

Abstract
Virus-induced activation of nuclear factor-kappa B (NF-κB) is required for Type 3 (T3) reovirus-induced apoptosis. We now show that NF-κB is also activated by the prototypic Type 1 reovirus strain Lang (T1L), which induces significantly less apoptosis than T3 viruses, indicating that NF-κB activation alone is not sufficient for apoptosis in reovirus-infected cells. A second phase of virus-induced NF-κB regulation, where NF-κB activation is inhibited at later times following infection with T3 Abney (T3A), is absent in T1L-infected cells. This suggests that inhibition of NF-κB activation at later times post infection also contributes to reovirus-induced apoptosis. Reovirus-induced inhibition of stimulus-induced activation of NF-κB is significantly associated with apoptosis following infection of HEK293 cells with reassortant reoviruses and is determined by the T3 S1 gene segment, which is also the primary determinant of reovirus-induced apoptosis. Inhibition of stimulus-induced activation of NF-κB also occurs following infection of primary cardiac myocytes with apoptotic (8B) but not non-apoptotic (T1L) reoviruses. Expression levels of the NF-κB-regulated cellular FLICE inhibitory protein (cFLIP) reflect NF-κB activation in reovirus-infected cells. Further, inhibition of NF-κB activity and cFLIP expression promote T1L-induced apoptosis. These results demonstrate that inhibition of stimulus-induced activation of NF-κB and the resulting decrease in cFLIP expression promote reovirus-induced apoptosis.
Keywords: cFLIP, myocytes, NF-κB, reovirus
Introduction
Reoviruses are non-enveloped viruses comprised of two concentric protein capsids surrounding a genome of 10 segments of double-stranded RNA. Mammalian reoviruses provide a well-established experimental system for studying mechanisms of virus-induced pathogenesis. Type 3 (T3) reoviruses can induce apoptosis in cultured cells in vitro and in target tissues in vivo, including the heart and central nervous system.1 In these key target organs, viral infection, tissue injury and apoptosis co-localize suggesting that apoptosis is a critical mechanism by which disease is triggered in the host.2–5 Further, additional support for this view comes from studies indicating that inhibition of apoptosis reduces reovirus-induced tissue injury.4
Reovirus-induced apoptosis is mediated by members of the tumor necrosis factor (TNF) family of ligands, which attach to cell surface death receptors to induce the activation of initiator (caspase 8) and effector (caspases 3 and 7) caspases.3,6,7 Mitochondrial pathways of apoptosis, initiated following the caspase 8-dependent cleavage of Bid, contribute to effector caspase activation in virus-infected cells.7,8 In addition reovirus can sensitize cells to death receptor induced apoptosis.7,9
The nuclear factor-kappa B (NF-κB) family of transcription factors plays a key role in the regulation of cell growth and survival. The prototypical form of NF-κB exists as a heterodimer of proteins p50 and p65 (RelA), which is sequestered in the cytoplasm of quiescent cells through its association with members of the IκB family of inhibitory proteins.10–13 Site specific phosphorylation, followed by ubiquitination and proteosomal degradation of IκB, allows NF-κB to translocate to the nucleus and activate cellular gene expression.14–17 Infection with T3 reoviruses induces the activation of NF-κB in a variety of cell types, including human embryonic kidney 293 (HEK293) and HeLa cells.18,19 This activation is required for T3 reovirus-induced apoptosis in these cells, which is inhibited by stable over-expression of an IκB super-repressor or treatment of cells with a proteosome inhibitor (Z-L3VS) that blocks IκB degradation.18,19 T3 reovirus-induced apoptosis is also inhibited in immortalized mouse embryo fibroblasts (MEFs) with targeted disruptions in the genes encoding the p50 or p65 subunits of NF-κB.18 Although required for apoptosis, T3 reovirus-induced activation of NF-κB is transient and at later times post infection (PI) the activation of NF-κB by a wide variety of stimuli is inhibited in infected cells.18,19
In this report we show that NF-κB is also activated following infection with the Type 1 reovirus strain, Lang (T1L), which induces apoptosis inefficiently in infected cells (APO−), and that this activation parallels that of the strongly apoptotic (APO+) T3 reovirus strain, Abney (T3A).20–22 In contrast, T1L does not inhibit NF-κB activation at later times PI, whereas T3A does. Our results demonstrate that inhibition of stimulus-induced activation of NF-κB is associated with apoptosis following infection of both HEK293 cells and primary cardiac myocytes. We further show that the T3 S1 gene segment, which is the primary determinant of reovirus-induced apoptosis, also determines the ability of reovirus strains to inhibit stimulus-induced activation of NF-κB.20,21,23 In addition, chemical inhibition of NF-κB was found to significantly enhance apoptosis in primary myocytes following infection with T1L (APO−). Taken together these results demonstrate that inhibition of stimulus-induced activation of NF-κB following T3-reovirus infection promotes apoptosis in both HEK293 cells and primary myocytes, and therefore contributes to viral pathogenesis. Virus-induced regulation of the NF-κB dependent gene encoding the cellular inhibitor of caspase 8 (also known as FLICE inhibitory protein, cFLIP) provides a potential mechanism for these findings.
Materials and methods
Cells and virus
HEK293 (ATCC CRL1573) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 100 U/ml each of penicillin and streptomycin and containing 10% fetal bovine serum (FBS) and were maintained at 37°C with 5% CO2. HEK293 cells expressing a dominant negative (DN) form of the Fas Associated Death Domain protein (FADD-DN) or the IκB super-repressor (IκBΔN2) were gifts from Dr. Gary Johnson (University of North Carolina School of Medicine) Neonatal rat cardiac myocytes were generated in the laboratory of Dr. Carlin Long (University of Colorado Health Sciences Center). Myocytes were cultured in DMEM with Hanks salts supplemented with 5% FBS, 1 mg/ml bovine serum albumin, 50 U/ml penicillin, 2 μg/ml vitamin B-12 (Sigma), 10 μg/ml transferrin (Sigma), 10 μg/ml insulin (Sigma) and 0.1 mM BrdU (Sigma) and were maintained at 37°C with 1% CO2. Reovirus strains Type 3 Abney (T3A) and Type 1 Lang (T1L) are laboratory stocks that have been plaque purified and passaged (twice) in L929 (ATCC CCL1) cells to generate working stocks. T1L × T3A reassortant viruses were grown from stocks originally isolated by Tricia Jandris, Lynda Morrison and Graeme Wilson in the laboratory of Bernard Fields.24 Virus 8B is a reassortant virus derived from a mouse infected with T1L and T3D.25 EB121 and E3 are reassortant viruses derived from T1L and T3 Dearing (T3D). All EW reassortants were derived from 8B × EB121, whereas all DW reassortants were derived from EW60 and E3. Reovirus EW and DW reassortants have been characterized for their myocarditic potential.25,26 All EW and DW reassortants and 8B were gifts from Dr. Barbara Sherry (North Carolina State University). Virus infections were performed using a multiplicity of infection (MOI) of 100.
Apoptosis assays
Apoptotic nuclear morphology and cell viability were determined by staining with acridine orange and ethidium bromide at a final concentration of 1 μg/ml each. Following staining, cells were examined by epifluorescence microscopy (Nikon Labophot-2: B-2A filter, excitation, 450−490 nm; barrier, 520 nm; dichroic mirror, 505 nm). The percentage of cells containing condensed nuclei and/or marginated chromatin in a population of 100 cells was recorded. The specificity of this assay has been previously established in reovirus-infected cells using DNA laddering techniques and electron microscopy.9,20 Cell populations were also analyzed by flow cytometry to determine (i) the cell surface exposure of phosphatidylserine, using Annexin V-FITC (Trevigen), and (ii) intracellular levels of active caspase 3, using a fluorochrome inhibitor of caspases (FLICA, Immunochemistry Technologies).
Reagents
Etoposide were purchased from Sigma and was used at a concentration of 100 μM respectively. TNFα was purchased from Invitrogen and was used at a concentration of 100 ng/ml. The cell permeable, synthetic, peptide inhibitors of caspase 3 (Ac-DEVD-CHO; Ac-Asp-Glu-Val-Asp-CHO) and caspase 8 (Ac-IETD-CHO; Ac-Ile-Asp-Thr-Glu-CHO) were purchased from Calbiochem and were used at a concentration of 10 μM, which we have shown to be effective at inhibiting caspase activity in HEK293 cells.7 The cell permeable inhibitor of NF-κB translocation, SN50, was purchased from Calbiochem and was used at a concentration of 18 μM. Staurosporine (Sigma) was used at a concentration of 5 μM. Antisense and sense oligonucleotides were prepared by Integrated DNA Technologies (IDT) at a concentration of 1 μM following HPLC purification. Phosphothionate bonds present between nucleotides were included to enhance cell permeability. Antisense cFLIP: 5′-gatttcagcagacatcctac-3′ and sense cFLIP: 5′-catcctacagacgacttcag-3′ sequences were used. 27,28 Oligonucleotides were added directly to the media (10 nM) and cells were incubated for 18 h prior to viral infection. Oligonucleotides were then added back to the media following infections.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were prepared from treated cells (5 × 106) by washing cells in PBS followed by incubation in hypotonic lysis buffer (10 mM HEPES {pH 7.9}, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM dithiothreitol, 0.5 mM phenylmethylsulfonylfluoride, and a protease inhibitor cocktail {Boehringer Mannheim}) at 4°C for 15 min. One-twentieth volume 10% NP-40 was added to the cell lysate and the sample was vortexed for 10 s and centrifuged at 10,000× g for 5 min. The nuclear pellet was washed once in hypotonic buffer, resuspended in high-salt buffer (25% glycerol, 20 mM HEPES {pH 7.9}, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM dithiothreitol, 0.5 mM phenylmethylsulfonylfluoride, and protease inhibitor cocktail), and incubated at 4°C for 2 to 3 h. Samples were centrifuged at 10,000 × g for 10 min and the supernatant was used as the nuclear extract.
Nuclear extracts were assayed for NF-κB activation by EMSA using a {32P}-labeled oligonucleotide consisting of the NF-κB consensus binding sequence (Santa Cruz Biotechnology). Nuclear extracts (5 to 10 μg total protein) were incubated with a binding reaction buffer containing 2 μg poly·dI·dC (Sigma) in the presence of 20 mM HEPES (pH 7.9), 60 mM KCl, 1 mM EDTA, 1 mM dithiothreitol, and 5% glycerol at 4°C for 20 min. Radiolabeled NF-κB consensus oligonucleotide (0.1 to 1.0 ng) was added, and the mixture was incubated at room temperature for 20 min. For competition experiments, ten-fold excess unlabeled consensus oligonucleotide or an oligonucleotide containing the SP-1 consensus site (Santa Cruz Biotechnology) were added to reaction mixtures. Nucleoprotein complexes were subjected to electrophoresis on native 5% polyacrylamide gels at 180 V, dried under vacuum, and exposed to Biomax MR film (Kodak).
Western blot analysis
Following infection with reovirus, cells were pelleted by centrifugation, washed twice with ice-cold phosphate-buffered saline and lysed by sonication in 200 μl of a buffer containing 15 mM Tris, pH 7.5, 2 mM EDTA, 10 mM EGTA, 20% glycerol, 0.1% NP-40, 50 mM β-mercaptoethanol, 100 μg/ml leupeptin, 2 μg/ml aprotinin, 40 μM Z-D-DCB, and 1 mM PMSF. The lysates were then cleared by centrifugation at 16,000 g for 5 min, normalized for protein amount, mixed 1:1 with SDS sample buffer (100 mM Tris, pH 6.8, 2% SDS, 300 mM β-mercaptoethanol, 30% glycerol, and 5% pyronine Y), boiled for 5 min and stored at −70°C. Proteins were electrophoresed by SDS-PAGE (10% gels) and probed with antibodies directed against IκBα (Santa Cruz # 203) and cFLIP (GeneTex # GTX26144) which recognizes the 55kD cFLIPL (FLIP alpha) protein. All lysates were standardized for protein concentration with antibodies directed against actin (Oncogene # CP01). Autoradiographs were quantitated by densitometric analysis using a Fluor-S MultiImager (BioRad Laboratories).
Immunocytochemistry
Primary cardiac myocytes were grown on 8-well chamber slides coated with rat-tail collagen (Becton Dickenson 354630). Cells were infected with reovirus 24−26 h prior to fixation with 3.7% formaldehyde/ phosphate-buffered saline (PBS) for 15 min at room temperature. Cells were subsequently permeablized and blocked with 5% normal goat serum (Vector S1000) in PBS with 0.1% Tween 20 for 2−4 h at room temperature. Cells were incubated overnight at 4°C with antibodies directed against NF-κB (Santa Cruz, sc-8008) at a 1:30 dilution in blocking solution. After washing in PBS/0.1% Tween 20, cells were incubated with secondary anti-rabbit IgG conjugated to FITC (Vector FI 1200) for 1 h at room temperature before being counterstained with Hoechst 33342 (Molecular Probes H 3570) for 3 min at room temperature. Cells were then washed in PBS/0.1% Tween 20, mounted with vectashield (Vector H1000) and digitally imaged using a Zeiss Axioplan2 epifluorescence microscope.
Statistical analysis
One-way analysis of variance (ANOVA) was performed using GraphPad InStat software.
Results
Activation of NF-κB is not sufficient for apoptosis induced by prototype reovirus strains
The T3 prototype reovirus strain, T3A (APO+) induces significantly more apoptosis in infected HEK293 cells than the T1 prototype reovirus strain, T1L, as determined by nuclear morphology (P < 0.001 at 48 h) and caspase 3 activation assays (P < 0.001) (Figure 1). We have previously shown that T3A infection induces the transient activation of NF-κB in infected HEK293 cells.19 Thus at early times PI with T3A NF-κB is activated and translocates to the nucleus. However, at later times PI activated NF-κB is no longer present in the nuclei of T3A-infected cells. The activation of NF-κB following reovirus infection is required for T3A-induced apoptosis in HEK293 cells.19 In addition, the ability of reovirus to inhibit NF-κB activation at later times PI may sensitize cells to TRAIL-induced apoptosis and may be required for apoptosis in TRAIL-resistant cells.19
Figure 1.
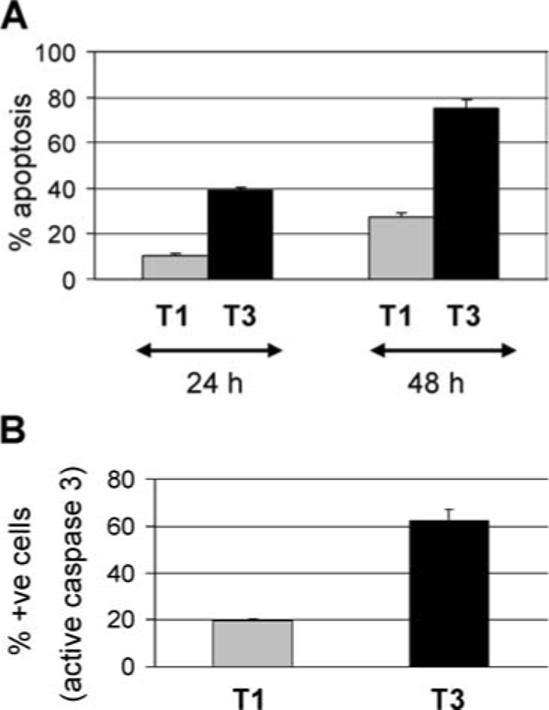
T3A induces significantly more apoptosis in infected HEK293 cells than T1L. HEK293 cells were infected with reovirus strains T3A (black bars) and T1L (gray bars). Cells were harvested 24 and 48 h PI. The graphs show the mean percentage of cells (above the percentage seen following mock-infection) containing apoptotic nuclei (A) and active caspase 3 (B) from three independent experiments. Error bars represent standard errors of the mean.
To determine the importance of NF-κB regulation in reovirus-induced apoptosis we investigated NF-κB activation following infection with T1L (APO−). EMSA analysis, using nuclear extracts from T1L-infected HEK293 cells, was used to demonstrate the presence of activated NF-κB in the nucleus of T1L (APO−)-infected cells as early as 2 h PI. As expected, activated NF-κB was also present in the nucleus of T3A (APO+)-infected cells at early times PI (Figure 2). However, at late time PI (24 h) levels of activated NF-κB were reduced in the nuclei of T3A (APO+)-infected cells whilst remaining high in the nuclei of T1 (APO−)-infected cells.
Figure 2.
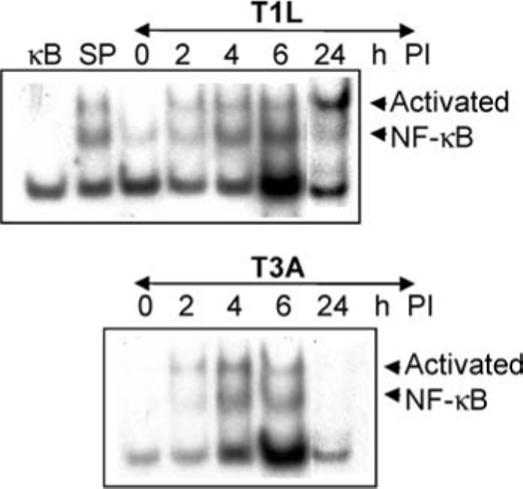
Activation of NF-κB following reovirus infection. HEK293 cells were infected with reovirus strains T3A or T1L for the indicated times. Nuclear extracts were then prepared and EMSA analysis was performed using a radiolabeled oligonucleotide probe comprising NF-κB binding sequences. Shifted bands, corresponding to activated NF-κB: DNA complexes are indicated. The specificity of the probe sequences was confirmed by including a 10-fold excess of cold oligonucleotides comprising NF-κB (κB) or SP6 (SP) binding sequences in reactions using nuclear extracts from T3A-infected cells (4 h PI).
These results demonstrate that T1L (APO−), in addition to T3A (APO+), activates NF-κB following viral infection of HEK293 cells. This indicates that NF-κB activity is required for, but is not sufficient for, reovirus-induced apoptosis. Our results also suggest that the inhibition of NF-κB seen at late times following infection with T3A (APO+), but not T1L (APO−), may be an additional requirement for reovirus-induced apoptosis.
T3A (APO+), but not TlL (APO−), inhibits stimulus-induced activation of NF-κB at later times post infection
The ability of T3A (APO+) to inhibit NF-κB activity at later times PI accounts for both the transient nature of NF-κB activation in reovirus-infected cells and the inhibition of stimulus-induced activation of NF-κB.19 Since reovirus-induced apoptosis is mediated by death ligands and since death ligand-induced apoptosis is enhanced in cells where NF-κB activity is blocked, the ability of reovirus to inhibit stimulus-induced activation likely promotes reovirus-induced apoptosis.19 Having shown that T1L (APO−)-induced activation of NF-κB is not followed by a later inhibition phase, we next investigated whether T1L (APO−) infection inhibited stimulus-induced activation of NF-κB. Both TNF and the topoisomerase-II inhibitor, etoposide, which are strong inducers of NF-κB, were used as stimuli for our experiments. HEK293 cells were infected with T1L (APO−) for 12 h before being treated with TNF and were harvested for EMSA analysis after a further 1 h (Figure 3A). T1L (APO−) did not inhibit the ability of TNF to activate NF-κB compared to mock-infected cells. In comparison T3A (APO+) completely inhibited TNF-induced activation of NF-κB (Figure 3A). Similar results were obtained using etoposide as the stimulus for NF-κB activation (not shown).
Figure 3.
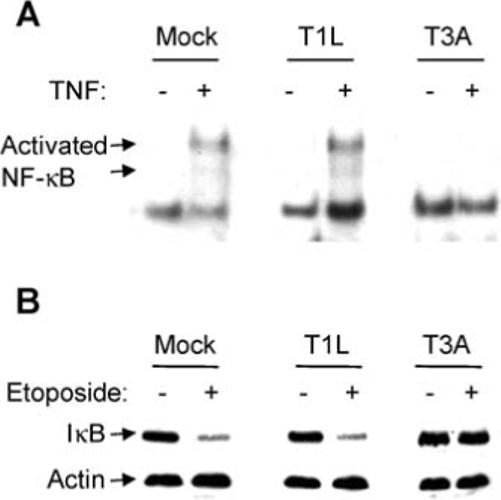
Inhibition of stimulus-induced activation of NF-κB following reovirus infection. HEK293 cells were infected with T3A and T1L strains of reovirus or were mock-infected. (A) Twelve h PI cells were treated with TNF or were left untreated. After a further 1 h cells were harvested for EMSA analysis using a radiolabelled oligonucleotide probe comprising NF-κB binding sequences. Shifted bands, corresponding to activated NF-κB: DNA complexes are indicated. (B) Twelve h PI cells were treated with etoposide or were left untreated. After a further 3 h cells were harvested for western blot analysis using antibodies directed against IκB. Antibodies directed against actin were used to control for protein loading.
The degradation of IκB is required for NF-κB activation. We therefore also investigated the ability of reovirus to inhibit stimulus-induced degradation of IκB. HEK293 cells were infected with reovirus for 12 h prior to treatment with etoposide and were harvested for western blot analysis after a further 3 h. T3A (APO+), but not T1L (APO−), was found to inhibit etoposide-induced degradation of IκB (Figure 3B). Similar results were obtained using TNF as the stimulus for IκB degradation (not shown).
Activation of NF-κB by two different stimuli is thus inhibited at later times PI in T3A (APO+), but not T1L (APO−)-infected HEK293 cells, again suggesting that the inhibition of NF-κB seen at late times following infection with T3A (APO+) may be required for reovirus-induced apoptosis.
Inhibition of stimulus-induced activation of NF-κB is determined by the T3A S1 gene segment and correlates with apoptosis in reovirus-infected cells
Having shown that T3A inhibits stimulus-induced activation of NF-κB, whereas T1L does not, we next wished to identify whether a specific viral gene determined this difference. HEK293 cells were infected with a panel of T1L × T3A reassortant reoviruses. Twelve h PI cells were treated with etoposide. Cells were then harvested after a further 3 h for western blot analysis using antibodies directed against IκB. The ability of different reassortant viruses to inhibit the etoposide-induced degradation of IκB seen in mock-infected cells and the derivation of the various genome segments of each virus is shown in Table 1. The S1 gene segment alone was identified as being significantly associated with strain-specific differences in virus-induced inhibition of etoposide-induced IκB degradation (t test, P = 0.0008; M-W test, P = 0.0159). Although statistical analysis identified the T3A S1 gene segment as an important determining factor in the ability of reoviruses to inhibit stimulus-induced degradation of IκB it is important to note that reassortant viruses containing the T3A S1 gene segment in the presence of other gene segments derived from T1L show reduced apoptosis. This indicates that it is likely that other T3A gene segments also play a role in apoptosis following viral infections.
Table 1.
Ability of reassortant reoviruses (T1L × T3A) to induce apoptosis and to inhibit stimulus-induced degradation of IκB.
|
Gene segment |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
L |
M |
S |
% Inhibition of etoposide-induced IκB degradation | |||||||||
| Virus | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 4 | % Apoptosis | |
| T3A | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 93 | 56 |
| GW7 | 1 | 1 | 1 | 1 | 3 | 3 | 3 | 1 | 3 | 3 | 88 | 49 |
| GW49 | 1 | 1 | 1 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 86 | 55 |
| GW12 | 3 | 1 | 3 | 3 | 1 | 3 | 3 | 3 | 3 | 1 | 74 | 47 |
| GW16 | 3 | 3 | 3 | 3 | 3 | 3 | 1 | 3 | 3 | 1 | 54 | 34 |
| GW10 | 1 | 1 | 3 | 1 | 1 | 3 | 1 | 3 | 1 | 1 | 47 | 38 |
| GW15 | 3 | 3 | 3 | 3 | 3 | 1 | 1 | 1 | 3 | 1 | 43 | 31 |
| GW26 | 1 | 1 | 3 | 3 | 1 | 1 | 1 | 1 | 1 | 1 | 42 | 23 |
| T1L | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 17 | 24 |
HEK293 cells were infected with reovirus (MOI 100). Twelve h following infection cells were treated with etoposide (100 μM). Cells were harvested after a further 3 h for western blot analysis. Results shown represent % inhibition of etoposide-induced IκB degradation in virus-infected cells compared to the amount of etoposide-induced degradation of IκB seen in mock-infected cells (column 3). This data is graphed against virus-induced apoptosis in Figure 4. Also shown is the mean % apoptosis for two independent experiments. Apoptosis was determined 48 h PI (MOI 100) by annexin assays.
Since the T3 S1 gene segment has previously been shown to be the primary determinant of reovirus-induced apoptosis we next investigated the correlation between inhibition of etoposide-induced degradation of IκB and apoptosis following infection of HEK293 cells with the T3A × T1L reassortant viruses (Table 1, Figure 4A).20,21 Using linear regression analysis we obtained an R2 value of 0.89 (P = 0.0001) indicating a significant correlation between apoptosis induction and inhibition of etoposide-induced degradation of IκB.
Figure 4.

Inhibition of stimulus-induced degradation of IκB is correlated with apoptosis in reovirus-infected cells. The ability of T3A × T1L (A) and 8B × T1L (B) reassortant reoviruses and the parental strains to inhibit etoposide-induced degradation of IκB was plotted against their ability to induce apoptosis. Values were obtained from Tables 1 and 2. Each point (square) represents a single virus strain or reassortant. Open squares indicate viruses with myocarditic potential.
Infection of neonatal mice with the reassortant reovirus 8B25 induces myocarditis.29,30 We have shown that apoptosis is a key mechanism by which 8B induces myocarditic cell death and tissue injury in infected animals.4,31 A second panel of reovirus reassortants that differ in myocarditic potential were therefore investigated for their ability to induce apoptosis and to inhibit stimulus-induced activation of NF-κB in HEK293 cells.24 These viruses again revealed a significant correlation (R2 = 0.91, P = 0.0004) between the ability to induce apoptosis and inhibition of stimulus-induced degradation of IκB (Table 2, Figure 4B). Among the reassortants tested a high level of apoptosis and a high degree of inhibition of stimulus-induced activation of NF-κB were required for the myocarditic phenotype (Figure 4B).
Table 2.
Ability of reassortant reoviruses (myocarditic and non-myocarditic) to induce apoptosis and to inhibit stimulus-induced degradation of IκB.
| Virus | % Apoptosis Annexin assay 40 h PI | % Apoptosis Nucl. Morph Assay 48 h PI | % Inhibition of etoposide-induced IκB degradation |
|---|---|---|---|
| T3A | 49 ± 2.0***c | 81 ± 3.7*** | 72 ± 4.8*** |
| 8B | 57 ± 3.2*** | 92 ± 3.2*** | 78 ± 4.4*** |
| EW60 | 59 ± 2.7*** | 87 ± 1.6*** | 90 ± 7.5*** |
| DB88 | 63 ± 2.1*** | 84 ± 2.4*** | 77 ± 8.7*** |
| DB181 | 55 ± 3.1*** | 93 ± 1.9*** | 89 ± 4.7*** |
| DB188 | 52 ± 1.5*** | 88 ± 3.2*** | 57 ± 3.7** |
| EW93A | 48 ± 1.7*** | 65 ± 1.7*** | 60 ± 6.4** |
| EW29B | 28 ± 2.0 | 32 ± 1.9 | 33 ± 6.0 |
| T1L | 20 ± 0.9 | 30 ± 1.4 | 22 ± 2.5 |
| Mock | 17 ± 1.2 | 7 ± 0.6 | 0 |
HEK293 cells were infected with reovirus (MOI 100). Twelve h following infection cells were treated with etoposide (100 μM). Cells were harvested after a further 3 h for western blot analysis. Results shown represent % inhibition of etoposide-induced IκB degradation in virus-infected cells compared to the amount of etoposide-induced degradation of IκB seen in mock-infected cells ± the standard error of the mean (column 4). Means were calculated from three independent experiments. Also shown is the mean % apoptosis for three independent experiments. Apoptosis values obtained by nuclear morphology assays are graphed against ability to inhibit stimulus-induced degradation of IκB (Figure 4B). Results showing statistical deviation from those seen in mock-infected cells are indicated
represents P < 0.001,
represents P < 0.01
These results demonstrate that inhibition of stimulus-induced activation of NF-κB is strongly correlated with apoptosis induction following reovirus-infection of HEK293 cells.
Reovirus-induced inhibition of stimulus-induced activation of NF-κB is associated with apoptosis in a pathogenic model of reovirus infection
Infection of primary cardiac myocytes has been used to investigate the mechanisms of cell death that result in viral-induced myocarditis.4,32–35 We used this model to explore the association of viral-induced inhibition of stimulus-induced activation of NF-κB and apoptosis during viral pathogenesis. Myocytes were mock-infected or were infected with either the strongly myocarditic reovirus reassortant 8B or the weakly myocarditic virus T1L. Twenty four h PI cells were then treated with staurosporine (NF-κB activating stimulus) and after a further 2 h the number of cells containing nuclear (active) NF-κB was determined by immunocytochemistry (Figure 5A). Following mock-infection staurosporine induced the nuclear translocation (activation) of NF-κB, as expected. Infection with 8B, but not T1L, inhibited staurosporine-induced activation of NF-κB. Consistent with our results in HEK293 cells, 8B, but not T1L, also induced apoptosis in primary cardiac myocytes (Figure 5B).
Figure 5.

The myocarditic reassortant 8B inhibits stimulus-induced activation of NF-κB and induces apoptosis in primary cardiac myocytes whereas the T1L (non-myocarditic) does not. Primary cardiac myocytes were mock-infected (clear bars) or were infected with either the myocarditic reovirus (Reo) reassortant 8B (black bars) or the non-myocarditic reovirus (Reo) T1L (gray bars). (A) Twenty four h PI cells were then treated with staurosporine (NF-κB activating stimulus) and after a further 2 h the number of cells containing nuclear (active) NF-κB was determined by immunocytochemistry. The graph shows the mean percentage of cells containing nuclear NF-κB for 3 different fields of view. Error bars represent standard deviations. (B) Forty eight h PI cells were assayed for apoptosis by annexin assays. The graph shows the mean percentage of annexin-positive cells (% apoptosis) for 3 individual populations of cells. Error bars represent standard deviations. Both graphs are representative of three independent experiments.
These results demonstrate that reovirus-induced inhibition of stimulus-induced activation of NF-κB is associated with apoptosis in primary cardiac myocytes.
Inhibition of NF-κB promotes apoptosis in reovirus-infected cells
Our results suggest that, in addition to the initial activation of NF-κB,18,19 reovirus-induced apoptosis also requires a later phase of NF-κB regulation where stimulus-induced activation of NF-κB is inhibited in infected cells. To confirm the contribution of this later, inhibitory, phase to reovirus-induced apoptosis we investigated the effect of a cell permeable inhibitor of NF-κB (SN50), which functions to inhibit nuclear translocation of NF-κB, on reovirus-induced apoptosis. Primary cardiac myocytes were infected with 8B (APO+) or T1L (APO−) in the presence or absence of SN50 and were assayed for apoptosis after 48 h by nuclear morphology assays. As expected, 8B (APO+) induced high levels of apoptosis in infected primary cardiac myocytes at 48 h PI. This apoptosis was not inhibited following treatment of cells with SN50 (Figure 6A), suggesting that reovirus-induced apoptosis in primary cardiac myocytes does not require NF-κB activation. In fact, SN50-treatment resulted in a small increase in 8B-induced apoptosis. Also as expected, T1L did not induce apoptosis in infected primary cardiac myocytes (Figure 6A). However, in SN50-treated cells the ability of T1L to induce apoptosis increased significantly (P < 0.05) from 11 to 42%. These results demonstrate that NF-κB inhibition promotes apoptosis following reovirus infection of primary cardiac myocytes and suggest that the ability of reoviruses to inhibit stimulus-induced activation of NF-κB is a pro-apoptotic event.
Figure 6.
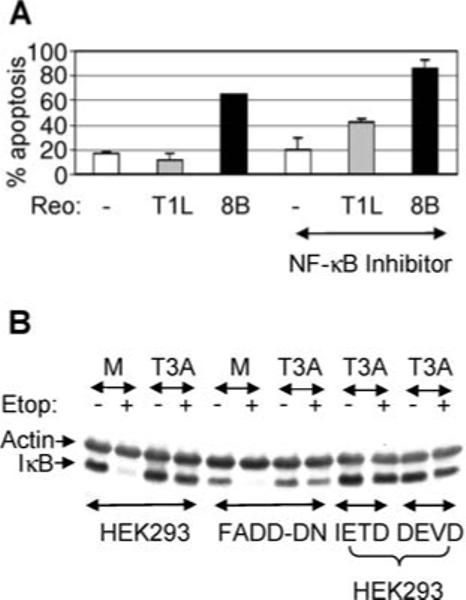
Inhibition of NF-κB promotes apoptosis following reovirus (Reo) infection. (A) Primary cardiac myocytes were either mock-infected (white bars) or were infected with T1L (gray bars) or 8B (black bars) with or without the NF-κB inhibitor SN50. Forty eight h PI cells were harvested and the percentage of cells with apoptotic nuclear morphology was determined. The graph shows the mean percentage of cells with apoptotic nuclear morphology (% apoptosis) for 3 individual populations of cells. Error bars represent standard deviations. The graph is representative of three independent experiments. (B) Untreated HEK293 cells, HEK293 cells expressing FADD-DN and HEK293 cells treated with inhibitors of caspase 3 (Ac-DEVD-CHO) or caspase 8 (Ac-IETD-CHO) were infected with T3A or were mock-infected. Twelve h PI cells were treated with etoposide and were harvested after a further 3 h for western blot analysis using antibodies directed against IκB. Antibodies directed against actin were used to control for protein loading.
In contrast, apoptosis is not required for inhibition of stimulus-induced activation of NF-κB following T3 reovirus infection (Figure 6B). HEK293 cells were infected with reovirus for 12 h prior to treatment with etoposide. After a further 3 h cells were then harvested for western blot analysis using antibody directed against IκB. Inhibition of apoptosis by expression of FADD-DN or treatment with inhibitors of caspase 3 (Ac-DEVD-CHO) or caspase 8 (Ac-IETD-CHO) did not block inhibition of stimulus-induced degradation of IκB following reovirus infection.
T3 reoviruses inhibit cFLIP expression
NF-κB regulates the expression of several cellular genes that function to inhibit apoptosis, including cFLIP, the cellular inhibitor of caspase 8.36 To determine whether the inhibition of NF-κB-regulated genes following reovirus-infection contributed to an apoptotic phenotype we investigated cFLIP expression in reovirus-infected cells. HEK293 cells were infected with reovirus and were harvested at various times post infection for western blot analysis using antibodies directed against cFLIP. Following infection with both T1L (APO−) and T3A (APO+)-infection cFLIP levels increased, as would be expected consequent to the early activation of NF-κB in reovirus-infected cells (Figure 7A). This increase in cFLIP activation was not seen following T3A-infection of cells expressing the NF-κB super-repressor, IκB N2. Since the expression of IκB N2 prevents NF-κB activation, this indicated that activation of NF-κB was required for reovirus induced increases in cFLIP. The requirement of NF-κB for reovirus-induced activation of cFLIP was confirmed using the NF-κB inhibitor, SN50, which also inhibited up-regulation of cFLIP in T3A-infected cells (Figure 7B). At later times PI cFLIP levels dropped in T3A-infected cells, consistent with the shut-off of NF-κB activation in these cells (Figure 7A). In contrast, following T1L (APO−) infection levels of cFLIP increased and then remained stable. These results suggest that falling levels of cFLIP are associated with enhanced apoptosis in reovirus-infected cells (Figure 7A).
Figure 7.
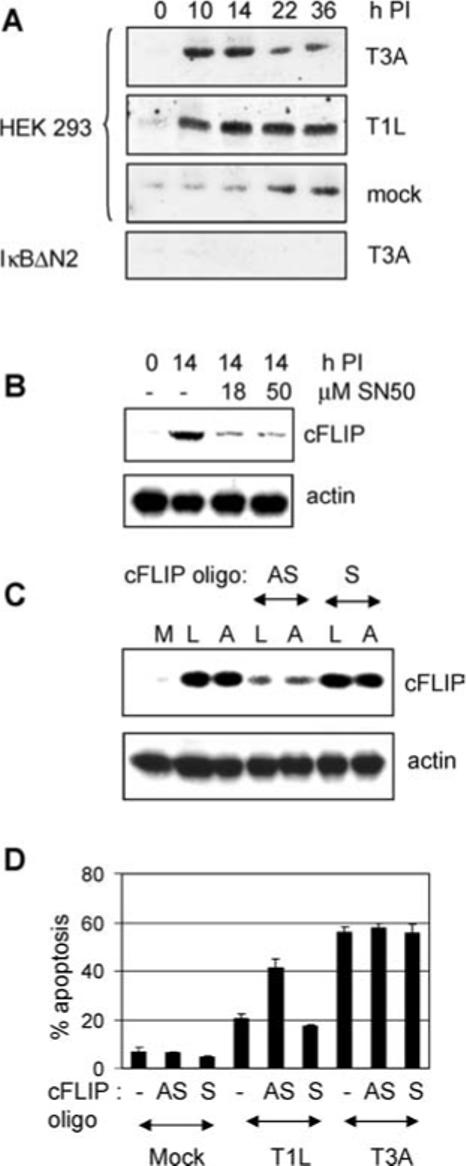
Expression of cFLIP following reovirus infection. (A) HEK293 cells or HEK293 cells expressing IκBΔN2 were either mock-infected or were infected with reovirus strains T1L or T3A. At various times PI cells were harvested for western blot analysis using antibodies directed against cFLIP. Antibodies directed against actin were used to control for protein loading (not shown). (B) HEK293 cells were mock-infected or were infected with T3A in the presence of the NF-κB inhibitor SN50. At 14 h PI cells were harvested for western blot analysis using antibodies directed against cFLIP and actin. HEK293 cells were also mock (M)-infected or were infected with T3A (A) or T1L (L) in the presence of antisense (AS) or sense (s) cFLIP oligonucleotides (cFLIP oligo). At 14 h PI cells were harvested for western blot analysis using antibodies directed against cFLIP and actin (C) and at 48 h PI cells for apoptosis assays (D). The graph shows the mean percentage of cells containing apoptotic nuclei from three independent experiments. Error bars represent standard errors of the mean.
We next used antisense oligonucleotides directed against cFLIP 28 to determine the effect of reduced cFLIP expression on reovirus-induced apoptosis. At a concentration of 10 μM antisense cFLIP oligonucleotides decreased T1L-induced up-regulation of cFLIP (Figure 7C) and significantly (P < 0.001) increased T1L induced-apoptosis from 21 to 41% (Figure 7D). In contrast sense cFLIP oligonucleotides did not decrease T1L-induced up-regulation of cFLIP and did not increase T1L-induced apoptosis. Neither sense nor antisense cFLIP oligonucleotides changed apoptosis induced by T3A (Figure 7D).
Discussion
The NF-κB pathway provides an attractive target to viral pathogens for modulating host cell events. Activation of NF-κB is a rapid immediate early response that occurs within minutes after exposure to a relevant inducer, which does not require de novo protein synthesis and which can promote the expression of over 100 cellular genes, including genes that participate in the host immune response, oncogenesis and regulation of apoptosis. NF-κB is activated by many viruses, including human immunodeficiency virus 1 (HIV-1),37 human T cell leukemia virus-1,38 hepatitis B virus,39 hepatitis C virus (HCV),40,41 Epstein Barr Virus,42 rotavirus43 and influenza virus44 to promote viral replication, prevent virus-induced apoptosis, and mediate the immune response to the invading pathogen.45 In contrast, activation of NF-κB by Sindbis46,47 and Dengue virus48 is associated with the induction of apoptosis, which may increase viral spread. In still other cases, proteins encoded by adenovirus,49 HCV50 and African swine fever virus51 inhibit NF-κB activity to enhance replication or contribute to viral pathogenicity.
T3 strains of reovirus (APO+) induce the activation of NF-κB in epithelial cell lines and this activation is required for apoptosis in infected cells.18,19 However, we now demonstrate that T1L, which induces significantly less apoptosis than T3A, activates NF-κB to a similar degree (Figure 2). This suggests that although required for apoptosis, reovirus-induced activation of NF-κB is not sufficient for apoptosis in infected cells.
Reovirus strain T3A (APO+) induces a second phase of NF-κB regulation in infected cells where the activation of NF-κB is inhibited at later times PI.19 This inhibition results in both the transient nature of NF-κB activation following infection with T3A (APO+) and in the inhibition of NF-κB activation following treatment of cells with external stimuli, such as TNF or etoposide.19 T1L (APO−) does not induce this second phase of NF-κB regulation. Thus T1L (APO−)-induced activation of NF-κB is sustained and activated NF-κB is present in the nucleus of infected cells at late times post infection (Figure 2). Further, T1L (APO−) does not inhibit stimulus-induced activation of NF-κB (Figure 3). These results suggest that reovirus-induced inhibition of NF-κB at later times PI is also required for apoptosis in infected cells and is supported by our demonstration that inhibition of stimulus-induced degradation of IκB is determined by the T3A S1 gene segment, which also determines reovirus-induced apoptosis.20,21 In addition, the ability of reoviruses to inhibit stimulus-induced degradation of IκB correlates with apoptosis following infection with two independent panels of reovirus reassortants (Figure 4).
The reovirus S1 gene segment encodes 2 viral proteins, the viral attachment protein σ 1 and the non-structural protein σ 1s both of which may contribute to apoptosis in reovirus-infected cells. σ 1s is the determinant of reovirus-induced G2/M cell cycle arrest, an effect that results from inhibition of the G2/M regulatory kinase p34cdc2.52,53 σ 1s contains a nuclear localization sequence and causes dramatic changes in nuclear architecture in infected cells.54 Although it is not required for reovirus-induced apoptosis of L929 or HEK293 cells,52 σ 1s enhances both the kinetics and extent of reovirus-induced apoptosis in vivo by as yet undefined mechanism.55
In virions, the reovirus σ 1 protein is a homotrimer comprised of an elongated fibrous tail, which inserts into the virion, and an externally facing globular head.56 The heads of both reovirus T1 and T3 σ 1 proteins contain a binding domain for junctional adhesion molecule (JAM), which serves as the primary reovirus receptor.57 In addition, the fibrous tail of the T3 reovirus σ 1 protein contains a domain that binds α-linked sialic acid.58 Type 3 reovirus binding to both JAM and sialic acid are required for reovirus-induced activation of NF-κB and apoptosis.23 The S1 gene segment of T3, but not T1L, reoviruses, is also associated with the ability to induce the activation of the c-Jun N-terminal kinase (JNK), which is also required for reovirus-induced apoptosis.59,60 Together, these results suggest that reovirus-induced apoptosis is induced by the activation of cellular signaling pathways early in viral infection. We therefore predict that signaling pathways induced by reovirus binding will bring about the inhibition of NF-κB seen following T3 infection.
Reovirus-induced apoptosis is mediated in epithelial cells by TNF related death-inducing ligand (TRAIL) and is blocked by reagents that inhibit TRAIL binding to its apoptosis-associated receptors, death receptors (DRs) 4 and 5.6 Reovirus-induced apoptosis is also blocked by reagents that inhibit signaling events downstream of TRAIL-receptor binding.6 NF-κB has the ability to influence TRAIL-signaling pathways in two ways. Firstly, NF-κB can act in a pro-apoptotic manner by up-regulating the expression of both TRAIL and its receptors.61–64 The increase in levels of DR5 protein expression seen following reovirus infection of HEK293 cells and the release of TRAIL from infected cells may thus reflect virus-induced activation of NF-κB.6
Death receptor signaling pathways are commonly used by viruses to induce apoptosis. For example, HIV infection increases the expression of TRAIL and sensitizes T-cells to TRAIL-mediated apoptosis.65 In addition, alteration of the cell surface expression of Fas may be involved in virus-induced, or viral regulation of, apoptosis in cells infected with influenza virus,66,67 herpes simplex virus type 2,68 bovine herpesvirus 4 (BHV 4),69 adenovirus70 and HIV-1.71,72 Similarly, apoptosis induced by Hepatitis B virus,73 HIV-1,74 BHV 469 and parvovirus H-175 may involve the TNF receptor signaling pathway. NF-κB regulation is thus likely to have implications for death ligand-mediated apoptosis and disease resulting from a variety of viral infections.
NF-κB also regulates many genes encoding proteins with anti-apoptotic properties, including cFLIP,36 which can inhibit DR-induced apoptosis. Our results show that cFLIP is regulated by NF-κB following reovirus infection and that inhibition of cFLIP can promote apoptosis in T1L-reovirus infected cells. We thus propose that following infection of HEK293 cells TRAIL-mediated apoptosis is first initiated by the activation of NF-κB and then enhanced by the later inhibition phase which results in the down-regulation of cFLIP. Inhibition of cFLIP in T1L infected cells does not increase apoptosis to levels seen following infection with T3A suggesting that additional pro-apoptotic mechanisms, are present in T3A, but not T1L-infected cells. These could include the down-regulation of additional NF-κB-dependent anti-apoptotic genes. Alternatively, it is possible that the sustained activation of NF-κB in T1L-infected cells, may prevent apoptosis through the up-regulation of as yet unidentified genes.
Reovirus infection of primary cardiac myocytes has been used to investigate the mechanisms of cell death that result in tissue injury.4,32–35 We have previously shown that apoptosis is a key mechanism by which reovirus induces myocarditic cell death and tissue injury in infected animals.4,31 We now show that in contrast to results in HEK293 cells, activation of NF-κB is not required for reovirus-induced apoptosis in primary cardiac myocytes (Figure 6). In addition, 8B induces apoptosis in the hearts of infected p50 −/− mice to a similar degree to that seen in wild type controls (not shown). The requirement of NF-κB activation for reovirus-induced apoptosis may therefore be cell-type specific. However, our demonstration that 8B (myocarditic), but not T1L (non-myocarditic), induces both the inhibition of stimulus-induced activation of NF-κB and apoptosis in infected primary cardiac myocytes (Figure 5) suggests that reovirus-induced inhibition of stimulus-induced activation of NF-κB contributes to viral-induced myocarditis. We further show that chemical inhibition of NF-κB enhances T1L-induced apoptosis in these cells (Figure 6).
Acknowledgments
Work supported by the National Institute of Health (5K08AI52261-03, R.L.D.), the Department of Veterans Affairs (Merit and REAP grants, K.L.T.), the U.S. Army Medical Research and Material Command (DAMD17-98-1-8614, K.L.T.), the Reuler-Lewin Family Professorship of Neurology (K.L.T.) and the Ovarian Cancer Research Fund (P.C.).
References
- 1.Clarke P, Tyler KL. Reovirus-induced apoptosis: A minireview. Apoptosis. 2003;8:141–150. doi: 10.1023/a:1022966508671. [DOI] [PubMed] [Google Scholar]
- 2.Oberhaus SM, Smith RL, Clayton GH, Dermody TS, Tyler KL. Reovirus infection and tissue injury in the mouse central nervous system are associated with apoptosis. J Virol. 1997;71:2100–2106. doi: 10.1128/jvi.71.3.2100-2106.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richardson-Burns SM, Kominsky DJ, Tyler KL. Reovirus-induced neuronal apoptosis is mediated by caspase 3 and is associated with the activation of death receptors. J Neuroviro. 2002;8:365–380. doi: 10.1080/13550280260422677. [DOI] [PubMed] [Google Scholar]
- 4.DeBiasi RL, Edelstein CL, Sherry B, Tyler KL. Calpain inhibition protects against virus-induced apoptotic myocardial injury. J Virol. 2001;75:351–361. doi: 10.1128/JVI.75.1.351-361.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richardson-Burns SM, Tyler KL. Regional differences in viral growth and central nervous system injury correlate with apoptosis. J Virol. 2004;78:5466–5475. doi: 10.1128/JVI.78.10.5466-5475.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clarke P, Meintzer SM, Gibson S, et al. Reovirus-induced apoptosis is mediated by TRAIL. J Virol. 2000;74:8135–8139. doi: 10.1128/jvi.74.17.8135-8139.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kominsky DJ, Bickel RJ, Tyler KL. Reovirus-induced apoptosis requires both death receptor- and mitochondrial-mediated caspase-dependent pathways of cell death. Cell Death Differ. 2002;9:926–933. doi: 10.1038/sj.cdd.4401045. [DOI] [PubMed] [Google Scholar]
- 8.Kominsky DJ, Bickel RJ, Tyler KL. Reovirus-induced apoptosis requires mitochondrial release of Smac/DIABLO and involves reduction of cellular inhibitor of apoptosis protein levels. J Virol. 2002;76:11414–11424. doi: 10.1128/JVI.76.22.11414-11424.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clarke P, Meintzer SM, Spalding AC, Johnson GL, Tyler KL. Caspase 8-dependent sensitization of cancer cells to TRAIL-induced apoptosis following reovirus-infection. Oncogene. 2001;20:6910–6919. doi: 10.1038/sj.onc.1204842. [DOI] [PubMed] [Google Scholar]
- 10.Baeuerle PA, Baltimore D. A 65-kappaD subunit of active NF-kappaB is required for inhibition of NF-kappaB by I kappaB. Genes Dev. 1989;3:1689–1698. doi: 10.1101/gad.3.11.1689. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh S, Gifford AM, Riviere LR, Tempst P, Nolan GP, Baltimore D. Cloning of the p50 DNA binding subunit of NF-kappa B: homology to rel and dorsal. Cell. 1990;62:1019–1029. doi: 10.1016/0092-8674(90)90276-k. [DOI] [PubMed] [Google Scholar]
- 12.Baeuerle PA, Baltimore D. I kappa B: A specific inhibitor of the NF-kappa B transcription factor. Science. 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- 13.Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-kappa B/I kappa B family: Intimate tales of association and dissociation. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 14.Brockman JA, Scherer DC, McKinsey TA, et al. Coupling of a signal response domain in I kappa B alpha to multiple pathways for NF-kappa B activation. Mol Cell Biol. 1995;15:2809–2818. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 16.Chen Z, Hagler J, Palombella VJ, et al. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 17.Traenckner EB, Pahl HL, Henkel T, Schmidt KN, Wilk S, Baeuerle PA. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J. 1995;14:2876–2883. doi: 10.1002/j.1460-2075.1995.tb07287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Connolly JL, Rodgers SE, Clarke P, et al. Reovirus-induced apoptosis requires activation of transcription factor NF-kappaB. J Virol. 2000;74:2981–2989. doi: 10.1128/jvi.74.7.2981-2989.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clarke P, Meintzer SM, Moffitt LA, Tyler KL. Two distinct phases of virus-induced nuclear factor kappa B regulation enhance tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in virus-infected cells. J Biol Chem. 2003;278:18092–18100. doi: 10.1074/jbc.M300265200. [DOI] [PubMed] [Google Scholar]
- 20.Tyler KL, Squier MK, Rodgers SE, et al. Differences in the capacity of reovirus strains to induce apoptosis are determined by the viral attachment protein sigma 1. J Virol. 1995;69:6972–6979. doi: 10.1128/jvi.69.11.6972-6979.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tyler KL, Squier MK, Brown AL, et al. Linkage between reovirus-induced apoptosis and inhibition of cellular DNA synthesis: role of the S1 and M2 genes. J Virol. 1996;70:7984–7991. doi: 10.1128/jvi.70.11.7984-7991.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodgers SE, Barton ES, Oberhaus SM, et al. Reovirus-induced apoptosis of MDCK cells is not linked to viral yield and is blocked by Bcl-2. J Virol. 1997;71:2540–2546. doi: 10.1128/jvi.71.3.2540-2546.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Connolly JL, Barton ES, Dermody TS. Reovirus binding to cell surface sialic acid potentiates virus-induced apoptosis. J Virol. 2001;75:4029–4039. doi: 10.1128/JVI.75.9.4029-4039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson GA, Morrison LA, Fields BN. Association of the reovirus S1 gene with serotype 3-induced biliary atresia in mice. J Virol. 1994;68:6458–6465. doi: 10.1128/jvi.68.10.6458-6465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sherry B, Fields BN. The reovirus M1 gene, encoding a viral core protein, is associated with the myocarditic phenotype of a reovirus variant. J Virol. 1989;63:4850–4856. doi: 10.1128/jvi.63.11.4850-4856.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sherry B, Blum MA. Multiple viral core proteins are determinants of reovirus-induced acute myocarditis. J Virol. 1994;68:8461–8465. doi: 10.1128/jvi.68.12.8461-8465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perlman H, Pagliari LJ, Georganas C, Mano T, Walsh K, Pope RM. FLICE-inhibitory protein expression during macrophage differentiation confers resistance to Fas-mediated apoptosis. J Exp Med. 1999;190:1679–1688. doi: 10.1084/jem.190.11.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okano H, Shiraki K, Inoue H, et al. Cellular FLICE/caspase-8-inhibitory protein as a principal regulator of cell death and survival in human hepatocellular carcinoma. Lab Invest. 2003;83:1033–1043. doi: 10.1097/01.lab.0000079328.76631.28. [DOI] [PubMed] [Google Scholar]
- 29.Sherry B. Pathogenesis of reovirus myocarditis. In: KL Tyler Oldstone MBA, editor. Reoviruses II: Cytopathogenicity and Pathogenesis. Springer-Verlag; Berlin, Germany: 1998. pp. 51–66. [DOI] [PubMed] [Google Scholar]
- 30.Sherry B, Schoen FJ, Wenske E, Fields BN. Derivation and characterization of an efficiently myocarditic reovirus variant. J Virol. 1989;63:4840–4849. doi: 10.1128/jvi.63.11.4840-4849.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeBiasi RL, Robinson BA, Sherry B, et al. Caspase inhibition protects against reovirus-induced myocardial injury in vitro and in vivo. J Virol. 2004;78:11040–11050. doi: 10.1128/JVI.78.20.11040-11050.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stewart MJ, Blum MA, Sherry B. PKR's protective role in viral myocarditis. Virology. 2003;314:92–100. doi: 10.1016/s0042-6822(03)00414-8. [DOI] [PubMed] [Google Scholar]
- 33.Azzam-Smoak K, Noah DL, Stewart MJ, Blum MA, Sherry B. Interferon regulatory factor-1, interferon-beta, and reovirus-induced myocarditis. Virology. 2002;298:20–29. doi: 10.1006/viro.2002.1470. [DOI] [PubMed] [Google Scholar]
- 34.Sherry B, Torres J, Blum MA. Reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures correlate with induction of myocarditis and are determined by viral core proteins. J Virol. 1998;72:1314–1323. doi: 10.1128/jvi.72.2.1314-1323.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noah DL, Blum MA, Sherry B. Interferon regulatory factor 3 is required for viral induction of beta interferon in primary cardiac myocyte cultures. J Virol. 1999;73:10208–10213. doi: 10.1128/jvi.73.12.10208-10213.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kreuz S, Siegmund D, Scheurich P, Wajant H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol. 2001;21:3964–3973. doi: 10.1128/MCB.21.12.3964-3973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roulston A, Lin R, Beauparlant P, Wainberg MA, Hiscott J. Regulation of human immunodeficiency virus type 1 and cytokine gene expression in myeloid cells by NF-kappa B/Rel transcription factors. Microbiol Rev. 1995;59:481–505. doi: 10.1128/mr.59.3.481-505.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun SC, Ballard DW. Persistent activation of NF-kappaB by the tax transforming protein of HTLV-1: Hijacking cellular IkappaB kinases. Oncogene. 1999;18:6948–6958. doi: 10.1038/sj.onc.1203220. [DOI] [PubMed] [Google Scholar]
- 39.Weil R, Sirma H, Giannini C, et al. Direct association and nuclear import of the hepatitis B virus X protein with the NF-kappaB inhibitor IkappaBalpha. Mol Cell Biol. 1999;19:6345–6354. doi: 10.1128/mcb.19.9.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.You LR, Chen CM, Lee YH. Hepatitis C virus core protein enhances NF-kappaB signal pathway triggering by lymphotoxin-beta receptor ligand and tumor necrosis factor alpha. J Virol. 1999;73:1672–1681. doi: 10.1128/jvi.73.2.1672-1681.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tai DI, Tsai SL, Chen YM, et al. Activation of nuclear factor kappaB in hepatitis C virus infection: Implications for pathogenesis and hepatocarcinogenesis. Hepatology. 2000;31:656–664. doi: 10.1002/hep.510310316. [DOI] [PubMed] [Google Scholar]
- 42.Sylla BS, Hung SC, Davidson DM, et al. Epstein-Barr virus-transforming protein latent infection membrane protein 1 activates transcription factor NF-kappaB through a pathway that includes the NF-kappaB-inducing kinase and the IkappaB kinases IKKalpha and IKKbeta. Proc Natl Acad Sci USA. 1998;95:10106–10111. doi: 10.1073/pnas.95.17.10106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Casola A, Garofalo RP, Crawford SE, et al. Interleukin-8 gene regulation in intestinal epithelial cells infected with rotavirus: Role of viral-induced IkappaB kinase activation. Virology. 2002;298:8–19. doi: 10.1006/viro.2002.1475. [DOI] [PubMed] [Google Scholar]
- 44.Pahl HL, Baeuerle PA. The ER-overload response: Activation of NF-kappa B. Trends Biochem Sci. 1997;22:63–67. doi: 10.1016/s0968-0004(96)10073-6. [DOI] [PubMed] [Google Scholar]
- 45.Hiscott J, Kwon H, Genin P. Hostile takeovers: Viral appropriation of the NF-kappaB pathway. J Clin Invest. 2001;107:143–151. doi: 10.1172/JCI11918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin KI, Lee SH, Narayanan R, Baraban JM, Hardwick JM, Ratan RR. Thiol agents and Bcl-2 identify an alphavirus-induced apoptotic pathway that requires activation of the transcription factor NF-kappa B. J Cell Biol. 1995;131:1149–1161. doi: 10.1083/jcb.131.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin KI, DiDonato JA, Hoffmann A, Hardwick JM, Ratan RR. Suppression of steady-state, but not stimulus-induced NF-kappaB activity inhibits alphavirus-induced apoptosis. J Cell Biol. 1998;141:1479–1487. doi: 10.1083/jcb.141.7.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jan JT, Chen BH, Ma SH, et al. Potential dengue virus-triggered apoptotic pathway in human neuroblastoma cells: Arachidonic acid, superoxide anion, and NF-kappaB are sequentially involved. J Virol. 2000;74:8680–8691. doi: 10.1128/jvi.74.18.8680-8691.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shao R, Hu MC, Zhou BP, et al. E1A sensitizes cells to tumor necrosis factor-induced apoptosis through inhibition of IkappaB kinases and nuclear factor kappaB activities. J Biol Chem. 1999;274:21495–21498. doi: 10.1074/jbc.274.31.21495. [DOI] [PubMed] [Google Scholar]
- 50.Shrivastava A, Manna SK, Ray R, Aggarwal BB. Ectopic expression of hepatitis C virus core protein differentially regulates nuclear transcription factors. J Virol. 1998;72:9722–9728. doi: 10.1128/jvi.72.12.9722-9728.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Powell PP, Dixon LK, Parkhouse RM. An IkappaB homolog encoded by African swine fever virus provides a novel mechanism for downregulation of proinflammatory cytokine responses in host macrophages. J Virol. 1996;70:8527–8533. doi: 10.1128/jvi.70.12.8527-8533.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poggioli GJ, Keefer C, Connolly JL, Dermody TS, Tyler KL. Reovirus-induced G2/M cell cycle arrest requires σ 1s and occurs in the absence of apoptosis. J Virol. 2000;74:9562–9570. doi: 10.1128/jvi.74.20.9562-9570.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poggioli GJ, Dermody TS, Tyler KL. Reovirus-induced σ 1s-dependent G2/M phase cell cycle arrest is associated with inhibition of p34cdc2. J Virol. 2001;75:7429–7434. doi: 10.1128/JVI.75.16.7429-7434.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoyt CC, Bouchard RJ, Tyler KL. Novel nuclear herniations induced by nuclear localization of a viral protein. J Virol. 2004;78:6360–6369. doi: 10.1128/JVI.78.12.6360-6369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoyt CC, Richardson-Burns SM, Goody RJ, Robinson BA, DeBiasi RL, Tyler KL. Nonstructural protein σ 1s is a determinant of reovirus virulence and influences the kinetics and severity of apoptosis induction in the heart and central nervous system. J Virol. 2005;79:2743–2753. doi: 10.1128/JVI.79.5.2743-2753.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chappell JD, Prota AE, Dermody TS, Stehle T. Crystal structure of reovirus attachment protein sigma 1 reveals evolutionary relationship to adenovirus fiber. EMBO J. 2002;15:1–11. doi: 10.1093/emboj/21.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barton ES, Forrest JC, Connolly JL, et al. Junction adhesion molecule is a receptor for reovirus. Cell. 2001;104:441–451. doi: 10.1016/s0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- 58.Chappell JD, Duong JL, Wright BW, Dermody TS. Identification of carbohydrate-binding domains in the attachment proteins of Type1 and Type 3 reoviruses. J Virol. 2000;74:8472–8479. doi: 10.1128/jvi.74.18.8472-8479.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clarke P, Meintzer SM, Widmann C, Johnson GL, Tyler KL. Reovirus infection activates JNK and the JNK-dependent transcription factor c-Jun. J Virol. 2001;75:11275–11283. doi: 10.1128/JVI.75.23.11275-11283.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clarke P, Meintzer SM, Wang Y, et al. JNK regulates the release of proapoptotic mitochondrial factors in reovirus-infected cells. J Virol. 2004;78:13132–13138. doi: 10.1128/JVI.78.23.13132-13138.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gibson SB, Oyer R, Spalding AC, Anderson SM, Johnson GL. Increased expression of death receptors 4 and 5 synergizes the apoptosis response to combined treatment with etoposide and TRAIL. Mol Cell Biol. 2000;20:205–212. doi: 10.1128/mcb.20.1.205-212.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ravi R, Bedi GC, Engstrom LW, et al. Regulation of death receptor expression and TRAIL/Apo2L-induced apoptosis by NF-kappaB. Nat Cell Biol. 2001;3:409–416. doi: 10.1038/35070096. [DOI] [PubMed] [Google Scholar]
- 63.Spalding AC, Jotte RM, Scheinman RI, et al. TRAIL and inhibitors of apoptosis are opposing determinants for NF-kappaB-dependent, genotoxin-induced apoptosis of cancer cells. Oncogene. 2002;21:260–271. doi: 10.1038/sj.onc.1205048. [DOI] [PubMed] [Google Scholar]
- 64.Rivera-Walsh I, Waterfield M, Xiao G, Fong A, Sun SC. NF-kappaB signaling pathway governs TRAIL gene expression and human T-cell leukemia virus-I Tax-induced T-cell death. J Biol Chem. 2001;276:40385–40388. doi: 10.1074/jbc.C100501200. [DOI] [PubMed] [Google Scholar]
- 65.Jeremias I, Herr I, Boehler T, Debatin KM. TRAIL/Apo-2-ligand-induced apoptosis in human T cells. Eur J Immunol. 1998;28:143–152. doi: 10.1002/(SICI)1521-4141(199801)28:01<143::AID-IMMU143>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 66.Takizawa T, Matsukawa S, Higuchi Y, Nakamura S, Nakanishi Y, Fukuda R. Induction of programmed cell death (apoptosis) by influenza virus infection in tissue culture cells. J Gen Virol. 1993;74:2347–2355. doi: 10.1099/0022-1317-74-11-2347. [DOI] [PubMed] [Google Scholar]
- 67.Takizawa T, Fukuda R, Miyawaki T, Ohashi K, Nakanishi Y. Activation of the apoptotic Fas antigen-encoding gene upon influenza virus infection involving spontaneously produced beta-interferon. Virology. 1995;209:288–296. doi: 10.1006/viro.1995.1260. [DOI] [PubMed] [Google Scholar]
- 68.Sieg S, Yildirim Z, Smith D, et al. Herpes simplex virus type 2 inhibition of Fas ligand expression. J Virol. 1996;70:8747–8751. doi: 10.1128/jvi.70.12.8747-8751.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang GH, Bertin J, Wang Y, et al. Bovine herpesvirus 4 BORFE2 protein inhibits Fas- and tumor necrosis factor receptor 1-induced apoptosis and contains death effector domains shared with other gamma-2 herpesviruses. J Virol. 1997;71:8928–8932. doi: 10.1128/jvi.71.11.8928-8932.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tollefson AE, Hermiston TW, Lichtenstein DL, et al. Forced degradation of Fas inhibits apoptosis in adenovirus-infected cells. Nature. 1998;392:726–730. doi: 10.1038/33712. [DOI] [PubMed] [Google Scholar]
- 71.Conaldi PG, Biancone L, Bottelli A, et al. HIV-1 kills renal tubular epithelial cells in vitro by triggering an apoptotic pathway involving caspase activation and Fas upregulation. J Clin Invest. 1998;102:2041–2049. doi: 10.1172/JCI3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kaplan D, Sieg S. Role of the Fas/Fas ligand apoptotic pathway in human immunodeficiency virus type 1 disease. J Virol. 1998;72:6279–6282. doi: 10.1128/jvi.72.8.6279-6282.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Su F, Schneider RJ. Hepatitis B virus HBx protein sensitizes cells to apoptotic killing by tumor necrosis factor alpha. Proc Natl Acad Sci USA. 1997;94:8744–8749. doi: 10.1073/pnas.94.16.8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Herbein G, Mahlknecht U, Batliwalla F, et al. Apoptosis of CD8+ T cells is mediated by macrophages through interaction of HIV gp120 with chemokine receptor CXCR4. Nature. 1998;395:189–194. doi: 10.1038/26026. [DOI] [PubMed] [Google Scholar]
- 75.Johnson DE, Gastman BR, Wieckowski E, et al. Inhibitor of apoptosis protein hILP undergoes caspase-mediated cleavage during T lymphocyte apoptosis. Cancer Res. 2000;60:1818–1823. [PubMed] [Google Scholar]