

Abstract
One mechanism of regulating V-ATPase activity in vivo involves reversible dissociation into its component V1 and V0 domains, which in yeast occurs in response to glucose depletion. V-ATPase complexes containing the Vph1p isoform of subunit a (VCC) are targeted to the vacuole whereas Stv1p-containing complexes (SCC) are targeted to the Golgi. Overexpression of Stv1p results in mistargeting of SCC to the vacuole. We have investigated the role of the a subunit isoform and cellular environment in controlling dissociation using vacuolar protein sorting (vps) mutants which accumulate proteins in either the prevacuolar compartment (PVC) (vps27Δ) or a post-Golgi compartment (PGC) (vps21Δ). Dissociation of both VCC and SCC depends upon cellular environment, with dissociation most complete in the vacuole and least complete in the PVC. The dependence of dissociation on V-ATPase activity was also investigated using both concanamycin and inactivating mutations. Concanamycin partly blocks dissociation of both VCC and SCC in all three compartments, with inhibition generally greater for SCC then VCC. The R735Q mutant of Vph1p results in loss of both ATPase and proton transport whereas the R735K mutant lacks proton transport but has 10% of wild type ATPase activity. For VCC in the vacuole, dissociation is completely blocked for the R735Q but not the R735K mutant. Significant dissociation of VCC is observed for both mutants in the PVC and PGC, indicating that V-ATPase activity is not absolutely required for dissociation. Similar results were obtained for SCC, although dissociation of SCC is again generally more sensitive to activity than VCC. These results suggest that the cellular environment is important both in controlling in vivo dissociation of the V-ATPase and the dependence of this process on catalytic activity. Moreover catalytic activity is not absolutely required for V-ATPase dissociation.
Introduction
The vacuolar (H+)-ATPases (V-ATPases1) are a family of ATP-dependent proton pumps that both acidify intracellular compartments and pump protons across the plasma membrane (1–4). Within intracellular compartments, V-ATPases function in receptor-mediated endocytosis, intracellular membrane traffic, degradation and processing of proteins, and coupled transport of small molecules, such as neurotransmitters. They also assist in the entry of envelope viruses, such as influenza virus, and the killing of cells by toxins, such as diphtheria toxin and anthrax toxin (5). V-ATPases are also present in the plasma membrane of cells, where they function in bone resorption (6), renal acidification (3), sperm maturation (7), pH homeostasis (8) and tumor metastasis (9). V-ATPases are thus potential targets in treating such diseases as osteoporosis and cancer.
The structure and mechanism of V-ATPases resembles that of the F-ATPase (10). V-ATPases are composed of a peripheral V1 complex consisting of eight subunits (A-H), that hydrolyzes ATP and an integral V0 complex consisting of six subunits (a, d, e, c, c′ and c″), that conducts protons (1–4). The V0 complex includes a ring of proteolipid subunits (c, c′ and c″) onto which subunit d sits (11). Subunit a is a 100 kDa protein composed of a hydrophilic amino-terminus and a hydrophobic carboxyl-terminus that is in contact with the proteolipid ring (12). V-ATPases operate by a rotary mechanism (13, 14) in which the c-ring rotates relative to subunit a, with proton transport driven by interactions at the subunit a-proteolipid interface (12).
Subunit a is also important for targeting the V-ATPase to different membranes in the cell. In yeast there are two isoforms of subunit a, Vph1p and Stv1p (15, 16). Vph1p targets V-ATPases to the vacuole whereas Stv1p causes V-ATPases to be retained in the Golgi (16, 17). The amino-terminus of subunit a contains the signal responsible for targeting of V-ATPases (18). In a strain disrupted in both Vph1p and Stv1p, over-expression of Stv1p results in the appearance of a significant number of Stv1p-containing complexes in the vacuole (16–18). V-ATPase complexes containing Vph1p and Stv1p also differ in assembly and activity. Stv1p-containing complexes localized to the vacuole show lower assembly of V0 and V1 as well as a 4-fold lower coupling of proton transport to ATP hydrolysis relative to Vph1p-containing complexes (17). In mammals there are four isoforms of subunit a (a1–a4) (6,19,20). The a3 isoform is present in the plasma membrane of osteoclasts and mutations in the a3 gene cause the disease osteopetrosis (21). The a4 isoform is expressed in the plasma membrane of renal intercalated cells and mutations cause the disease renal tubule acidosis (22).
An important mechanism of controlling V-ATPase activity in vivo involves reversible dissociation of the V1 and V0 domains (1,2). Reversible dissociation has been shown to regulate V-ATPase activity in both yeast and insect cells (23,24), and has recently been implicated in control of acidification in renal cells (25) and in dendritic cells (26), where V-ATPases are essential for antigen processing. A distinctive feature of dissociation of V-ATPases is that the free V1 domain does not hydrolyze MgATP (27) and free V0 is not passively permeable to protons (28). This property is essential to avoid generation of an uncoupled ATPase activity in the cytosol or an unregulated passive proton conductance in cellular membranes.
In yeast dissociation of V-ATPase complexes is induced by glucose depletion, occurs rapidly and reversibly and does not require new protein synthesis (23). Various signaling pathways involved in response to glucose depletion appear not to be involved in this process (29), although dissociation (but not reassembly) requires an intact microtubular network (30). By contrast, reassembly (but not dissociation) requires a novel cytoplasmic complex termed RAVE, which is also important for normal assembly of the V-ATPase (31,32). Thus dissociation and reassembly appear to be independently controlled processes. Dissociation has also been shown to require catalytic activity (29) and to be blocked by chloroquine which neutralizes internal acidic compartments (33).
Previous results from our lab have shown that Vph1p-containing complexes localized to the vacuole undergo glucose-dependent dissociation whereas Stv1p-containing complexes localized to the Golgi do not (17). By contrast, Stv1p-containing complexes localized to the vacuole do show dissociation. Trafficking of Vph1p-containing complexes to the vacuole can be blocked using vacuolar protein sorting (vps) mutants. Strains disrupted in VPS27 accumulate proteins (including Vph1p) in a prevacuolar compartment (PVC) whereas vps21 mutants retain proteins (including Vph1p) in a post-Golgi compartment (34). Vph1p-containing complexes localized to the PVC or post-Golgi compartments showed glucose-dependent dissociation, but less completely than when localized to the vacuole (17). In order to determine whether the a subunit isoform plays any role in controlling in vivo dissociation, we have now examined the dissociation behavior of both Vph1p and Stv1p-containing complexes in wild type, vps21Δ and vps27Δ strains. The dependence of dissociation on activity has also been examined in these strains. The results suggest that the intracellular environment plays an important role in controlling both dissociation behavior and its dependence on activity.
Experimental Procedures
Materials and Strains
Zymolyase 100T was obtained from Seikagaku America, Inc. Concanamycin A were purchased from Fluka. The mouse monoclonal antibody 8B1 against the yeast V-ATPase subunit A was purchased from Molecular Probes; the monoclonal antibody 3F10 against the HA antigen conjugated to horseradish peroxidase and protease inhibitors were purchased from Roche. Restriction endonucleases, T4 DNA ligase, and other molecular biology reagents were from Invitrogen and New England Biolabs. Dithiobis [succinimidyl propionate] was purchased from Pierce. SDS, nitrocellulose membranes, Tween 20 and horseradish peroxidase-conjugated goat anti-mouse/rabbit IgG were purchased from Bio-Rad. The chemiluminescence substrate for horseradish peroxidase was from Kirkegaard & Perry Laboratories. Protein A-Sepharose, protein G-agarose and most other chemicals such as dithiothreitol were purchased from Sigma. Materials for E. coli and yeast culture and carrier DNA were purchased from BD Biosciences.
Yeast strain MM112 (MATa vph1::LEU2 stv1::LYS2 his3-Δ 200 leu2 lys2 ura3-52) (16) was used for VPS21 or VPS27 gene knockout and for HA-Vph1p or HA-Stv1p expression. Yeast cells were grown in yeast extract-peptone-dextrose medium or synthetic dropout medium (35).
Yeast Gene Replacement
The strains MM112 vps21Δ and MM112 vps27Δ were constructed following the protocol of two-step gene disruption (36). The VPS21 and VPS27 genes were replaced by homologous recombination in MM112 by the marker gene Kanr and the strains MM112 vps21Δ and MM112 vps27Δ were selected using media containing the antibiotic G418.
Transformation and Selection
The single copy plasmid pRS316 carrying the HA-VPH1 sequence (pRS316-VPH1::HA, also called pSKN12) or the HA-STV1 sequence (pRS316-STV1::HA, also called pSKN11) and the high copy plasmid YEp352 carrying HA-Stv1p (YEp352-STV1::HA, also called pSKN14) were constructed previously (17), and were transformed into yeast strain MM112 by the lithium acetate method (37). The transformants were selected on uracil minus (Ura−) plates. For co-transformation of yeast strains with HA-Stv1p and untagged Vph1p, the VPH1 gene was subcloned into pRS413, a vector containing the HIS3 selectable marker. A yeast strain carrying pSKN14 was transformed with pRS413-Vph1p and then selected on uracil and histidine minus (Ura−His−) plates.
Construction of Mutants
Site-directed mutagenesis was performed to introduce mutations R795K and R795Q on EcoRI-BamHI fragments of the HA-Stv1p using the Altered Sites II In Vitro Mutagenesis System (Promega). The mutagenic oligonucleotides are designed as 5′-CATCATATCTACAACTTTGGGCACTA-3′ and 5′-AGCATCATATCTAAAACTTTGGG CACTAT-3′ for R795Q and R795K respectively. Mutations were confirmed by DNA sequencing. Fragments containing the indicated mutations were then substituted back into the pSKN14 (YEp352-STV1::HA). Cloned plasmids were again confirmed by DNA sequencing. The EcoRI-BamHI region of HA-Vph1p was substituted by the corresponding sequence from either the R735Q or R735K mutant of Vph1p constructed previously (38).
In Vivo Dissociation of Yeast V-ATPases in Response to Glucose Deprivation
In vivo dissociation of yeast V-ATPases was induced by glucose deprivation as described before (23) with some modification. The yeast strains MM112, MM112 vps21Δ and MM112 vps27Δ expressing either normal levels of Vph1p (using pSKN12) or high levels of Stv1p (using pSKN14) were grown overnight, diluted in fresh media to an absorbance at 600 nm of 0.1–0.2 and grown to an absorbance of 0.6–0.8. Cells were harvested and converted to spheroplasts by addition of 0.3 mg/ml Zymolyase in lysis buffer (YEPD, 0.1 M Tris Mes pH7.5, 0.7 M sorbitol, 2 mM dithiothreitol) and incubated for 30 min at 30° C. Cells were then split into 2 aliquots and incubated in YEP (without glucose) and YEPD medium (with 2% glucose) at 30 °C for 15 min. Spheroplasts were pelleted and lysed in PBS pH7.2 containing 1% C12E9, protease inhibitors and 1 mM dithiobis [succinimidyl propionate]. For concanamycin A inhibition experiments, the inhibitor was added to the medium to a final concentration 1 μM in the final 10 min of spheroplasting and then kept at this concentration until spheroplasts were lysed.
V-ATPase complexes were immunoprecipitated from the lysate using the antibody 8B1 against subunit A and protein A-Sepharose followed by electrophoresis on 7.5% acrylamide gels and transfer to nitrocellulose. Western blotting was then performed using the horseradish peroxidase-conjugated monoclonal antibody 3F10 against HA to blot the a subunit of the V0 domain or the antibody 8B1 against the A subunit to blot the V1 domain followed by a horseradish peroxidase-conjugated secondary antibody. Dissociation of the V-ATPase complex is reflected as a reduction in the amount of subunit a (in V0 complexes) co-immunoprecipitated by the antibody directed against subunit A (in the V1 complexes). Densitometry analysis was performed using the software ImageJ (http://rsb.info.nih.gov/ij/).
To determine the linearity of the signal obtained in the Western blotting experiments, a titration was performed analyzing the signal obtained with varying amounts of the immunoprecipitates using both the anti-HA and anti-A subunit antibodies. Over the intensity range employed in these experiments (relative intensities of approximately 1,000 to 16,000 units), the signal obtained was linear with the amount of HA-Vph1p loaded but showed some deviation from linearity for subunit A. This may be because detection of subunit A involved both a primary antibody (8B1) and an HRP-conjugated secondary antibody whereas detection of HA-Vph1p was done directly using an HRP-conjugated anti-HA antibody (3F10), eliminating the necessity of a secondary antibody. Quantitation of assembly was done by measuring the amount of subunit a precipitated, which was linear. The slight non-linearity of signal with the amount of subunit A was not a problem because samples were matched for the amount of subunit A within a given experiment and nearly matched between experiments.
Immunoisolation of compartments carrying HA-Stv1p
The strains MM112, MM112 vps27Δ and MM112 vps21Δ were co-transformed with pPRS413-VPH1 and YEp352-STV1::HA, cultured, harvested and converted to spheroplasts as described under the procedure for measurement of in vivo dissociation. Spheroplasts were lysed by osmotic shock in the absence of detergent in solubilization buffer (50 mM Tris-HCl pH 7.5, 1 mM EDTA, and 1% glycerol), followed by sedimentation twice for 1 min at 3,000×g to remove unbroken cells and nuclei. Supernatants were precleared by addition of protein G agarose, incubation for 1 hour and sedimentation at 3,000×g for 1 min. Supernatants were then incubated with the anti-HA antibody (3F10) plus protein G agarose overnight at 4°C. The compartments carrying HA-Stv1p were then precipitated by sedimentation and the proteins eluted from the beads with SDS sample buffer and separated by SDS-PAGE. The presence of both HA-Stv1p and Vph1p in the immunoprecipitates were detected by Western blotting using the monoclonal antibodies against HA (3F10) and Vph1p (10D7), respectively.
Other Procedures
Vacuolar membrane vesicles were isolated using the protocol described previously (39). Quinacrine staining of yeast was performed as previously described (40). Protein concentrations were determined by the method of Lowry (41). ATPase activity was measured using a coupled spectrophotometric assay in the presence or absence of 1 μM concanamycin A as described previously (33). ATP-dependent proton transport was measured in transport buffer (25 mM MES-Tris, pH 7.2, 5 mM MgCl2) using the fluorescence probe 9-amino-6-chloro-2-methoxyacridine in the presence or absence of 1 μM concanamycin, as described previously (42).
Results
Stv1p-containing complexes localized to the Golgi show reduced assembly with V1 relative to Vph1p-containing complexes localized to the vacuole
Previous results from our laboratory have demonstrated that V0 complexes containing the Stv1p isoform of subunit a, when localized to the vacuole through overexpression, showed significantly lower levels of assembly with the V1 domain than V0 complexes containing Vph1p, thus giving rise to the approximately 10-fold difference in ATPase activity for these two complexes (17). Because the vacuolar environment is different in a number of respects from that of the Golgi, it was of interest to determine whether this reduced assembly with V1 was also observed for Stv1p-containing complexes present in their normal cellular environment, namely the Golgi. HA-tagged Stv1p was expressed at low levels in the yeast strain MM112 using the single-copy plasmid pRS316. As a control HA-tagged Vph1p was also expressed in MM112 using the same plasmid. Complexes containing either HA-Vph1p or HA-Stv1p were immunoprecipitated using the antibody 3F10 directed against the HA tag. After SDS-PAGE, the precipitants were Western blotted using the anti-HA antibody and an antibody directed against subunit A of the V1 domain. Since subunit A is co-immunoprecipitated with subunit a only when the V1 and V0 domains are assembled, the amount of subunit A precipitated using the anti-HA antibody is a reflection of the degree of assembly of the V1 and V0 domains. As can be seen in Fig. 1 (left two lanes), Vph1p-containing complexes localized to the vacuole show much better assembly with V1 than do Stv1p-containing complexes localized to the Golgi. Thus, the difference in assembly behavior of Vph1p and Stv1p-containing complexes is not an artifact of mislocalization of Stv1p to the vacuole. For comparison, the degree of assembly of Stv1p localized to the vacuole through overexpression is also shown (Fig. 1, lane3). For this experiment, the amount of antibody used to precipitate HA is limiting, such that no significant difference between the HA signal in lanes 2 and 3 is observed. To determine whether Stv1p-containng complexes localized to the Golgi might be unstable during solubilization and immunopreciptation, the experiments were repeated in the presence of the specific V-ATPase inhibitor concanamycin, which has been shown to inhibit both Vph1p and Stv1p-containing V-ATPases (17). As can be seen in Fig. 1 (lanes 4 and 5), the difference in assembly of Vph1p-containing complexes in the vacuole and Stv1p-containing complexes localized to the Golgi was observed even in the presence of concanamycin, although Stv1p-containing complexes in the vacuole show much higher assembly with V1 in the presence of concanamycin (lane 6). This latter result rules out the possibility that the HA epitope in the HA-Stv1p-containing complexes is not accessible in intact V1V0.
Figure 1. Assembly properties of subunit a isoforms localized to their normal cellular environment.

Yeast strain MM112 (vph1Δ stv1Δ) containing plasmids pSKN12 (pRS316-VPH1::HA) or pSKN11(pRS316-STV1::HA) were grown to log phase, harvested, and converted to spheroplasts with Zymolyase. Whole cell lysates were prepared by treatment with C12E9 (with or without 1 μM concanamycin) and V0 complexes were immunoprecipitated using the antibody 3F10 (Roche) against the HA epitope. Following SDS-PAGE, Western blotting was performed using the antibody 8B1-F3 (Molecular Probes) against subunit A (Vma1p) as well as 3F10 against HA. The amount of subunit A co-precipitated with the antibody against HA is a reflection of the degree of assembly of V1 and V0.
Cellular environment influences glucose-dependent dissociation of V-ATPase complexes containing either isoform of subunit a
In order to examine the dependence of in vivo dissociation of the V-ATPase on cellular environment, experiments were performed using yeast strains disrupted in two genes of the CPY (carboxypeptidase Y) pathway. V-ATPase complexes containing Vph1p employ the CPY pathway for delivery to the vacuole (34,43,44). Yeast strains disrupted in the VPS27 gene accumulate proteins in a PVC as a result of a defect in budding of vesicles from this compartment (43). In strains disrupted in VPS21, proteins accumulate in Golgi-derived vesicles due to the inability of these vesicles to fuse with the PVC (34). The ability of HA-tagged Vph1p and Stv1p-containing complexes to undergo dissociation in response to glucose depletion was examined in wild type cells as well as cells disrupted in either VPS27 or VPS21. HA-Stv1p was overexpressed using a high copy plasmid, under which conditions a significant amount of HA-Stv1p is mistargeted to the vacuole in a strain that is wild type for the CPY pathway (17).
To test whether Vph1p and overexpressed Stv1p are localized to the same compartments in the vps21Δ and vps27Δ strains, HA-tagged Stv1p and untagged Vph1p were co-expressed in the same yeast strain. Following cell lysis by osmotic shock in the absence of detergent, the compartments containing HA-Stv1p were immunoprecipitated using the monoclonal antibody 3F10 against the HA tag plus protein G agarose. The immunoprecipitates were then analyzed by Western blot for both HA and Vph1p using the respective monoclonal antibodies. As can be seen in Fig. 2, lanes 1–3, Vph1p is co-immunoprecipitated with HA-Stv1p in wild type cells as well as the vps21Δ and vps27Δ strains. By contrast, no significant co-immunoprecipitation was observed if immunoprecipitation was performed on cell lysates from cells expressing either Vph1p or HA-Stv1p which were mixed before immunoprecipitation (Fig. 2, lane 4). These results suggest that Vph1p and overexpressed HA-Stv1p are present in the same compartments in both the wild type cells and the vps21Δ and vps27Δ mutant strains.
Figure 2. Presence of Vph1p in cellular compartments containing HA-Stv1p immunoisolated using the antibody against HA.

Yeast strains co-expressing Vph1p and HA-Stv1p (expressed using a high copy plasmid) were lysed by osmotic shock in the absence of detergent to maintain the integrity of intracellular compartments. Following removal of unbroken cells and nuclei by sedimentation, immunoprecipitation was performed using the monoclonal antibody (3F10) against HA and protein G agarose. Following SDS-PAGE, Western blotting was performed using both 3F10 (directed against HA) and 10D7 (specific for Vph1p). Lane 1–3: immunoisolated samples from strains MM112, MM112 vps27Δ and MM112 vps21Δ, respectively, co-transformed with pRS413-VPH1 and YEp352-STV1::HA; lane 5–7: whole cell lysates of the same strains. As a negative control, whole cell lysates from the cells separately expressing either Vph1p or HA-Stv1p were mixed in vitro followed by immunoprecipitation and Western blotting as described above (lane 4).
To determine the amount of glucose-dependent dissociation occurring in each strain, spheroplasts were incubated in the presence or absence of glucose for 15 min at 30° C followed by detergent solubilization and immunoprecipitation of V1 and V1V0 complexes using an antibody specific for subunit A. Following SDS-PAGE, Western blotting was performed using both the antibody against subunit A and an antibody against the HA epitope tag present in subunit a. The amount of HA-Vph1p or HA-Stv1p co-immunoprecipitated with subunit A thus reflects the degree of association between the V1 and V0 domains. Representative results are shown for both HA-Vph1p (Fig. 3a) and HA-Stv1p (Fig. 3c). Band intensities were determined by densitometry and the results from at least three independent determinations are shown in the bar graphs (Figs. 3b and 3d). The values shown represent the ratio of the degree of assembly observed in the absence of glucose divided by the degree of assembly observed in the presence of glucose. As can be seen, Vph1p-containing complexes showed the greatest degree of glucose-dependent dissociation when localized to the vacuole and the least amount of dissociation when localized to the PVC. These results are in agreement with earlier findings using non-tagged Vph1p (17). Stv1p-containing complexes show nearly the same dependence of dissociation on cellular environment, with dissociation being most complete in the vacuole and least complete in the PVC. Thus Stv1p-containing complexes appear to show the same behavior with respect to glucose-dependent dissociation as Vph1p-containing complexes, not only when localized to the vacuole (17), but also when present in other cellular compartments.
Figure 3. Dependence of glucose-dependent dissociation of Vph1p or Stv1p-containing complexes on intracellular environment and the presence or absence of concanamycin.
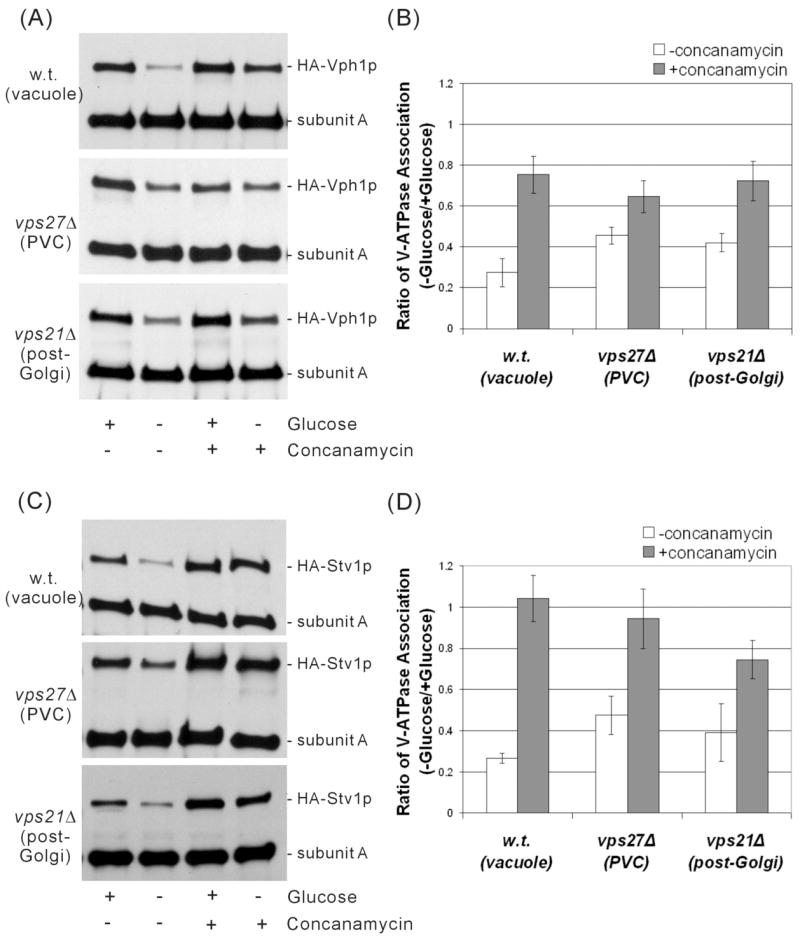
Yeast strains MM112, MM112 vps21Δ and MM112 vps27Δ expressing either HA-Vph1p or HA-Stv1p were grown to an absorbance of 0.6–0.8, converted to spheroplasts, and incubated in the presence or absence of 2% glucose for 15 min. Spheroplasts were lysed in the presence of C12E9 and both V1 and V1V0 complexes were immunoprecipitated using the antibody 8B1 against subunit A. Following SDS-PAGE, Western blotting was performed using both the antibody against subunit A and the anti-HA antibody (3F10) directed against the HA-tagged subunit a. The degree of assembly of V1 and V0 is reflected in the amount of subunit a co-precipitated with the antibody against subunit A. Where indicated V-ATPase activity was inhibited by addition of 1 μM concanamycin A during the final 10 min of spheroplasting and kept at this concentration through lysis. The results of a representative experiment are shown on the left (panel A for Vph1p and panel C for Stv1p) and the average of three independent determinations are shown on the right (panel B for Vph1p and panel D for Stv1p). Band intensities were determined by densitometry and the results are expressed as the ratio of assembly observed in the absence of glucose divided by the assembly observed in the presence of glucose, where assembly is quantitated as the ratio of the intensity of subunit a divided by the intensity of subunit A. Error bars correspond to average deviations.
Cellular environment affects the dependence of in vivo dissociation on catalytic activity
Previous results have demonstrated that glucose-dependent dissociation of Vph1p-containing V-ATPase complexes localized to the vacuole depends upon catalytic activity (29). In order to determine whether this dependence of dissociation on activity extends to Stv1p-containing complexes and to both Vph1p and Stv1p-containing complexes localized to other compartments, the effect of the specific V-ATPase inhibitor concanamycin on dissociation was examined. As can be seen in Fig. 3a and b, concanamycin was able to partially block dissociation of Vph1p-containing complexes localized to the vacuole, the PVC and the post-Golgi compartments, with the greatest effect observed for V-ATPases present in the vacuole. Similar results were obtained for Stv1p-containing complexes (Fig. 3c and d), although more complete inhibition of dissociation was observed for Stv1p-containing complexes relative to Vph1p-containing complexes in all compartments except the post-Golgi compartment. In particular, complete inhibition of dissociation was only observed for Stv1p-containing complexes localized to both the vacuole and the PVC. These results suggest that the dependence of dissociation on activity is influenced by both the cellular environment and, to some extent, by the a subunit isoform present.
It should also be noted that treatment with concanamycin partly stabilized Stv1p-containing complexes even in the presence of glucose, as seen from the ratio of staining of subunits a and A in the presence and absence of concanamycin (Fig. 3c). These results may suggest that Stv1p-containing complexes are partly destabilized and that this instability can be partly overcome by inhibition of the complex.
Dissociation behavior of inactive mutants of the V-ATPase when localized to different intracellular compartments
As noted above, in most cases concanamycin did not show complete inhibition of in vivo dissociation of the V-ATPase, particularly for Vph1p-conaining complexes. Because of the possibility that concanamycin may not have equal accessibility to V-ATPases in all compartments, it was of interest to determine the effect of inactivating mutations on glucose-dependent dissociation. We had previously shown that R735 in TM7 of Vph1p is essential for proton transport by the V-ATPase (38). Any substitution of the arginine at this position (including the conservative R735K mutation) leads to complete loss of proton transport activity (38). Moreover, all mutants except R735K completely lacked ATPase activity (the latter mutant retained some residual uncoupled ATP hydrolysis). Quinacrine staining of cells expressing the R735K and R735Q mutants of Vph1p confirms that no detectable acidification of intracellular compartments is observed in these strains (Fig. 4). Dissociation of V-ATPase complexes containing Vph1p bearing these two mutations were compared, first for V-ATPases localized to the vacuole. Under these conditions, complete inhibition of dissociation for the R735Q mutant was observed whereas significant dissociation of the R735K mutant occurred (Fig. 5a,b). These results suggest that ATPase activity rather than proton transport is more critical for glucose-dependent dissociation, at least for Vph1p-containing complexes localized to the vacuole. By contrast, both Vph1p mutants showed significant dissociation when localized to either the PVC or the post-Golgi compartment. These results indicate that catalytic activity (either proton transport or ATPase activity) is not absolutely required for in vivo dissociation. Moreover, the cellular environment affects the dependence of in vivo dissociation on catalytic activity.
Figure 4. Quinacrine staining of yeast cells expressing wild type and mutant forms of Vph1p localized to different cellular compartments.
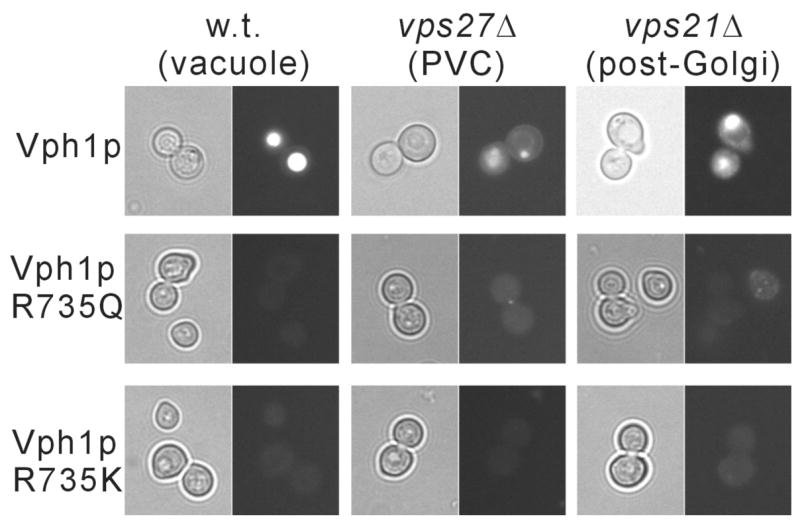
Yeast strains MM112, MM112 vps21Δ and MM112 vps27Δ expressing either HA-Vph1p (wild type ) or HA-Vph1p containing the R735Q or R735 K mutations were stained with quinacrine and observed by either phase contrast (left panels) or fluorescence microscopy (right panels) as described under Experimental Procedures.
Figure 5. Dependence of glucose-dependent dissociation V-ATPase complexes containing mutant forms Vph1p or Stv1p on intracellular environment.

Yeast strains MM112, MM112 vps21Δ and MM112 vps27Δ expressing either the R735K or R735Q mutant forms of HA-Vph1p (top panels) or the R795Q or R795K mutant forms of HA-Stv1p (bottom panels) were tested for glucose-dependent dissociation as described in the legend to Fig. 2. The results of a representative experiment are shown on the left (panel A for Vph1p and panel C for Stv1p) and the average of three independent determinations are shown on the right (panel B for Vph1p and panel D for Stv1p). Results are again expressed as the ratio of assembly observed in the absence of glucose divided by the assembly observed in the presence of glucose, with error bars corresponding to average deviations.
For Stv1p, we observed that mutation of R795 (the residue that corresponds to R735 in Vph1p) to lysine leads to loss of 93% of wild type ATPase activity whereas mutation to glutamine leads to loss of 100% of ATPase activity. Dissociation of complexes containing these mutant forms of Stv1p were then compared in the wild type, vps21Δ and vps27Δ strains. As can be seen in Fig. 5c and d, the mutant forms of Stv1p dissociated somewhat less completely than Vph1p in almost all cases, although only for the R795Q mutant in the vacuole and PVC was dissociation nearly completely blocked. Thus some degree of dissociation of Stv1p-containing complexes was observed even for completely inactive mutants, consistent with the concanamycin results described above.
Discussion
Reversible dissociation of V-ATPase complexes represents an important but as yet incompletely understood process for regulating acidification in vivo. Because V-ATPases reside in multiple cellular compartments, it might be predicted that not all of the V-ATPases in the cell would show the same degree of dissociation in response to a given stimulus. This prediction is supported by the observation that the degree of dissociation of the V-ATPase in yeast in response to glucose depletion does depend upon the cellular environment. Thus, V-ATPase complexes localized to the vacuole show the greatest degree of dissociation, those localized to the PVC or post-Golgi compartments show intermediate dissociation behavior, while those localized to the Golgi (at least for complexes containing Stv1p) do not dissociate at all (Figure 2 and reference 17). The failure of Golgi-localized V-ATPases to dissociate in response to glucose depletion is not due to an intrinsic property of Stv1p, since Stv1p localized to compartments other than the Golgi dissociates efficiently. It might be argued that Stv1p-containing complexes are already associated to such a low level that no further dissociation is possible. However, because Stv1p shows similarly low levels of assembly in both the Golgi and the vacuole, even in the presence of glucose (Fig. 1 and reference 17), but shows dissociation when localized to the vacuole but not the Golgi, this possibility is ruled out. The ability of the cell to retain Golgi-localized V-ATPases in an assembled state, even in the absence of glucose, may indicate that the Golgi V-ATPase, even though less active than that present in the vacuole, nevertheless serves an indispensable function.
In addition to providing a more alkaline pH to the Golgi compartment, does the lower level of assembly of Stv1p-containing V-ATPases localized to the Golgi serve any additional function? One possibility is that the free V0 domains present as a result of this lower assembly participate in membrane fusion reactions occurring in this compartment. The V0 domain has been postulated to participate directly in membrane fusion based upon studies of homotypic vacuole fusion in yeast (45,46). In this model, V0 domains in adjacent membranes pair in a trans complex that follows trans-SNARE pairing but that promotes fusion of the tethered membranes. Recent studies of vesicle fusion in Drosophila and C.elegans (47,48) suggest that this role may not be restricted to yeast. The presence of additional free V0 domains in Golgi membranes may reflect the high degree of membrane fusion occurring in this compartment. Additional studies will be required to determine whether free V0 domains participate in fusion of vesicles involved in movement of proteins between Golgi stacks.
It was previously observed that in vivo dissociation of V-ATPase complexes depends upon catalytic activity. This was demonstrated for Vph1p-containing complexes localized to the vacuole (29). In the present paper we demonstrate that this dependence of dissociation on activity is, in most cases, not absolute. Thus, while treatment with concanamycin in all cases at least partially blocks dissociation of both Vph1p and Stv1p-containing complexes, in most cases this inhibition of dissociation is incomplete. For Vph1p-containing complexes, in particular, 20–40% of dissociation is still observed, even in the presence of concanamycin. This is also true for inactivating mutants of Vph1p, where only the R735Q mutant localized to the vacuole shows complete inhibition of dissociation. For Stv1p, both concanamycin and inactivating mutations generally lead to more complete inhibition of dissociation than for Vph1p. Even for Stv1p-containing complexes, however, partial dissociation is observed when these complexes are localized to the post-Golgi compartment. These results clearly indicate that catalytic activity is not absolutely required for in vivo dissociation of the V-ATPase complex.
The molecular basis for the dependence of dissociation on cellular environment remains uncertain. It has previously been observed that neutralization of acidic compartments with chloroquine leads to at least partial inhibition of dissociation (33). This suggests that one environmental factor affecting dissociation is the lumenal pH, such that when the lumenal pH becomes too alkaline, dissociation is inhibited. Inconsistent with this conclusion is the observation that Stv1p, when localized to the vacuole, shows very low levels of proton transport, yet still dissociates efficiently in response to glucose depletion. Moreover, completely inactive mutants of Vph1p still show dissociation when localized to compartments other than the vacuole. The absence of quinacrine staining of these compartments in strains expressing mutant forms of Vph1p suggests that they are not being acidified by some other mechanism. Thus, while lumenal pH may partly control dissociation in certain cases, this cannot be the only environmental factor affecting this process. It is likely that specific proteins or lipids localized to different cellular compartments help control in vivo dissociation.
It is important to note that the studies described employ yeast mutant strains that are disrupted in normal intracellular trafficking, and that it is therefore possible that some of the differences observed in dissociation behavior are a secondary consequence of disruption of trafficking, such as the failure of an important regulatory molecule to reach its correct cellular destination. While this possibility cannot be ruled out, it is important to note that the vps21Δ and vps27Δ mutants used in these studies have fewer pleiotropic effects than other yeast trafficking mutants. For example, class C vps mutants, which are defective in proteins involved in multiple membrane fusion events (49,44), are characterized by the absence of identifiable vacuoles. By contrast, vps21Δ and vps27Δ mutants can form and maintain identifiable vacuoles. Moreover, although CPY cannot be targeted to the vacuole in these mutants, processing of CPY in the ER and Golgi is normal (43,34). In addition, targeting of ALP to the vacuole via a pathway distinct from the CPY pathway as well as vacuolar processing of ALP is normal in the vps21Δ and vps27Δ mutants (50,34). These results suggest that it is less likely that the observed differences in dissociation are a secondary consequence of defective trafficking.
An additional question emerging from these results is why the dependence of dissociation on activity is in turn a function of the cellular environment. One possibility is that for dissociation to occur, the V-ATPase must adopt a particular conformational state, and that by inhibiting activity, the enzyme is prevented from achieving the necessary conformational state and is hence blocked in its dissociation. It is possible, however, that as a result of environmental factors within the cell (interaction with unique proteins or lipids localized to particular organelles), the V-ATPase adopts different stable conformations in the inhibited state. Thus the inhibited conformation in the vacuole may be incompatible with dissociation whereas the inhibited state in the PVC may be identical to the state necessary for dissociation to occur. This may also explain why Vph1p and Stv1p show a different dependence of dissociation on activity. Additional probes of the conformational state of the complex are required to address this possibility
In summary, our results suggest that both dissociation of the V-ATPase complex and the dependence of dissociation on activity is a function of the cellular environment and the a subunit isoform present, and that dissociation of the V-ATPase is not absolutely dependent upon catalytic activity.
Acknowledgments
The authors thank Dr. Daniel Jay for the use of his fluorescence microscope, Dr. Takao Inoue for helpful discussions and Drs. Daniel Cipriano, Ayana Hinton and Yanru Wang as well as Sarah Bond, Kevin Jefferies and Kathleen Forgac for careful reading of the manuscript and helpful discussions.
This work was supported by National Institutes of Health Grant GM 34478 (to M.F.). E.coli strains were provided through NIH Grant DK34928.
Footnotes
The abbreviations used are: V-ATPase, vacuolar proton-translocating adenosine 5′-triphosphatase; F-ATPase, F1F0 ATP synthase; VCC, Vph1p-containing complex; SCC, Stv1p-containing complex; PVC, pre-vacuolar compartment; PGC, post-Golgi compartment; PAGE, polyacrylamide gel electrophoresis; MES, 4-morpholine ethanesulfonic acid; C12E9, polyoxyethylene 9-lauryl ether; HA, influenza hemagglutinin; YEPD, yeast extract-peptone-dextrose.
References
- 1.Nishi T, Forgac M. Nat Rev Mol Cell Biol. 2002;3(2):94–103. doi: 10.1038/nrm729. [DOI] [PubMed] [Google Scholar]
- 2.Kane PM. Microbiol Mol Biol Rev. 2006;70:177–191. doi: 10.1128/MMBR.70.1.177-191.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wagner CA, Finberg KE, Breton S, Marshansky V, Brown D, Geibel JP. Physiol Rev. 2004;84:1263–1314. doi: 10.1152/physrev.00045.2003. [DOI] [PubMed] [Google Scholar]
- 4.Nelson N. J Bioenerg Biomembr. 2003;35:281–289. doi: 10.1023/a:1025768529677. [DOI] [PubMed] [Google Scholar]
- 5.Abrami L, Lindsay M, Parton RG, Leppla SH, van der Goot FG. J Cell Biol. 2004;166:645–651. doi: 10.1083/jcb.200312072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toyomura T, Murata Y, Yamamoto A, Oka T, Sun-Wada GH, Wada Y, Futai M. J Biol Chem. 2003;278:22023–22030. doi: 10.1074/jbc.M302436200. [DOI] [PubMed] [Google Scholar]
- 7.Pietrement C, Sun-Wada GH, Silva ND, McKee M, Marshansky V, Brown D, Futai M, Breton S. Biol Reprod. 2006;74:185–194. doi: 10.1095/biolreprod.105.043752. [DOI] [PubMed] [Google Scholar]
- 8.Nanda A, Brumell JH, Nordstrom T, Kjeldsen L, Sengelov H, Borregaard N, Rotstein OD, Grinstein S. J Biol Chem. 1996;271:15963–15970. doi: 10.1074/jbc.271.27.15963. [DOI] [PubMed] [Google Scholar]
- 9.Sennoune SR, Bakunts K, Martinez GM, Chua-Tuan JL, Kebir Y, Attaya MN, Martinez-Zaguilan R. Am J Physiol Cell Physiol. 2004;286(6):C1443–1452. doi: 10.1152/ajpcell.00407.2003. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida M, Muneyuki E, Hisabori T. Nat Rev Mol Cell Biol. 2001;2:669–677. doi: 10.1038/35089509. [DOI] [PubMed] [Google Scholar]
- 11.Iwata M, Imamura H, Stambouli E, Ikeda C, Tamakoshi M, Nagata K, Makyio H, Hankamer B, Barber J, Yoshida M, Yokoyama K, Iwata S. Proc Natl Acad Sci U S A. 2004;101:59–64. doi: 10.1073/pnas.0305165101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawasaki-Nishi S, Nishi T, Forgac M. J Biol Chem. 2003;278:41908–41913. doi: 10.1074/jbc.M308026200. [DOI] [PubMed] [Google Scholar]
- 13.Imamura H, Nakano M, Noji H, Muneyuki E, Ohkuma S, Yoshida M, Yokoyama K. Proc Natl Acad Sci U S A. 2003;100:2312–2315. doi: 10.1073/pnas.0436796100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirata T, Iwamoto-Kihara A, Sun-Wada GH, Okajima T, Wada Y, Futai M. J Biol Chem. 2003;278:23714–23719. doi: 10.1074/jbc.M302756200. [DOI] [PubMed] [Google Scholar]
- 15.Manolson MF, Proteau D, Preston RA, Stenbit A, Roberts BT, Hoyt MA, Preuss D, Mulholland J, Botstein D, Jones EW. J Biol Chem. 1992;267(20):14294–14303. [PubMed] [Google Scholar]
- 16.Manolson MF, Wu B, Proteau D, Taillon BE, Roberts BT, Hoyt MA, Jones EW. J Biol Chem. 1994;269(19):14064–14074. [PubMed] [Google Scholar]
- 17.Kawasaki-Nishi S, Nishi T, Forgac M. J Biol Chem. 2001;276(21):17941–17948. doi: 10.1074/jbc.M010790200. [DOI] [PubMed] [Google Scholar]
- 18.Kawasaki-Nishi S, Bowers K, Nishi T, Forgac M, Stevens TH. J Biol Chem. 2001;276(50):47411–47420. doi: 10.1074/jbc.M108310200. [DOI] [PubMed] [Google Scholar]
- 19.Nishi T, Forgac M. J Biol Chem. 2000;275(10):6824–6830. doi: 10.1074/jbc.275.10.6824. [DOI] [PubMed] [Google Scholar]
- 20.Oka T, Murata Y, Namba M, Yoshimizu T, Toyomura T, Yamamoto A, Sun-Wada GH, Hamasaki N, Wada Y, Futai M. J Biol Chem. 2001;276(43):40050–40054. doi: 10.1074/jbc.M106488200. [DOI] [PubMed] [Google Scholar]
- 21.Frattini A, Orchard PJ, Sobacchi C, Giliani S, Abinun M, Mattsson JP, Keeling DJ, Andersson AK, Wallbrandt P, Zecca L, Notarangelo LD, Vezzoni P, Villa A. Nat Genet. 2000;25(3):343–346. doi: 10.1038/77131. [DOI] [PubMed] [Google Scholar]
- 22.Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, Hulton SA, Sanjad SA, Al-Sabban EA, Lifton RP, Scherer SW, Karet FE. Nat Genet. 2000;26:71–75. doi: 10.1038/79208. [DOI] [PubMed] [Google Scholar]
- 23.Kane PM. J Biol Chem. 1995;270(28):17025–17032. [PubMed] [Google Scholar]
- 24.Sumner JP, Dow JA, Earley FG, Klein U, Jager D, Wieczorek H. J Biol Chem. 1995;270(10):5649–5653. doi: 10.1074/jbc.270.10.5649. [DOI] [PubMed] [Google Scholar]
- 25.Sautin YY, Lu M, Gaugler A, Zhang L, Gluck SL. Mol Cell Biol. 2005;25(2):575–589. doi: 10.1128/MCB.25.2.575-589.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I. Science. 2003;299(5611):1400–1403. doi: 10.1126/science.1080106. [DOI] [PubMed] [Google Scholar]
- 27.Parra KJ, Keenan KL, Kane PM. J Biol Chem. 2000;275(28):21761–21767. doi: 10.1074/jbc.M002305200. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Myers M, Forgac M. J Biol Chem. 1992;267(14):9773–9778. [PubMed] [Google Scholar]
- 29.Parra KJ, Kane PM. Mol Cell Biol. 1998;18(12):7064–7074. doi: 10.1128/mcb.18.12.7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu T, Forgac M. J Biol Chem. 2001;276:24855–24861. doi: 10.1074/jbc.M100637200. [DOI] [PubMed] [Google Scholar]
- 31.Seol JH, Shevchenko A, Shevchenko A, Deshaies RJ. Nat Cell Biol. 2001;3:384–391. doi: 10.1038/35070067. [DOI] [PubMed] [Google Scholar]
- 32.Smardon AM, Tarsio M, Kane PM. J Biol Chem. 2002;277:13831–13839. doi: 10.1074/jbc.M200682200. [DOI] [PubMed] [Google Scholar]
- 33.Shao E, Forgac M. J Biol Chem. 2004;279:48663–48670. doi: 10.1074/jbc.M408278200. [DOI] [PubMed] [Google Scholar]
- 34.Gerrard SR, Bryant NJ, Stevens TH. Mol Biol Cell. 2000;11(2):613–626. doi: 10.1091/mbc.11.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sherman F. Methods Enzymol. 2002;350:3–41. doi: 10.1016/s0076-6879(02)50954-x. [DOI] [PubMed] [Google Scholar]
- 36.Johnston M, Riles L, Hegemann JH. Methods Enzymol. 2002;350:290–315. doi: 10.1016/s0076-6879(02)50970-8. [DOI] [PubMed] [Google Scholar]
- 37.Gietz D, St Jean A, Woods RA, Schiestl RH. Nucleic Acids Res. 1992;20(6):1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawasaki-Nishi S, Nishi T, Forgac M. Proc Natl Acad Sci U S A. 2001;98(22):12397–12402. doi: 10.1073/pnas.221291798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Inoue T, Forgac M. J Biol Chem. 2005;280(30):27896–27903. doi: 10.1074/jbc.M504890200. [DOI] [PubMed] [Google Scholar]
- 40.Conibear E, Stevens TH. Methods Enzymol. 2002;351:408–432. doi: 10.1016/s0076-6879(02)51861-9. [DOI] [PubMed] [Google Scholar]
- 41.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. J Biol Chem. 1951;193(1):265–275. [PubMed] [Google Scholar]
- 42.Feng Y, Forgac M. J Biol Chem. 1992;267(9):5817–5822. [PubMed] [Google Scholar]
- 43.Piper RC, Cooper AA, Yang H, Stevens TH. J Cell Biol. 1995;131(3):603–617. doi: 10.1083/jcb.131.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bowers K, Stevens TH. Biochim Biophys Acta. 2005;1744(3):438–454. doi: 10.1016/j.bbamcr.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 45.Peters C, Bayer MJ, Buhler S, Andersen JS, Mann M, Mayer A. Nature. 2001;409(6820):581–588. doi: 10.1038/35054500. [DOI] [PubMed] [Google Scholar]
- 46.Bayer MJ, Reese C, Buhler S, Peters C, Mayer A. J Cell Biol. 2003;162(2):211–222. doi: 10.1083/jcb.200212004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hiesinger PR, Fayyazuddin A, Mehta SQ, Rosenmund T, Schulze KL, Zhai RG, Verstreken P, Cao Y, Zhou Y, Kunz J, Bellen HJ. Cell. 2005;121(4):607–620. doi: 10.1016/j.cell.2005.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liegeois S, Benedetto A, Garnier JM, Schwab Y, Labouesse M. J Cell Biol. 2006;173(6):949–961. doi: 10.1083/jcb.200511072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peterson MR, Emr SD. Traffic. 2001;2(7):476–486. doi: 10.1034/j.1600-0854.2001.20705.x. [DOI] [PubMed] [Google Scholar]
- 50.Piper RC, Bryant NJ, Stevens TH. J Cell Biol. 1997;138(3):531–545. doi: 10.1083/jcb.138.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]