and MnSOD restore NO-induced inhibition of migration in VSMC exposed to high glucose
The antioxidant SOD mimetic, Tempol, was used to test the hypothesis that oxidants were responsible for preventing the inhibition of migration by NO in VSMC exposed to high glucose. As shown in Figure 3A, Tempol did not affect the migration rate of VSMC exposed to HG for 3 d, but it preserved the ability of SNAP to inhibit migration.
To further test the role of oxidants induced by high glucose in the abnormal response to iNOS derived NO in VSMC migration, Cu/Zn SOD and MnSOD were overexpressed with adenoviral vectors during the 3 d exposure to HG. Expression of neither antioxidant enzyme affected migration of VSMC exposed to HG, however, both preserved the ability of IL-1β induced iNOS to inhibit migration (Figure 3B). In similar experiments, adenoviral expression of neither catalase nor glutathione peroxidase restored inhibition of cell migration by IL-1β suggesting that hydrogen peroxide is not involved (data not shown).
Based on studies shown in Figure 2A and B, we hypothesized that Tempol preserves the ability of NO to inhibit migration by protecting the reactive thiol on cysteine-674 from being oxidized during exposure to high glucose. The data in Figure 3C shows that labeling of cysteine-674 with b-IAM that is markedly decreased by exposure to HG for 3 d is maintained by Tempol similar to that in cells exposed to normal glucose. This indicates that Tempol preserves the reduced thiol on cysteine-674 during exposure to HG.
High glucose prevents the ability of NO to decrease calcium-dependent association of calmodulin and PP2B
To detect the effects of HG on the ability of NO to mediate a prolonged decrease in intracellular free calcium in VSMC, the calcium-dependent association between calmodulin (CaM) and the calcium-dependent phosphatase, calcineurin (PP2B), was assessed because it requires a sustained increase in intracellular calcium levels. As presented in Figure 4, 1 h exposure of VSMC to NO donor significantly decreased the association of CaM with PP2B. In contrast, NO failed to significantly affect the CaM-PP2B association in cells exposed to HG for 3 d. This result is consistent with HG causing a disruption in the ability of NO to regulate SERCA-dependent decreases in intracellular calcium levels.
Figure 4.
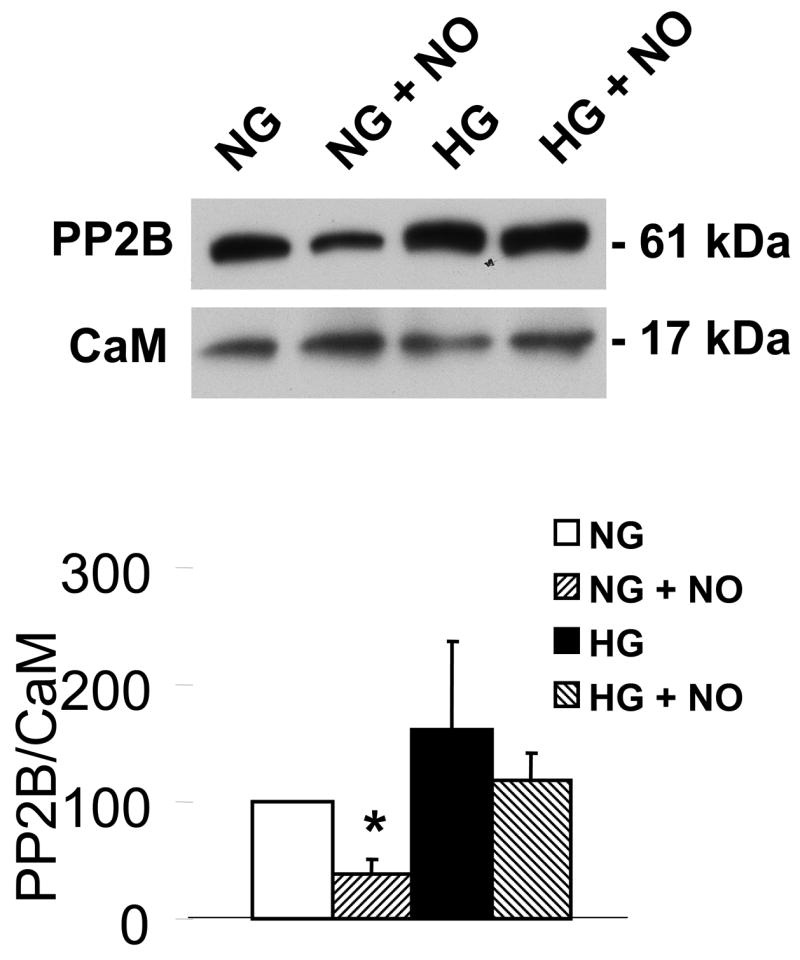
High glucose prevents the ability of NO to decrease calcium-dependent association of calmodulin (CaM) with PP2B. The upper panel shows that immuno-precipitated CaM is associated with less PP2B in VSMC treated with DETA-NONOate when the cells were exposed to NG, but not HG for 3 d. Immuno-blot showed that similar amounts of CaM were contained in the immuno-precipitate. The bar graph in the lower panel summarizes the ratio of the densitometric values detected by the PP2B antibody with that by the CaM antibody in 3 independent experiments. These data show that NO decreases the association of CaM-PP2B in NG, but not in HG. *P<0.05, two-way ANOVA.
Discussion
VSMC migration is an important pathological process in several vascular occlusive diseases, including atherosclerosis and restenosis, both of which are accelerated in diabetes. Using a cell culture model of smooth muscle cell migration, the results of the present study suggest a mechanism whereby oxidants generated by high glucose prevent NO-induced S-glutathiolation of the most reactive cysteine in SERCA, thereby interfering with a mechanism that normally inhibits migration. The most important findings of the current study are that in uninfected or Ad-GFP infected VSMC exposed to high glucose, the NO-induced inhibition of migration was absent, while overexpression of SERCA WT or SOD, or the antioxidant Tempol preserved the response to NO. The normal response to NO was associated with S-glutathiolation of SERCA cysteine-674, which we previously showed increases SERCA activity, lowers intracellular Ca2+, and inhibits cell migration by a cyclic GMP-independent mechanism [8,10]. Of relevance to the migration of smooth muscle cells in vivo, NO produced by cytokine-induced iNOS also failed to inhibit migration of smooth muscle cells exposed to high glucose. These results suggest that oxidation of SERCA may be a key mechanism by which high glucose promotes neointimal lesions whose growth is normally impeded by iNOS expressed in the lesions [7,16].
Dedifferentiation of smooth muscle cells precedes their migration from the media to the intima, where their proliferation leads to the formation of the occlusive neointima. The smooth muscle cells used in this study have undergone phenotypic changes from a differentiated contractile phenotype in the rat aorta to a dedifferentiated, synthetic phenotype, thus creating a model in cell culture of the phenotype of the migrating cell type in the neointima. Studies by others indicate that an important feature of the dedifferentiated VSMC phenotype in cell culture as well as the neointima is downregulation of cyclic GMP signaling [17]. In such cells, the cyclic GMP-independent regulation of SERCA by NO is likely to be more important than in well-differentiated VSMC.
Notably, overexpression of SERCA 2b in this study and our previous study [8] failed to influence VSMC migration; it only altered regulation of migration by NO. These results are in contrast with those of Lipskaia et al [9] who found that overexpression of SERCA 2a inhibits VSMC proliferation. While we do not know if overexpression of SERCA 2a inhibits VSMC migration, differences in the ability of the two SERCA2 isoforms to inhibit proliferation might be due to differences in calcium sensitivity that have been described [18]. However, we would expect similar NO-dependent regulation of SERCA 2a and 2b, because of the identical amino acid sequence in the region of the protein containing cysteine-674, and because SERCA from heart, in which SERCA2a predominates, is S-glutathiolated and stimulated by NO [10].
We previously proposed that chronic elevations of oxidants in atherosclerosis irreversibly oxidized the thiol of SERCA cysteine-674, thereby inhibiting its NO-induced S-glutathiolation and impairing NO-induced arterial relaxation [10]. Decreased biotinylated iodoacetamide binding to SERCA in smooth muscle cells exposed to high glucose indicates that cysteine-674, which accounts for more than 75% of IAM binding, [8, 19] is oxidized. An increase in oxidants associated with high glucose is also indicated by the fact that treatment with the antioxidant, Tempol, prevented the decrease in b-IAM binding. The irreversible oxidization SERCA cysteine-674 was confirmed directly by detecting the sulfonic acid form of cysteine-674 with a sequence-specific antibody in a membrane preparation of cells exposed to high glucose. The oxidation of SERCA cysteine-674 also explains the decreased biotinylated S-glutathiolation of SERCA, which we have shown also occurs predominantly on cysteine-674 [8, 10]. Having shown that the C674S SERCA mutant is not S-glutathiolated or stimulated by NO, and in addition, that NO does not inhibit migration of cells overexpressing this mutant [8], the oxidation of cysteine-674 in high glucose exposed cells also explains why NO does not inhibit serum-induced migration. Thus, although other amino acid residues of SERCA, including other cysteines may be affected during the high glucose exposure, these studies point to oxidation of cysteine-674 rather than to other potential modifications of the enzymes as the most important for the loss of response to NO.
Overexpression of wild type human SERCA 2b restored the ability of NO to inhibit VSMC migration despite exposure to high glucose. This is not because the expressed SERCA cysteine-674 thiol is not oxidized. In fact, b-IAM labeling showed that while the total amount of cysteine-674 binding increased with SERCA expression, there still was a relative decrease in b-IAM binding in cells overexpressing WT SERCA that had been exposed to high glucose. These results can be interpreted to indicate that the reason why overexpressing wild type SERCA preserves the ability of NO to inhibit VSMC migration is that the total amount of SERCA that is capable of responding to NO by S-glutathiolation of cysteine-674 is increased compared with uninfected cells. The essential nature of cysteine-674 is reemphasized by the fact that overexpression of the cysteine-674 mutant fails to increase b-IAM binding or the response to NO. Our results indicate that endogenous rat SERCA that may remain in C674S mutant infected cells is not sufficient to retain a response to NO. Possibly for reasons related to protein turnover during the prolonged time course of these studies, the expressed human protein may suppress expression of endogenous rat SERCA. Because we lack an antibody that detects rat SERCA to the exclusion of human SERCA, we have not been able to assess residual rat SERCA in VSMC that overexpress the human protein. Because expression of WT SERCA did not affect migration in response to serum itself, and the C674S mutant had no effect on the ability of VSMC to respond to NO despite similar levels of expression and similar calcium uptake activity [10] as the WT protein, these results raise the possibility that therapeutic effects of increasing SERCA expression [9] may in part be due to increasing expression of SERCA protein that possesses the redox-active cysteine-674 that is responsive to NO.
Cells in the neointima including smooth muscle cells [20] express iNOS, and although its role in regulation of neointimal growth is controversial [6, 21], several studies agree that overexpression of iNOS inhibits neointimal expansion [7, 16]. For these reasons, and to assess the role of endogenously produced NO on migration of smooth muscle cells exposed to high glucose, iNOS was induced by IL-1β, a cytokine relevant to the induction of iNOS and neointimal formation [22]. The inhibition of migration by endogenous NO released from iNOS was also inhibited by high glucose, indicating the potential pathophysiological importance of oxidation of SERCA cysteine-674. This occurred despite a slight increase in SERCA expression in cells stimulated by the cytokine. Once again, the failed response to NO could be restored with wild type SERCA, but not the SERCA mutant indicating the importance of oxidation of cysteine-674 demonstrated to occur in uninfected cells.
In this dedifferentiated VSMC model, NO regulates intracellular calcium levels mainly through its stimulation of SERCA activity which pumps cytosolic Ca2+ into the SR/ER, lowers cytosolic Ca2+ levels, and inhibits cell migration [4]. In uninfected VSMC NO decreased the calcium-dependent association of CaM with PP2B in normal glucose, but not in high glucose, indicating that NO fails to decrease intracellular Ca2+ in cells exposed to HG (Figure 5). Because we have previously demonstrated that SERCA cysteine-674 is essential for NO to normally decrease intracellular Ca2+, it is logical to conclude that oxidation SERCA cysteine-674 in high glucose is key in preventing the inhibition of migration by NO.
Figure 5.

The proposed mechanism of the regulation of NO of cell migration through redox regulation of SERCA2b in normal and high glucose. In normal glucose, NO released from exogenous NO donor or from iNOS induced by IL-1β induces the reversible S-glutathiolation of SERCA, predominantly on the most reactive thiol on cysteine 674, which increases SERCA activity and intracellular calcium uptake into SR/ER, decreases calcium influx, and inhibits cell migration. High glucose interrupts this cascade by the oxidation of the reactive thiol on SERCA cysteine 674, blocks the NO-induced S-glutathiolation of SERCA, and abolishes the inhibitory effect of NO on cell migration.
Preventing the oxidation of cysteine-674 with antioxidants restored the response of migrating smooth muscle cells to NO, demonstrating not only the importance of oxidants, but also providing an explanation for the observed therapeutic effect of Tempol on neointimal formation [23]. These studies indicate that irreversible oxidation of the most reactive cysteine in SERCA can explain the loss of response to NO in arteries exposed to hyperglycemia and accelerated neointimal formation [24] and atherosclerosis [25] observed in diabetes.
Supplementary Material
Supplemental figure 1. A: The overexpression level of SERCA2b and SERCA2b C674S in normal glucose detected by two different antibodies. GFP infected and uninfected VSMC serve as controls. 15 μg protein in each group was loaded and detected by K30/A43 (1:500) and IID8 (1:2,000) separately. K30/A43 is a polyclone antibody recognizing both rat and human SERCA. IID8 (910) is a monoclone antibody which recognizes human SERCA but not rat SERCA. B: Overexpression of WT or C674S mutant SERCA in VSMC in normal glucose (NG), high mannose (MG), or high glucose (HG). Equal amounts of SERCA were expressed in VSMC infected with Ad-SERCA2b (WT) or Ad-C674S mutant SERCA detected by IID8 monoclonal antibody (910, 1:2,000). GAPDH acts as a loading control. C: SERCA and iNOS expression was similar in VSMC infected with WT or C674 mutant SERCA and treated with or without IL-1β (IL-1β, 5 ng/mL) regardless of exposure to NG or HG. iNOS was detected with an anti-iNOS antibody (1:1000). GAPDH acts as a loading control.
Supplemental figure 2. iNOS inhibitor L-NIL prevented the inhibition of migration caused by IL-1β in SERCA WT infected VSMC. Cells infected with Ad-WT SERCA and exposed to high glucose were treated with IL-1β (5 ng/mL ) with or without the iNOS inhibitor, N6-(1-iminoethyl)-L-lysine (L-NIL, 10 μmol/L) for 24 h before the migration assay. N=4, *P<0.05, one-way ANOVA.
Acknowledgments
The work was supported by NIH R01’s HL31607 and AG27080, P01 HL68758, and the NHLBI sponsored Boston University Cardiovascular Proteomics Center (Contract No. N01-HV-28178).
Footnotes
Disclosures: As investigators on these projects XT and RAC receive in excess of $10,000 salary support from these sources.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yasunari K, Kohno M, Kano H, Yokokawa K, Minami M, Yoshikawa J. Mechanisms of action of troglitazone in the prevention of high glucose-induced migration and proliferation of cultured coronary smooth muscle cells. Circ Res. 1997;81:953–62. doi: 10.1161/01.res.81.6.953. [DOI] [PubMed] [Google Scholar]
- 2.Yasunari K, Kohno M, Kano H, Yokokawa K, Minami M, Yoshikawa J. Antioxidants improve impaired insulin-mediated glucose uptake and prevent migration and proliferation of cultured rabbit coronary smooth muscle cells induced by high glucose. Circ. 1999;99(10):1370–8. doi: 10.1161/01.cir.99.10.1370. [DOI] [PubMed] [Google Scholar]
- 3.Scherberich A, Campos-Toimil M, Ronde P, Takeda K, Beretz A. Migration of human vascular smooth muscle cells involves serum-dependent repeated cytosolic calcium transients. J Cell Sci. 2000;113:653–62. doi: 10.1242/jcs.113.4.653. [DOI] [PubMed] [Google Scholar]
- 4.Cohen RA, Weisbrod RM, Gericke M, Yaghoubi M, Bierl C, Bolotina VM. Mechanism of nitric oxide-induced vasodilatation. Refilling of intracellular stores by sarcoplasmic reticulum Ca2+ ATPase and inhibition of store-operated Ca2+ influx. Circ Res. 1999;84:210–9. doi: 10.1161/01.res.84.2.210. [DOI] [PubMed] [Google Scholar]
- 5.Touyz RM. Reactive oxygen species as mediators of calcium signaling by angiotensin II: implications in vascular physiology and pathophysiology. Antioxid Redox Signal. 2005;7(9–10):1302–14. doi: 10.1089/ars.2005.7.1302. [DOI] [PubMed] [Google Scholar]
- 6.Mayr U, Zou Y, Zhang Z, Dietrich H, Hu Y, Xu Q. Accelerated arteriosclerosis of vein grafts in inducible NO synthase(−/−) mice is related to decreased endothelial progenitor cell repair. Circ Res. 2006;98(3):412–20. doi: 10.1161/01.RES.0000201957.09227.6d. [DOI] [PubMed] [Google Scholar]
- 7.Barbato JE, Zuckerbraun BS, Overhaus M, Raman KG, Tzeng E. Nitric oxide modulates vascular inflammation and intimal hyperplasia in insulin resistance and the metabolic syndrome. Am J Physiol Heart Circ Physiol. 2005;289(1):H228–H236. doi: 10.1152/ajpheart.00982.2004. [DOI] [PubMed] [Google Scholar]
- 8.Ying J, Tong XY, Weisbrod RM, Pimentel DR, Trucillo M, Adachi T, Cohen RA. Cysteine-674 of the sarco/endoplasmic reticulum calcium ATPase is required for the inhibition of cell migration by nitric oxide. Arterioscler Thromb Vasc Biol. 2007;27(4):783–90. doi: 10.1161/01.ATV.0000258413.72747.23. [DOI] [PubMed] [Google Scholar]
- 9.Lipskaia L, del Monte F, Capiod T, Yacoubi S, Hadri L, Hours M, Hajjar RJ, Lompre AM. Sarco/endoplasmic reticulum Ca2+-ATPase gene transfer reduces vascular smooth muscle cell proliferation and neointima formation in the rat. Circ Res. 2005;97(5):488–95. doi: 10.1161/01.RES.0000180663.42594.aa. [DOI] [PubMed] [Google Scholar]
- 10.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10(11):1200–7. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 11.Jiang B, Brecher P. N-Acetyl-L-cysteine potentiates interleukin-1beta induction of nitric oxide synthase : role of p44/42 mitogen-activated protein kinases. Hypertension. 2000;35(4):914–8. doi: 10.1161/01.hyp.35.4.914. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki H, Obara M, Kuwayama H, Kanazawa T. A Conformational Change of N-Iodoacetyl-N′-(5-Sulfo-1-Naphthyl)Ethylenediamine-Labeled Sarcoplasmic-Reticulum Ca-2+-ATPase Upon ATP Binding to the Catalytic Site. Journal of Biological Chemistry. 1987;262(32):15448–56. [PubMed] [Google Scholar]
- 13.Yamashita T, Kawakita M. Reactive Sulfhydryl-Groups of Sarcoplasmic-Reticulum ATPase. 2 Site of Labeling with Iodoacetamide and Its Fluorescent Derivative. Journal of Biochemistry. 1987;101(2):377–85. doi: 10.1093/oxfordjournals.jbchem.a121922. [DOI] [PubMed] [Google Scholar]
- 14.Clavreul N, Adachi T, Pimentel DR, Ido Y, Schoneich C, Cohen RA. S-glutathiolation by peroxynitrite of p21ras at cysteine-118 mediates its direct activation and downstream signaling in endothelial cells. FASEB J. 2006;20(3):518–20. doi: 10.1096/fj.05-4875fje. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt K, Desch W, Klatt P, Kukovetz WR, Mayer B. Release of nitric oxide from donors with known half-life: A mathematical model for calculating nitric oxide concentrations in aerobic solutions. Naunyn-Schmiedebergs Archives of Pharmacology. 1997;355:457–462. doi: 10.1007/pl00004969. [DOI] [PubMed] [Google Scholar]
- 16.Moore ZW, Hui DY. Apolipoprotein E inhibition of vascular hyperplasia and neointima formation requires inducible nitric oxide synthase. J Lipid Res. 2005;46(10):2083–90. doi: 10.1194/jlr.M500177-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson PG, Boerth NJ, Liu M, McNamara DB, Cornwell TL, Lincoln TM. Cyclic GMP-dependent protein kinase expression in coronary arterial smooth muscle in response to balloon catheter injury. Arterioscler Thromb Vasc Biol. 2000;20(10):2192–7. doi: 10.1161/01.atv.20.10.2192. [DOI] [PubMed] [Google Scholar]
- 18.Vangheluwe P, Schuermans M, Raeymaekers L, Wuytack F. Tight interplay between the Ca2+ affinity of the cardiac SERCA2 Ca2+ pump and the SERCA2 expression level. Cell Calcium. 2007;42(3):281–9. doi: 10.1016/j.ceca.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 19.Bishop JE, Squier TC, Bigelow DJ, Inesi G. (Iodoacetamido)fluorescein labels a pair of proximal cysteines on the Ca2+-ATPase of sarcoplasmic reticulum. Biochemistry. 1988;27(14):5233–40. doi: 10.1021/bi00414a043. [DOI] [PubMed] [Google Scholar]
- 20.Sirsjo A, Lofving A, Hansson GK, Wagsater D, Tokuno S, Valen G. Deficiency of nitric oxide synthase 2 results in increased neointima formation in a mouse model of vascular injury. J Cardiovasc Pharmacol. 2003;41(6):897–902. doi: 10.1097/00005344-200306000-00010. [DOI] [PubMed] [Google Scholar]
- 21.Chyu KY, Dimayuga P, Zhu J, Nilsson J, Kaul S, Shah PK, Cercek B. Decreased neointimal thickening after arterial wall injury in inducible nitric oxide synthase knockout mice. Circ Res. 1999;85(12):1192–8. doi: 10.1161/01.res.85.12.1192. [DOI] [PubMed] [Google Scholar]
- 22.Chamberlain J, Evans D, King A, Dewberry R, Dower S, Crossman D, Francis S. Interleukin-1beta and signaling of interleukin-1 in vascular wall and circulating cells modulates the extent of neointima formation in mice. Am J Pathol. 2006;168(4):1396–403. doi: 10.2353/ajpath.2006.051054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jagadeesha DK, Lindley TE, Deleon J, Sharma RV, Miller F, Bhalla RC. Tempol therapy attenuates medial smooth muscle cell apoptosis and neointima formation after balloon catheter injury in carotid artery of diabetic rats. Am J Physiol Heart Circ Physiol. 2005;289(3):H1047–H1053. doi: 10.1152/ajpheart.01071.2004. [DOI] [PubMed] [Google Scholar]
- 24.Salzberg SP, Filsoufi F, Anyanwu A, von HK, Karlof E, Carpentier A, Dansky HM, Adams DH. Increased neointimal formation after surgical vein grafting in a murine model of type 2 diabetes. Circ. 2006;114(1 Suppl):I302–I307. doi: 10.1161/CIRCULATIONAHA.105.001339. [DOI] [PubMed] [Google Scholar]
- 25.Zuccollo A, Shi C, Mastroianni R, Maitland-Toolan KA, Weisbrod RM, Zang M, Xu S, Jiang B, Oliver-Krasinski JM, Cayatte AJ, Corda S, Lavielle G, Verbeuren TJ, Cohen RA. The thromboxane A2 receptor antagonist S18886 prevents enhanced atherogenesis caused by diabetes mellitus. Circ. 2005;112(19):3001–8. doi: 10.1161/CIRCULATIONAHA.105.581892. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure 1. A: The overexpression level of SERCA2b and SERCA2b C674S in normal glucose detected by two different antibodies. GFP infected and uninfected VSMC serve as controls. 15 μg protein in each group was loaded and detected by K30/A43 (1:500) and IID8 (1:2,000) separately. K30/A43 is a polyclone antibody recognizing both rat and human SERCA. IID8 (910) is a monoclone antibody which recognizes human SERCA but not rat SERCA. B: Overexpression of WT or C674S mutant SERCA in VSMC in normal glucose (NG), high mannose (MG), or high glucose (HG). Equal amounts of SERCA were expressed in VSMC infected with Ad-SERCA2b (WT) or Ad-C674S mutant SERCA detected by IID8 monoclonal antibody (910, 1:2,000). GAPDH acts as a loading control. C: SERCA and iNOS expression was similar in VSMC infected with WT or C674 mutant SERCA and treated with or without IL-1β (IL-1β, 5 ng/mL) regardless of exposure to NG or HG. iNOS was detected with an anti-iNOS antibody (1:1000). GAPDH acts as a loading control.
Supplemental figure 2. iNOS inhibitor L-NIL prevented the inhibition of migration caused by IL-1β in SERCA WT infected VSMC. Cells infected with Ad-WT SERCA and exposed to high glucose were treated with IL-1β (5 ng/mL ) with or without the iNOS inhibitor, N6-(1-iminoethyl)-L-lysine (L-NIL, 10 μmol/L) for 24 h before the migration assay. N=4, *P<0.05, one-way ANOVA.