

Summary
Osteoporosis and the associated risk of fracture are major clinical challenges in the elderly. Telomeres shorten with age in most human tissues, including bone, and because telomere shortening is a cause of cellular replicative senescence or apoptosis in cultured cells, including mesenchymal stem cells (MSCs) and osteoblasts, it is hypothesized that telomere shortening contributes to the aging of bone. Osteoporosis is common in the Werner (Wrn) and dyskeratosis congenita premature aging syndromes, which are characterized by telomere dysfunction. One of the targets of the Wrn helicase is telomeric DNA, but the long telomeres and abundant telomerase in mice minimize the need for Wrn at telomeres, and thus Wrn knockout mice are relatively healthy. In a model of accelerated aging that combines the Wrn mutation with the shortened telomeres of telomerase (Terc) knockout mice, synthetic defects in proliferative tissues result. Here, we demonstrate that deficiencies in Wrn−/− Terc−/− mutant mice cause a low bone mass phenotype, and that age-related osteoporosis is the result of impaired osteoblast differentiation in the context of intact osteoclast differentiation. Further, MSCs from single and Wrn−/− Terc−/− double mutant mice have a reduced in vitro lifespan and display impaired osteogenic potential concomitant with characteristics of premature senescence. These data provide evidence that replicative aging of osteoblast precursors is an important mechanism of senile osteoporosis.
Keywords: aging, dyskeratosis congenita, osteoblast differentiation, osteoporosis, telomerase, Werner syndrome
Introduction
Clues to human aging mechanisms have come from segmental progeroid disorders, genetic diseases that hasten certain features of aging. Among these are dyskeratosis congenita (DC) and Werner syndrome (WS). Although the phenotypic spectra of the two diseases overlap only partially, with DC primarily affecting rapidly proliferating tissues [bone marrow (BM), epidermis] and WS primarily affecting slowly dividing mesenchymal tissues (dermis, fat, vasculature, bone), both diseases are characterized by premature osteoporosis. In DC, the osteoporosis is an accelerated form of that seen with natural aging, while the osteoporosis of WS has an unusual distribution, affecting the limbs more than the axial skeleton (Hofer et al., 2005; Mason et al., 2005). The two most common forms of DC, the X-linked and autosomal dominant forms, are caused by mutations in DKC1 and TERC (the RNA template component of telomerase), respectively. Each of these mutations reduces telomerase activity, causing individuals with DC to have prematurely shortened telomeres, and thus making it highly likely that defects in telomere and/or telomerase status contribute to the pathology of DC (Mason et al., 2005). Most cases of WS are caused by loss-of-function mutations in Wrn, which encodes a DNA helicase of the RecQ family that functions in the recombinational repair of stalled replication forks or double-strand breaks (Ozgenc & Loeb, 2005). It is also apparent that WRN plays important roles in telomere maintenance (Wyllie et al., 2000; Johnson et al., 2001; Opresko et al., 2002; Crabbe et al., 2004) and might be particularly important under conditions, such as cellular aging, where telomeres become dysfunctional.
In mice lacking telomerase and with shortened telomeres, Wrn mutation results in an acceleration of defects seen in Terc mutants (in rapidly dividing tissues like skin, testes, gastrointestinal tract) and also the appearance of pathology typical of WS (e.g. osteoporosis, insulin resistance, and cataracts) (Chang et al., 2004; Du et al., 2004).
It is generally recognized that senile osteoporosis is characterized by a decrease in bone-forming capacity mediated by defects in the number and function of osteoblasts, observations that implicate the senescence of osteoblasts and their precursors as a possible causative mechanism (Majors et al. 1997). In this respect, it seemed possible that the Wrn−/− Terc−/− mutant mouse model may recapitulate the mechanism of senile bone loss, with dysfunctional telomeres as the basis for putative proliferative defects in bone-forming cells and their precursors. Here, we show that deficiencies in Wrn−/− Terc−/− mutant mice cause a low bone mass phenotype and that age-related osteoporosis is indeed the result of impaired osteoblast differentiation associated with accelerated cell senescence of mesenchymal stem cells.
Results
Deficiencies in telomere maintenance molecules cause osteoporosis
Because laboratory strains of mice have telomeres that are approximately five times longer than human telomeres, mice must lack telomerase for several generations (3–6, depending on genetic background) before telomeres become critically short and phenotypic defects can be easily detected (Herrera et al., 1999; Rudolph et al., 1999). Individual or combined mutations in Wrn and the related Blm RecQ helicase-encoding genes can accelerate and accentuate the appearance of age-related phenotypes caused by mutation in Terc (Chang et al., 2004; Du et al., 2004). Our original experiments involved mutations in both Wrn and Blm (using a Wrn null allele and the BlmM3 hypomorphic allele) (McDaniel et al., 2003) in an effort to uncover possible functional redundancy between the helicases.
We have previously reported that among third generation (G3) young (4.5-month-old) animals, only triple mutant Wrn−/−Blm−/− Terc−/− mice show an abnormal bone phenotype consisting of kyphosis and reduced bone mineral density (BMD) (Du et al., 2004). However, in older G3 mice (mean age: 14.7 months), osteoporosis in the spine is also evident in the Wrn−/− Terc−/− double mutants (Fig. 1a,b). Among G3 mice, 70% of wild-type (WT) animals had BMDs by small animal DXA corresponding to greater than or equal to the 50th percentile of BMDs of animals from both genotypes. In comparison, only 17% of the Wrn−/−Terc−/− double mutants had BMDs greater than or equal to the 50th percentile (Fig. 1a). Spine BMD in the Wrn−/− Terc−/− mutants was about 19% lower relative to WT animals (Fig. 1b). Whole body BMD in the Wrn−/− Terc−/− mutant showed a statistically nonsignificant decrease of about 1.2% compared to WT (P = 0.4092). There was also a statistically nonsignificant decrease in body mass index in the mutant group (P = 0.1023).
Fig. 1.
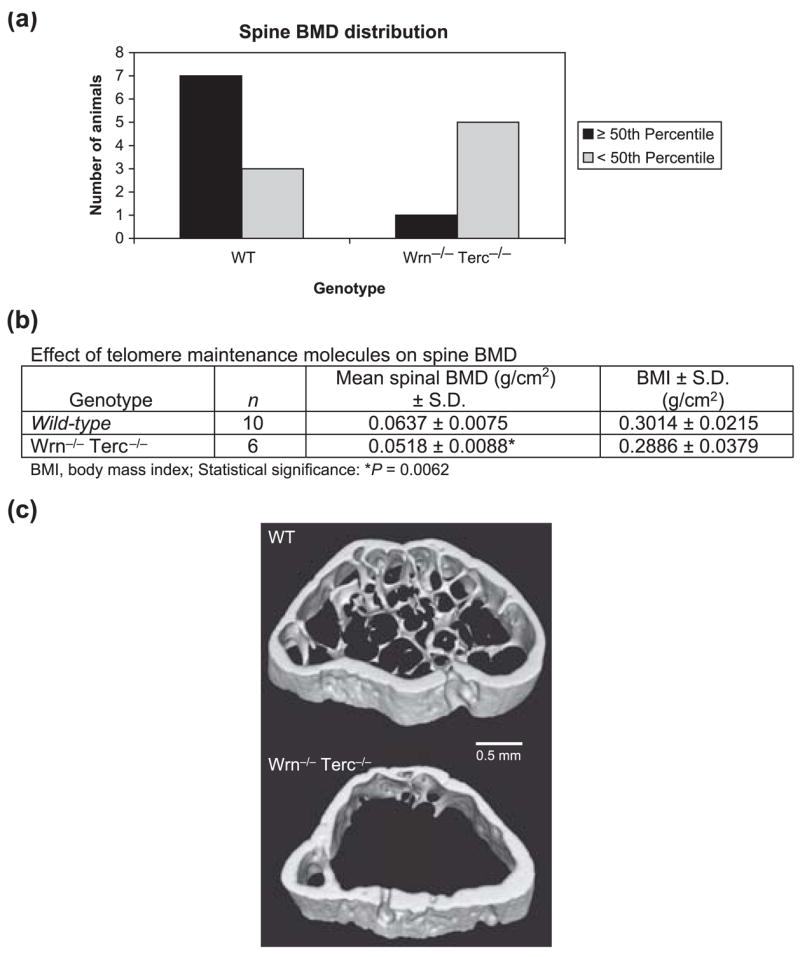
Deficiencies in telomere maintenance molecules cause osteoporosis. (a) Spinal bone mineral density (BMD) distribution by small animal DXA is shown for wild-type (WT) mice and for those with deficiencies in Wrn and Terc (Wrn−/− Terc−/−). (b) Table comparing mean spinal BMD among WT and mutant mice. Average age among WT and G3 Wrn−/− Terc−/− mutants was 14.7 months. (c) Volume-rendered cross-sectional slabs from the distal metaphyses of two mouse femurs, revealing dramatic differences in trabecular architecture between a WT (top) and a Wrn−/− Terc−/− mutant (bottom) mouse. Quantitative analysis of these slabs shows BMD differences are trabecular rather than cortical in nature (see text).
Detailed quantitative analysis of micro-CT data from a small subset of mice (two WT, two Wrn−/−Terc−/−) was performed in a narrow region of the distal femoral metaphysis (Fig. 1c). Trabecular BMD (Tb.BMD) in the distal femur was found to be significantly lower (P = 0.026) in Wrn−/−Terc−/− distal femurs (76 ± 7 mg cc−1) than in WT (185 ± 37 mg cc−1). However, cortical BMDs (C.BMDs) for Wrn−/− Terc−/− (1217 ± 48 mg cc−1) and WT (1203 ± 30 mg cc−1) distal femurs were not significantly different (P = 0.38). These analyses suggest that trabecular bone loss is a major source of differences in areal BMD in Wrn−/− Terc−/− mutants compared to WT.
Age-related osteoporosis is the result of impaired osteoblast differentiation
We hypothesized that declines in BMD seen in mice lacking Terc and Wrn were related to impairment of osteoprogenitor or BM stromal cell (BMSC) differentiation into osteoblasts. We found that measures of osteoblast differentiation, such as alkaline phosphatase (Alk P) activity and mineralization, are less robust in BMSC derived from 10-month-old G3 single mutant Wrn−/−mice, and barely detectable in G3 single mutant Terc−/− and double mutant mice (Fig. 2a). As a percentage of total colonies, BMSCs from any mutant genotype never formed more than 4.4% alizarin red positive colonies (P < 0.0001) or more than 25.9% Alk P positive colonies (P < 0.0001) (Fig. 2b). Wild-type BMSCs formed about 88% and 89% positive colonies, respectively (Fig. 2b). In each case, the Wrn−/− mutant scored the highest percentage of positive colonies among all the mutant genotypes.
Fig. 2.
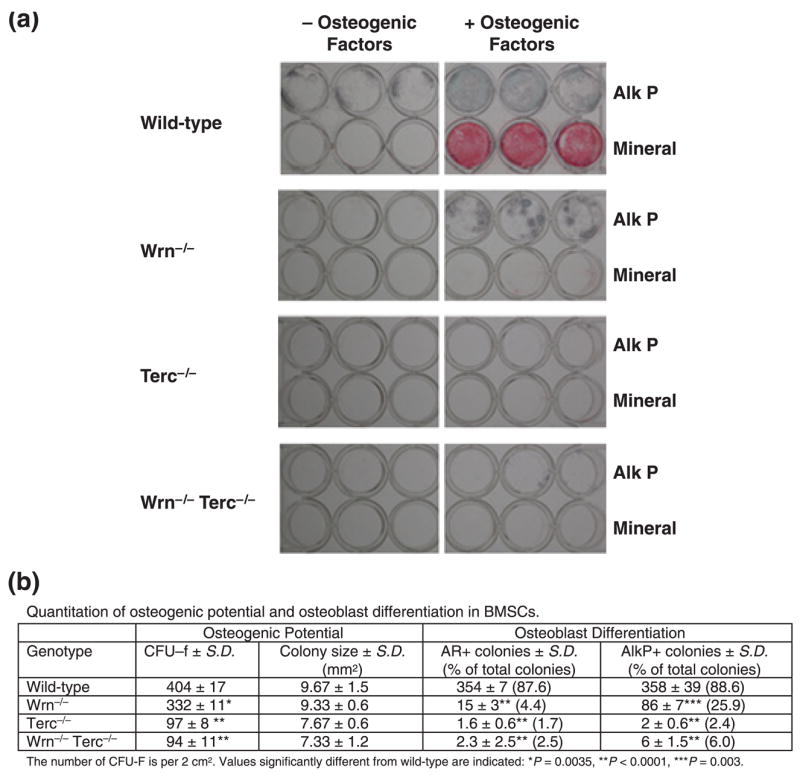
Osteoblast differentiation is impaired in mouse bone marrow stromal cells (BMSCs) derived from G3 single (Wrn−/− or Terc−/−) and double (Wrn−/−/Terc−/−) mutants. (a) Detection of alkaline phosphatase (Alk P) activity and mineralization (Mineral) is shown for BMSCs isolated from 10-month-old animals. (b) Quantitation of osteogenic potential and osteoblast differentiation in BMSCs. Shown are the number of colony-forming units–fibroblast (CFU–F), colony size, alizarin red S positive colonies (AR+), and alkaline phosphatase positive (AlkP+) colonies for the indicated genotypes.
Using the relative percentages of colony-forming units–fibroblast (CFUs–F) as an indicator of numbers of mesenchymal stem cells (MSCs), and the percentages of positive colonies as an indicator of osteoblast differentiation, defects in osteogenic potential as well as differentiation, respectively, occur among all the mutant phenotypes (Fig. 2b). The Terc−/− and Wrn−/−Terc−/− mutants, however, appear to have greater detriments in both MSCs and the capacity for MSCs to differentiate into osteoblasts. There is also a statistically nonsignificant trend toward smaller average colony sizes among the mutant mice (Fig. 2b).
Other markers of osteoblast differentiation, including osteopontin and osteocalcin, were also analyzed in WT and mutant mice with similar findings. Figure 3 shows the reduced expression of osteopontin in BMSCs derived from Wrn−/− mice and the relative absence of osteopontin in Terc−/− mice as well as in the double mutant mice. Expression of osteocalcin followed a similar pattern among the mutant genotypes (see Supplementary Fig. S1). It is of interest that young mutant animals (4.5 months) with normal BMD also demonstrated impairment in osteoblast differentiation (see Supplementary Fig. S2), suggesting that the defects in osteogenic precursor cells imparted by deficiencies in genomic maintenance precede any demonstrable change in bone phenotype. It is possible that these changes in osteoblast differentiation in vitro are unmasked by the relatively more harsh culture conditions compared to the in vivo environment. Furthermore, at this earlier time, osteoblast differentiation in vitro is more severely compromised in the double mutant compared to either single mutant, demonstrating the individual contributions of Wrn and Terc to the impaired differentiation in the double mutant.
Fig. 3.
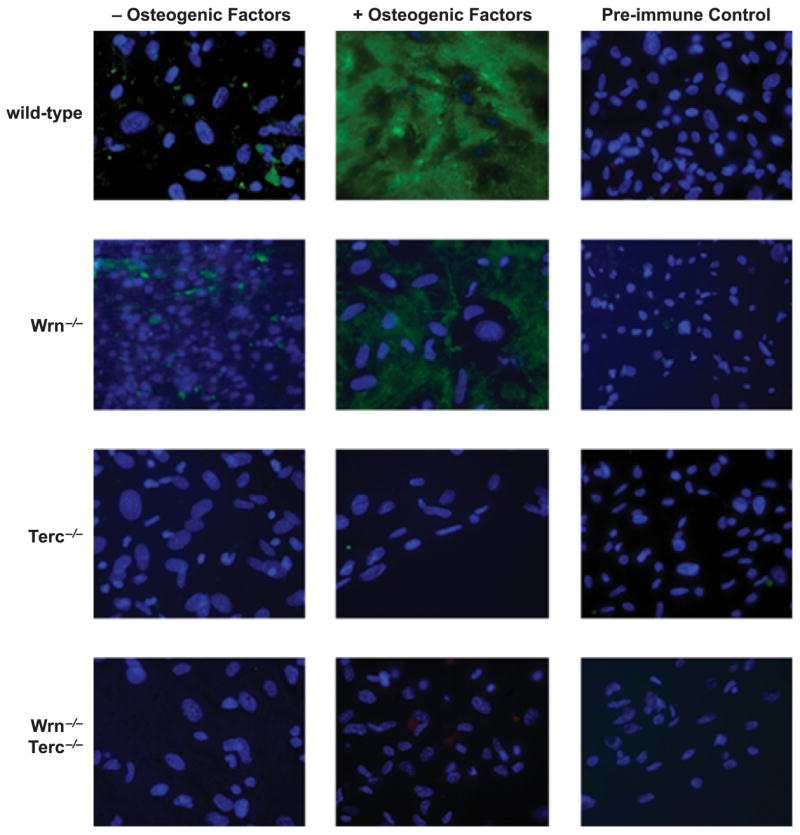
Osteopontin expression in mouse bone marrow stromal cells (BMSCs) derived from single (Wrn−/− or Terc−/−) and double (Wrn−/−/Terc−/−) mutants by immunofluorescent staining. The percentages of positive cells are as follows: 98%, wild type (WT); 42%, Wrn−/−; 0.3%, Terc−/−; 0.2% Wrn−/− Terc−/−. BMSCs were derived from 10-month-old animals.
In contrast to the defects in osteoblast differentiation, we have found no differences in the ability of any mutant mice to undergo osteoclast differentiation (Fig. 4a,b), and so the lower BMD seen in older mutant mice is likely the result of bone resorption uncoupled to formation. In this respect, the Wrn−/−Terc−/− model recapitulates the gross mechanism of bone loss seen with aging in humans.
Fig. 4.
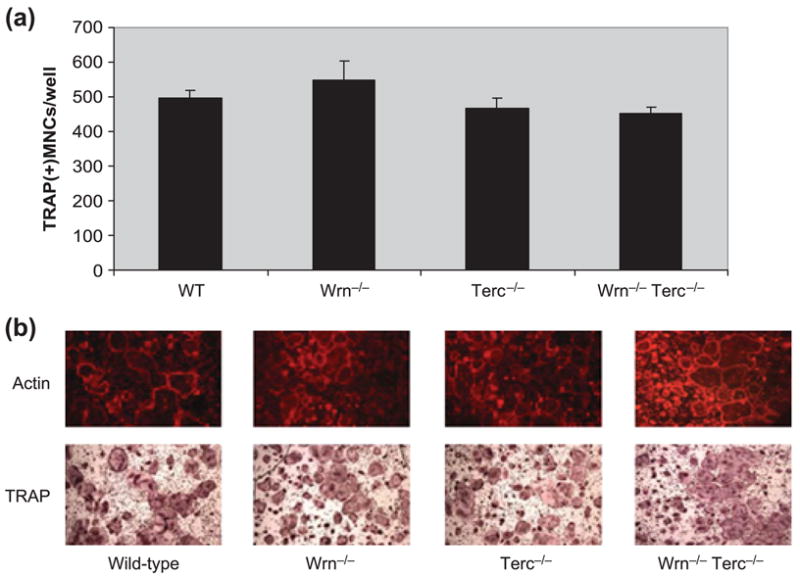
Osteoclast differentiation is intact in mouse bone marrow-derived macrophages derived from G3 single (Wrn−/− or Terc−/−) and double (Wrn−/−/Terc−/−) mutants. (a) Quantitation of tartrate-resistant acid phosphatase (TRAP) positive multinuclear cells (MNCs) is shown. (b) Representative examples of actin and TRAP-positive osteoclasts derived from bone marrow cells of the indicated genotype are shown. Mice were 15–18 months of age. WT, wild type.
Accelerated replicative senescence occurs concomitantly with impaired osteoblast differentiation
Telomere shortening is a cause of cellular replicative senescence in osteoblasts and in MSCs (Yudoh et al., 2001; Simonsen et al., 2002). Ectopic expression of telomerase in MSCs extends proliferative capacity in vitro, enhances osteoblast differentiation, and increases the capacity for bone formation in vivo (Yudoh et al., 2001; Simonsen et al., 2002). Based on these observations, we evaluated whether accelerated replicative senescence in Terc−/− BMSCs occurred concomitantly with the loss of markers for osteoblast differentiation, as a basis for providing strong evidence that telomerase and/or telomere status in MSCs is likely a critical component of bone formation. Relative to WT animals, BMSCs isolated from Terc−/− mice, Wrn−/− mice, and Terc−/−Wrn−/− mice display a reduced capacity to divide in culture (Fig. 5). The approximately 20% difference in cumulative population doublings (PDs) is statistically significant (P < 0.0001).
Fig. 5.
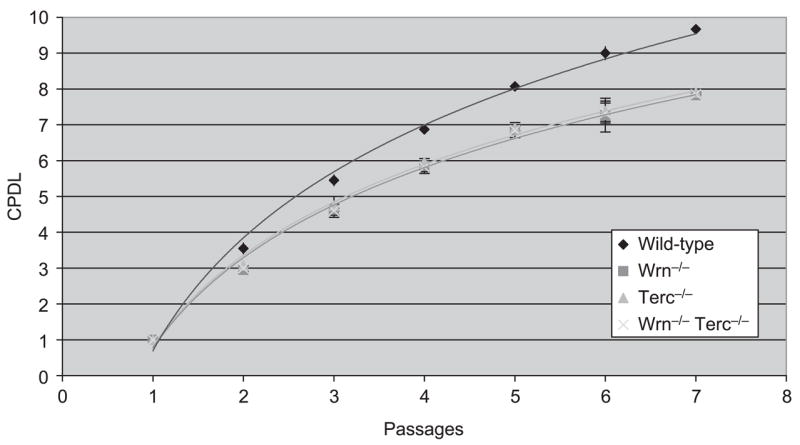
Accelerated cellular replicative senescence in bone marrow stromal cells (BMSCs) lacking Terc and Wrn. BMSCs cultured from two G3 mutants per genotype were serially propagated, and the cumulative population doubling level (CPDL) was measured at each passage. Mean values and standard errors are indicated. Curves represent the best-fit logarithmic plots.
Measures of osteoblast differentiation described earlier were evaluated at the second or third in vitro passage, when mutant BMSCs consistently displayed a 15–20% decrease in PDs with each subcultivation relative to WT BMSCs. Thus, BMSCs from single and Wrn−/− Terc−/− double mutant mice have a reduced in vitro lifespan and display impaired osteogenic potential concomitant with characteristics of premature senescence.
Discussion
A fundamental aspect of age-related bone loss is the uncoupling of bone formation and resorption. Decreased proliferative capacity and impaired differentiation of osteoblasts in the bone of older individuals likely contribute to this process (Kassem et al., 1997; Martinez et al., 1999, 2001; Chan & Duque, 2002). Furthermore, deficits in osteoblast function are accompanied by at least a relative increase in osteoclast activity (Chan & Duque, 2002).
There are conflicting reports regarding the effects of age on human MSCs with the weight of evidence in favor of modest declines in several measures associated with osteogenic potential, particularly after the age of 40 (Long et al., 1999; Nishida et al., 1999; Muschler et al., 2001; Stenderup et al., 2001). Mice, like humans, demonstrate osteopenic changes in bone with advanced age (Silberberg & Silberberg, 1962). The osteogenic potential of murine MSCs also declines with increasing donor age (Bergman et al., 1996).
Age-related telomere dysfunction is one potential cause of impaired osteoblast differentiation and may thus contribute to osteoporosis. Although there is insufficient evidence to be certain of the causal roles that telomere dysfunction may play in human aging, there are several indications of its importance (Cawthon et al., 2003; Hofer et al., 2005; Herbig et al., 2006; Minamino & Komuro, 2007). With regard to the aging of bone, evidence that telomere dysfunction plays a significant role includes observations that over-expression of telomerase in human MSCs extends proliferative capacity in vitro and enhances bone formation when these MSCs are transplanted into mice (Yudoh et al., 2001; Shi et al., 2002; Simonsen et al., 2002). Telomerase not only extends the lifespan of osteogenic precursors, but accelerates the osteogenic differentiation of MSCs (Gronthos et al., 2003).
Our data strongly suggest that deficiencies in genome maintenance molecules, such as TERC and WRN, are related to a low bone mass phenotype as the result of impairment in osteogenic potential (decreased MSCs) and osteoblast differentiation (decreased expression of osteoblast markers). We also show that BMSCs/MSCs derived from Wrn−/−, Terc−/−, and Wrn−/−Terc−/− mutant mice have a reduced in vitro lifespan concomitant with impaired osteogenic potential and osteoblast differentiation. While it has been reported that telomerase can increase the in vitro lifespan and differentiation capacity of BM MSCs, as mentioned earlier, here we show that the converse is also true. The absence of WRN, either alone or in combination with TERC deficiency, also confers a shortened in vitro lifespan, consistent with the finding that serially passaged human WS fibroblasts senesce prematurely (Salk et al., 1981). These cellular mechanisms are likely to be responsible for age-related bone loss in DC and WS, and depending on the extent of telomere dysfunction, may also be important in normally aging bone.
The relationship between the capacity for osteoblast differentiation and telomere maintenance/senescence may be complex. Bergman et al. (1996) demonstrated that osteogenic potential in WT BALB/c murine BMSCs declines with increasing donor age and in the absence of Wrn/Terc deletion. With normal aging, murine BMSCs decline in number and proliferative potential, suggesting that BMSCs have reduced telomerase activity and perhaps dysfunctional telomeres. In the Wrn−/− Terc−/−mutant, telomere dysfunction occurs, but at an accelerated rate. While BMSCs decrease in number and replicative capacity with both normal aging and with accelerated aging in Wrn−/− Terc−/− mice, the latter appears also to have impairment in osteoblast differentiation. It is possible that normal mice which are exceptionally aged will also have impaired differentiation of bone-forming cells similar to what is seen in the double mutant.
Neurofibromin 1 (NF1) haploinsufficient (+/−) mice have mesenchymal progenitor cells that demonstrate increased telomerase expression, reduced senescence in culture, and increased CFU–F capacity, but unexpectedly, decreased osteoblast differentiation capacity (Wu et al., 2006). It is possible that the NF1 tumor suppressor gene affects mesenchymal lineage determination independent of telomerase regulation. Although Wu et al. (2006) found no effect of NF1 haploinsufficiency on chondrocyte differentiation, it would be important to know whether adipocyte differentiation is altered, especially given that adipocytes and osteoblasts may share a common bipotential precursor. It is also possible that telomerase expression is needed early in osteogenesis, but its down-regulation is later required for terminal osteoblast differentiation. Alternatively, the high telomerase activity in the NF1+/− mutants may simply be a marker that osteoblast differentiation does not occur efficiently (i.e. telomerase activity may naturally decline with osteoblast differentiation, and because the mutant mesenchymal precursors do not differentiate well into osteoblasts, they maintain the earlier telomerase profile).
Interestingly, there are no obvious differences in replicative lifespan among the BMSCs isolated from any of the mutant genotypes. This may be the result of pooling BMSCs from three mice of the same genotype, thus minimizing the effect of short-lived cell strains from a single animal and always selecting for the longest-lived strain from a different mouse. Alternatively, it might reflect differential sensitivity of the various mutant strains to the cell culture environment (e.g. an unphysiologically high oxygen concentration), and as such may be an indication of their relative propensities to senescence in vivo under conditions of stress.
Osteoblast differentiation is impaired in mutant BMSCs concomitant with a decline in proliferation of approximately 20%. This suggests that only relatively modest declines in replicative capacity are needed to impair this differentiation, or that limited telomere-based dysfunction, which precedes replicative senescence, is sufficient to reduce osteoblast formation. In the case of the latter, decreased replicative capacity would then be a surrogate marker for the more physiologically relevant telomere dysfunction. Either or both mechanisms may apply and, regardless, the decline in mutant CFUs–F that undergo osteoblast differentiation suggests that continued telomere dysfunction and/or subsequent replicative senescence impair the osteoblast phenotype.
Replicative lifespan in vitro may be different from that in vivo, and so cumulative PDs should only be considered indirect evidence for in vivo cell senescence; thus, the physiological significance of accelerated senescence in mutant BMSCs is complicated by interpretation of both the degree of replicative aging in vitro and whether it is directly correlative to in vivo findings which may manifest as pathological changes with age, in this case osteoporotic changes. Despite these caveats, the percent decrease in cumulative PDs of the Wrn−/− Terc−/− mutant BMSCs is on the same order of the BMD loss in the spine of double mutant mice. Further, BMSC differentiation may be more sensitive to defects caused by loss of WRN and TERC than is replicative lifespan.
There appears to be a greater difference in the in vitro replicative capacity between WT and Wrn−/− Terc−/− fibroblast strains compared to BMSC strains. These differences are most likely explained by differences in generation after telomere loss. For example, Chang et al. (2004) saw almost no reduction in mouse embryonic fibroblast (MEF) replicative lifespan in G3 Wrn−/− Terc−/−mutants. However, they did indeed see a dramatic reduction in proliferative capacity in G5 MEFs. In our study, we used G3 mutants, and thus it is not so surprising that the difference in replicative lifespans was minimized. Another possibility is that differences in cell type may contribute to the effect of WRN and TERC dysfunction on replicative aging.
Markers of bone formation are reduced or absent in bone-marrow-derived BMSCs/MSCs from Wrn−/−, Terc−/−, and Wrn−/−Terc−/− mutant mice, while osteoclast differentiation is unchanged. These data support the association between age-related bone loss and stem cell defects. These defects are likely to be proliferative, suggesting that replicative senescence is perhaps a causative etiology for limiting stem cell pools and resulting in impairment of osteogenic differentiation. Telomere shortening and/or telomere uncapping may then account for both limited proliferative capacity and impaired osteoblast function. Evidence that adequate replicative capacity is required for osteoblast differentiation also implies that proliferation and differentiation are not mutually exclusive.
In summary, we show that genome maintenance molecules play functional roles in bone homeostasis, and that defects in these molecules highlight the importance of osteoprogenitor cells in preventing or delaying osteoporosis.
Experimental procedures
Animals
The University of Pennsylvania Institutional Animal Care and Use Committee approved the experimental use of WT and mutant mice described in this paper. Mice were generated as described previously (Du et al., 2004). Unless otherwise stated, all animals used in experiments were from G3 lineages.
Dissection and processing of femurs
After mice were euthanized by CO2 asphyxiation, and skin was removed along the back between the axilla and hind legs, femurs were carefully removed by cutting through the patellar tendon with a #15 scalpel blade at their distal ends and with small curved scissors where they articulate with the hip joint. Soft tissue was removed along the femoral shaft before isolation of BM cells.
Isolation of BMSCs
Femurs were dissected as described earlier, metaphyses removed, and BM plugs expelled by insertion of a 21-gauge needle into the marrow cavity and flushing the cavity with 10 mL of α-minimal essential medium (α-MEM) with nucleosides (Gibco/Invitrogen, Carlsbad, CA, USA). The BM cells (marrow plugs) from animals of the same genotype were collected, pooled, and dispersed by repeat pipetting before centrifugation at 1000 × g for 10 min. The supernatant was removed and the pellet resuspended in α-MEM containing 10% fetal calf serum (FCS) (HyClone, Logan, UT, USA). Cells were plated at a density of four marrow plugs per 25 cm2 of tissue culture growth surface and refed every 3 days until confluent. For subsequent experiments, cells were used at the first or second in vitro passage, after no more than about three PDs.
Cell culture and determination of replicative lifespan
Bone marrow stromal cells were cultured in α-MEM with nucleosides plus 10% FCS. For lifespan studies, cells were seeded at a density of 1 × 104 cells cm−2 at each passage and grown until confluent, usually by 10 days. Cultures were defined as being at the end of their proliferative lifespan when they were unable to complete one PD during a 4-week period that included three consecutive weeks of refeeding with fresh medium containing 10% FCS.
Osteoblast differentiation
For experiments evaluating osteogenic potential, BMSC cultures were seeded at 2 × 104 cell cm−2 and refed three times weekly with α-MEM + 15% FCS ± 50 μg mL−1 ascorbic acid (Sigma Chemical Co., St. Louis, MO, USA), 10 mM β-glycerophosphate (Sigma Chemical Co.), and 300 ng mL−1 bone morphogenetic protein 2 (Genetics Institute/Wyeth Pharmaceuticals, Madison, NJ, USA). The medium was replaced at regular intervals as preceding until mineralization was detected, usually within 2–4 weeks. All cultures were derived from BMSCs pooled from the femurs of three mice of the same genotype. All experiments were performed in duplicate. CFU–F were quantitated by light microscopy as the number of colonies containing at least 10 cells at the same time in vitro when WT cultures demonstrated evidence of mineralization. Colony size was measured using Image J software (Rasband, W.S., US National Institutes of Health, Bethesda, MD, USA; http://rsb.info.nih.gov/ij/, 1997–2006), and expressed as average colony size in mm2.
Osteoclast differentiation and tartrate-resistant acid phosphatase (TRAP) histochemistry
Bone marrow cells were isolated from femurs as described earlier. To prepare BM-derived macrophages, 1 × 107 BM cells were cultured with α-MEM/10% FCS with M-CSF (30 ng mL−1) using Corning 100 mm non-tissue culture-treated dishes (Corning Incorporated Life Science, Acton, MA, USA). After 3 days of culture, floating cells were removed and attached cells used as osteoclast precursors. To generate osteoclasts, precursors were cultured with TRANCE (300 ng mL−1) and M-CSF (30 ng mL−1) in 96-well culture plates (Corning; 2 × 104 cells/0.2 mL per well). After 3 days of culture, the medium was changed with fresh α-MEM/10% FCS containing TRANCE (300 ng mL−1) and M-CSF (30 ng mL−1), and cultured for 1 day more. Cells were stained for F-actin with rhodamine phalloidin (Molecular Probes, Eugene, OR, USA) followed by TRAP staining. After cells were washed in phosphate-buffered saline (PBS), and fixed in 10% formalin, they were stained for acid phosphatase in the presence of 0.05 M sodium tartrate (Sigma Chemical Co.). The substrate used was naphthol AS-MX phosphate (Sigma Chemical Co.). TRAP(+) cells containing more than three nuclei were considered to be osteoclasts.
Immunoflourescence
Adherent cells grown on glass coverslips or in chamber slides to less than 70% confluence were washed twice with 1× PBS and fixed with 0.2% paraformaldehyde for 10 min. After rinsing with PBS, the cells were placed in blocking solution containing 1% bovine serum albumin in PBS for 30 min. Primary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were diluted 1:200 to 1:500, depending on the manufacturer’s recommendation and experience from previous use, and incubated with cells in a humidified container at 4 °C for 18 h with protection from light. Unbound primary antibody was washed out with three rinses of PBS before incubation of cells with 4′,6-diamidino-2-phenylindole (DAPI) alone (to label nuclei) or, if the primary antibody lacked a fluorescent tag, DAPI and a fluorescently tagged secondary antibody (Invitrogen/Molecular Probes, Carlsbad, CA). DAPI labeling, with or without a tagged secondary antibody, was performed in the dark for 2 h. After rinsing cells three times with PBS, coverslips or chamber slides were air-dried before mounting onto slides or removing the chambers, respectively. Unless otherwise stated, all steps were performed at room temperature. Pre-immune serum was used as a negative control, at a dilution that normalized the concentration of IgG to that of the primary antibody. DAPI was used at a dilution of 1:1000 to 1:2000. All dilutions were made in blocking solution. Slides were stored at 4 °C in the dark. Cells were visualized at 200× using a Leica DMR microscope equipped with epi-fluorescence and Leica HC PL Fluotar objectives (Leica, Bannockburn, IL, USA), and images were obtained with a Hamamatsu C4742-95 high-resolution digital camera (Hamamatsu, Bridgewater, NJ, USA) and Openlab image acquisition software (Improvision, Lexington, MA, USA).
Images were stored in Openlab format and converted to TIFF format before transferring to Adobe Photoshop. The exposure time was 100 ms. Markers were quantitated as the percentage of positive cells, after counting a minimum of 200 cells per replicate.
Histology
A specific Alk P substrate 5-bromo-4-chloro-3-indolyl-phosphate (Moss, Inc., Pasadena, MD, USA) in the presence of p-nitro blue tetrazolium chloride was used to detect Alk P activity (cells are stained blue/purple). Alizarin red S staining was used to detect a mineralized matrix. Positive colonies were counted under light microscopy, and only those colonies with at least 10 cells were scored.
Small animal DXA scan
DXA scans were performed with a Lunar PIXImus 2, calibrated using a phantom model with 0.058 g cm−2 BMD.
Micro-CT analysis
The micro-CT system is an explore Locus-SP micro-CT specimen scanner (GE Healthcare, Waukesha, WI, USA). Three-dimensional (3-D) volumes with 16.4 μm isotropic voxels were reconstructed from the raw projection data of distal femurs using the vendor’s software (MicroView, GE Healthcare). A calibration phantom provided proper scaling into Hounsfield units and the means to quantify BMD (BMD, mg cc−1). The distal metaphyses of four femurs (two WT, two Wrn−/−Terc−/−) were analyzed at the same level by first reorienting each volume to align with the principal axes of each femur. To separate trabecular bone from cortical bone, a spline-interpolated 3-D ROI then was manually drawn circumscribed within the cortical bone, starting from the plane just proximal to the proximal tip of the femoral chondyles and ending 0.5 mm (31 slices) proximally from that plane. This produced a cross-sectional slab 0.5 mm thick which could be volume-rendered. Because the image-intensity histogram is bimodal, auto-thresholding provided adequate bone/background segmentation, both within the 3-D ROI for Tb.BMD and outside the 3-D ROI for C.BMD. BMD was calculated as the sum of scaled voxel intensities above the threshold, using the vendor’s software (MicroView, GE Healthcare).
Statistics
The t-test (Student’s t-test; one-sided and unpaired) was used to determine whether the mean value for bone density (continuous variable) in the Wrn−/− Terc−/− group differed significantly from that in the WT group. Statistical significance was set to P = 0.05. In the case of multiple comparisons to the WT group, one-sided unpaired t-tests were used with statistical significance set to P = 0.008 (Bonferroni adjustment). All statistics were performed by Graphpad software (www.graphpad.com).
Supplementary Material
The following supplementary material is available for this article:
Fig. S1 Osteocalcin expression in mouse bone marrow stromal cells (BMSCs) derived from single (Wrn−/− or Terc−/−) and double (Wrn−/−/Terc−/−) mutants. Immunofluorescent staining of osteocalcin is shown in BMSCs derived from 10-month-old mice of the indicated genotype. The percentages of positive cells are as follows: 87%, wild type; 16%, Wrn−/−; 0.2%, Terc−/−; 0.2% Wrn−/− Terc−/−.
Fig. S2 Osteoblast differentiation of mouse bone marrow stromal cells (BMSCs) derived from young G3 single (Wrn−/− or Terc−/−) and double (Wrn−/−/Terc−/−) mutants is impaired. Detection of alkaline phosphatase (Alk P) activity and mineralization (Mineral) is shown for BMSCs isolated from 4.5-month-old animals. The percentages of Alk P positive colonies are as follows: 82%, wild type (WT); 64%, Wrn−/−; 35%, Terc−/−; 2.4%, Wrn−/− Terc−/−. The percentages of alizarin red positive colonies are: 86%, WT; 16%, Wrn−/−; 0.2%, Terc−/−; 0.2%, Wrn−/− Terc−/−.
Acknowledgments
This work was funded by grants from The John A. Hartford Foundation/American Federation for Aging Research Academic Fellowship Award, the University of Pennsylvania Institute on Aging, the Penn Center for Musculoskeletal Disorders, and the National Institutes of Health/National Institute on Aging to R.J.P., and a Paul Beeson Scholars in Aging award to F.B.J. We would like to thank Stephanie Goldman for her technical assistance.
Footnotes
This material is available as part of the online article from:
http://www.Blackwellsynergy.com/doi/abs/10.1111/j.1474-9726.2007.00350.x
(This link will take you to the article abstract).
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Bergman RJ, Gazit D, Kahn AJ, Gruber H, McDougall S, Hahn TJ. Age-related changes in osteogenic stem cells in mice. J Bone Miner Res. 1996;11:568–577. doi: 10.1002/jbmr.5650110504. [DOI] [PubMed] [Google Scholar]
- Cawthon RM, Smith KR, O’Brien E, Sivatchenko A, Kerber RA. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet. 2003;361:393–395. doi: 10.1016/S0140-6736(03)12384-7. [DOI] [PubMed] [Google Scholar]
- Chan GK, Duque G. Age-related bone loss: old bone, new facts. Gerontology. 2002;48:62–71. doi: 10.1159/000048929. [DOI] [PubMed] [Google Scholar]
- Chang S, Multani AS, Cabrera NG, Naylor ML, Laud P, Lombard D, Pathak S, Guarente L, DePinho RA. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet. 2004;36:877–882. doi: 10.1038/ng1389. [DOI] [PubMed] [Google Scholar]
- Crabbe L, Verdun RE, Haggblom CI, Karlseder J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004;306:1951–1953. doi: 10.1126/science.1103619. [DOI] [PubMed] [Google Scholar]
- Du X, Shen J, Kugan N, Furth EE, Lombard DB, Cheung C, Pak S, Luo G, Pignolo RJ, DePinho RA, Guarente L, Johnson FB. Telomere shortening exposes functions for the mouse Werner and Bloom syndrome genes. Mol Cell Biol. 2004;24:8437–8446. doi: 10.1128/MCB.24.19.8437-8446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronthos S, Chen S, Wang CY, Robey PG, Shi S. Telomerase accelerates osteogenesis of bone marrow stromal stem cells by upregulation of CBFA1, osterix, and osteocalcin. J Bone Miner Res. 2003;18:716–722. doi: 10.1359/jbmr.2003.18.4.716. [DOI] [PubMed] [Google Scholar]
- Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311:1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- Herrera E, Samper E, Martin-Caballero J, Flores JM, Lee HW, Blasco MA. Disease states associated with telomerase deficiency appear earlier in mice with short telomeres. EMBO J. 1999;18:2950–2960. doi: 10.1093/emboj/18.11.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer AC, Tran RT, Aziz OZ, Wright W, Novelli G, Shay J, Lewis M. Shared phenotypes among segmental progeroid syndromes suggest underlying pathways of aging. J Gerontol A Biol Sci Med Sci. 2005;60:10–20. doi: 10.1093/gerona/60.1.10. [DOI] [PubMed] [Google Scholar]
- Johnson FB, Marciniak RA, McVey M, Stewart SA, Hahn WC, Guarente L. The Saccharomyces cerevisiae WRN homolog Sgs1p participates in telomere maintenance in cells lacking telomerase. EMBO J. 2001;20:905–913. doi: 10.1093/emboj/20.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassem M, Ankersen L, Eriksen EF, Clark BF, Rattan SI. Demonstration of cellular aging and senescence in serially passaged long-term cultures of human trabecular osteoblasts. Osteoporos Int. 1997;7:514–524. doi: 10.1007/BF02652556. [DOI] [PubMed] [Google Scholar]
- Long MW, Ashcraft EK, Normalle D, Mann KG. Age-related phenotypic alterations in populations of purified human bone precursor cells. J Gerontol A Biol Sci Med Sci. 1999;54:B54–B62. doi: 10.1093/gerona/54.2.b54. [DOI] [PubMed] [Google Scholar]
- Majors AK, Boehm CA, Nitto H, Midura RJ, Muschler GF. Characterization of human bone marrow stromal cells with respect to osteoblastic differentiation. J Orthop Res. 1997;15:546–557. doi: 10.1002/jor.1100150410. [DOI] [PubMed] [Google Scholar]
- Martinez ME, del Campo MT, Medina S, Sanchez M, Sanchez-Cabezudo MJ, Esbrit P, Martinez P, Moreno I, Rodrigo A, Garces MV, Munuera L. Influence of skeletal site of origin and donor age on osteoblastic cell growth and differentiation. Calcif Tissue Int. 1999;64:280–286. doi: 10.1007/s002239900619. [DOI] [PubMed] [Google Scholar]
- Martinez P, Moreno I, De Miguel F, Vila V, Esbrit P, Martinez ME. Changes in osteocalcin response to 1,25-dihydroxyvitamin D(3) stimulation and basal vitamin D receptor expression in human osteoblastic cells according to donor age and skeletal origin. Bone. 2001;29:35–41. doi: 10.1016/s8756-3282(01)00479-3. [DOI] [PubMed] [Google Scholar]
- Mason PJ, Wilson DB, Bessler M. Dyskeratosis congenita – a disease of dysfunctional telomere maintenance. Curr Mol Med. 2005;5:159–170. doi: 10.2174/1566524053586581. [DOI] [PubMed] [Google Scholar]
- McDaniel LD, Chester N, Watson M, Borowsky AD, Leder P, Schultz RA. Chromosome instability and tumor predisposition inversely correlate with BLM protein levels. DNA Repair (Amst) 2003;2:1387–1404. doi: 10.1016/j.dnarep.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res. 2007;100:15–26. doi: 10.1161/01.RES.0000256837.40544.4a. [DOI] [PubMed] [Google Scholar]
- Muschler GF, Nitto H, Boehm CA, Easley KA. Age- and gender-related changes in the cellularity of human bone marrow and the prevalence of osteoblastic progenitors. J Orthop Res. 2001;19:117–125. doi: 10.1016/S0736-0266(00)00010-3. [DOI] [PubMed] [Google Scholar]
- Nishida S, Endo N, Yamagiwa H, Tanizawa T, Takahashi HE. Number of osteoprogenitor cells in human bone marrow markedly decreases after skeletal maturation. J Bone Miner Metab. 1999;17:171–177. doi: 10.1007/s007740050081. [DOI] [PubMed] [Google Scholar]
- Opresko PL, von Kobbe C, Laine JP, Harrigan J, Hickson ID, Bohr VA. Telomere-binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicases. J Biol Chem. 2002;277:41110–41119. doi: 10.1074/jbc.M205396200. [DOI] [PubMed] [Google Scholar]
- Ozgenc A, Loeb LA. Current advances in unraveling the function of the Werner syndrome protein. Mutat Res. 2005;577:237–251. doi: 10.1016/j.mrfmmm.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96:701–712. doi: 10.1016/s0092-8674(00)80580-2. [DOI] [PubMed] [Google Scholar]
- Salk D, Bryant E, Au K, Hoehn H, Martin GM. Systematic growth studies, cocultivation, and cell hybridization studies of Werner syndrome cultured skin fibroblasts. Hum Genet. 1981;58:310–316. doi: 10.1007/BF00294930. [DOI] [PubMed] [Google Scholar]
- Shi S, Gronthos S, Chen S, Reddi A, Counter CM, Robey PG, Wang CY. Bone formation by human postnatal bone marrow stromal stem cells is enhanced by telomerase expression. Nat Biotechnol. 2002;20:587–591. doi: 10.1038/nbt0602-587. [DOI] [PubMed] [Google Scholar]
- Silberberg M, Silberberg R. Osteoarthrosis and osteoporosis in senile mice. Gerontologia. 1962;6:91–101. doi: 10.1159/000211110. [DOI] [PubMed] [Google Scholar]
- Simonsen JL, Rosada C, Serakinci N, Justesen J, Stenderup K, Rattan SI, Jensen TG, Kassem M. Telomerase expression extends the proliferative life-span and maintains the osteogenic potential of human bone marrow stromal cells. Nat Biotechnol. 2002;20:592–596. doi: 10.1038/nbt0602-592. [DOI] [PubMed] [Google Scholar]
- Stenderup K, Justesen J, Eriksen EF, Rattan SI, Kassem M. Number and proliferative capacity of osteogenic stem cells are maintained during aging and in patients with osteoporosis. J Bone Miner Res. 2001;16:1120–1129. doi: 10.1359/jbmr.2001.16.6.1120. [DOI] [PubMed] [Google Scholar]
- Wu X, Estwick SA, Chen S, Yu M, Ming W, Nebesio TD, Li Y, Yuan J, Kapur R, Ingram D, Yoder MC, Yang FC. Neurofibromin plays a critical role in modulating osteoblast differentiation of mesenchymal stem/progenitor cells. Hum Mol Genet. 2006;15:2837–2845. doi: 10.1093/hmg/ddl208. [DOI] [PubMed] [Google Scholar]
- Wyllie FS, Jones CJ, Skinner JW, Haughton MF, Wallis C, Wynford-Thomas D, Faragher RG, Kipling D. Telomerase prevents the accelerated cell ageing of Werner syndrome fibroblasts. Nat Genet. 2000;24:16–17. doi: 10.1038/71630. [DOI] [PubMed] [Google Scholar]
- Yudoh K, Matsuno H, Nakazawa F, Katayama R, Kimura T. Reconstituting telomerase activity using the telomerase catalytic subunit prevents the telomere shorting and replicative senescence in human osteoblasts. J Bone Miner Res. 2001;16:1453–1464. doi: 10.1359/jbmr.2001.16.8.1453. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following supplementary material is available for this article:
Fig. S1 Osteocalcin expression in mouse bone marrow stromal cells (BMSCs) derived from single (Wrn−/− or Terc−/−) and double (Wrn−/−/Terc−/−) mutants. Immunofluorescent staining of osteocalcin is shown in BMSCs derived from 10-month-old mice of the indicated genotype. The percentages of positive cells are as follows: 87%, wild type; 16%, Wrn−/−; 0.2%, Terc−/−; 0.2% Wrn−/− Terc−/−.
Fig. S2 Osteoblast differentiation of mouse bone marrow stromal cells (BMSCs) derived from young G3 single (Wrn−/− or Terc−/−) and double (Wrn−/−/Terc−/−) mutants is impaired. Detection of alkaline phosphatase (Alk P) activity and mineralization (Mineral) is shown for BMSCs isolated from 4.5-month-old animals. The percentages of Alk P positive colonies are as follows: 82%, wild type (WT); 64%, Wrn−/−; 35%, Terc−/−; 2.4%, Wrn−/− Terc−/−. The percentages of alizarin red positive colonies are: 86%, WT; 16%, Wrn−/−; 0.2%, Terc−/−; 0.2%, Wrn−/− Terc−/−.