

Abstract
The UV radiation in sunlight is the primary cause of non-melanoma skin cancer. Moreover, UV-exposure induces immune suppression. Early steps in the cascade of events leading to immune suppression are the binding of UV-induced platelet activating factor (PAF) to its receptor and the binding of cis-urocanic acid, a photoreceptor for UVB radiation, to the serotonin (5-HT2A) receptor. Here we tested the hypothesis that blocking the binding of PAF and 5-HT2A to their receptors would also block skin cancer induction. Hairless mice were injected with PAF or serotonin receptor antagonists and then exposed to solar simulated UV radiation. We noted a significant and substantial decrease in skin cancer incidence in mice treated with the PAF or 5-HT2A receptor antagonists. Also, the PAF and/or serotonin receptor antagonists blocked skin cancer progression. The PAF and serotonin receptor antagonists worked in a synergistic fashion to block skin cancer induction. We also measured the effect that injecting PAF and 5-HT2A receptor antagonists had on UV-induced skin damage after a single UV exposure. We noted a significant decrease in UV-induced hypertrophy, sunburn cell formation, and apoptosis when the mice were injected with PAF and/or 5-HT2A receptor antagonists. These data indicate that treating UV-irradiated mice with PAF and 5-HT2A receptor antagonists blocks skin cancer induction in vivo, in part by reversing UV-induced damage to the skin and by preventing the induction of immune suppression.
Keywords: UV radiation, skin cancer, inflammation, apoptosis, therapy
Introduction
The UV radiation found in sunlight has a number of adverse effects on the health and well being of humans. It is the primary cause of non-melanoma skin cancer (1) and is implicated in the induction of malignant melanoma (2). Skin cancer is the most prevalent form of human cancer. Statistics complied by the American Cancer Society indicate that over one-half of all cancers diagnosed in the United States last year were skin cancer. Approximately 1.3 million new cases of skin cancer were diagnosed last year, and approximately 10,000 deaths were attributed to skin cancer during the past year 3. The annual cost of treating non-melanoma skin cancer in the United States alone is estimated to be in excess of $800 million (3, 4), clearly illustrating that skin cancer represents a major public health problem.
In addition to its carcinogenic potential, UV radiation is also immune suppressive. Kripke and colleagues demonstrated that UV exposure suppressed the immune response (5) and suggested that there is an association between immune suppression and skin cancer induction (6). Subsequent studies with biopsy proven skin cancer patients (7) and immunosuppressed transplant patients (8) have confirmed that UV-induced immune suppression is a major risk factor for skin cancer induction. Because of the association between UV-induced immune suppression and carcinogenesis, and in light of the fact that exposure to UV radiation occurs daily and may be increasing due to changes in patterns of human behavior (i.e., increased sun exposure due to changes in dress styles and more leisure time spent in the sun) it is critically important to understand the mechanisms underlying UV-induced immune suppression.
To induce immune suppression the electromagnetic energy of UV radiation must first be absorbed by an epidermal photoreceptor and then converted into a biologically recognizable signal. One prominent example is urocanic acid (UCA), which is located superficially in the stratum corneum. Upon UV exposure, naturally occurring trans-UCA converts to the cis-isomer. Although UCA was first recognized as a UV-photoreceptor 20 years ago (9), and many have documented the ability of cis-UCA to initiate immune suppression (10), its exact mode of action remains elusive. Recently we reported that cis-UCA binds to the serotonin (5-HT2A) receptor, and that blocking the binding of cis-UCA to the 5-HT2A receptor with a series of selective serotonin receptor antagonists blocked immune suppression (11).
Another early UV-induced event that leads to immune suppression is the production of the lipid mediator of inflammation, platelet-activating factor (PAF). PAF is secreted by epidermal cells aμmost immediately following UV exposure (12). UV-induced PAF activates cytokine production and initiates UV-induced immune suppression. Both UV exposure and PAF-treatment activated the transcription of cyclooxygenase-2 (COX-2) and interleukin (IL)-10. Treating keratinocytes with a specific PAF receptor antagonist prior to UV exposure, suppressed the transcription of the COX-2 and IL-10 genes. In addition, PAF-like lipids such as UV-irradiated oxidized phosphatidylcholine induced COX-2 and IL-10 transcription. PAF mimicked the effects of UV by suppressing the induction of delayed type hypersensitivity in vivo. Immune suppression was abrogated when UV-irradiated mice were injected with PAF receptor antagonists (13). Furthermore, no immune suppression was observed when PAF receptor knockout mice were exposed to UV radiation (14).
Because the binding of PAF and/or cis-UCA to their receptors is an early and essential step in the cascade of events leading to UV-induced immunosuppression, we asked if treating UV-irradiated mice with selective PAF and/or serotonin receptor antagonist could prevent photocarcinogenesis. We find that both block UV-induced skin cancer. In addition we find a significant decrease in UV-induced skin damage (hypertrophy, sunburn cell formation, and apoptosis) when mice exposed to a single dose of UV were injected with PAF and/or 5-HT2A receptor antagonists. Our results suggest and PAF and 5-HT2A receptor antagonists may be considered as new therapeutic agents to prevent sunlight induced skin cancer.
Materials and Methods
Mice
Young-adult, specific pathogen free, female, hairless, SKH-hr1 (Charles River Laboratory) were used in these experiments. All animals were maintained with alternating 12-hour light and dark cycles and controlled temperature and humidity in facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care International, in accordance with current regulations of the United States Department of Health and Human Services. The University of Texas M.D. Anderson Cancer Center Institutional Animal Care and Use Committee approved all the animal procedures described here.
Reagents
The metabolically stable analogue of PAF, carbamyl-PAF (c-PAF) and the PAF-receptor antagonists, PCA-4248, CV-3988 and the 5-HT2A receptor antagonist, ketanserin were purchased from Biomol Research Labs. The 5-HT2A receptor antagonist 1-(1-Naphthyl) piperazine (1-NPZ) was purchased from Sigma Chemical Co. Stock solutions of PCA-4248, CV-3988 and ketanserin were prepared in 50% DMSO/PBS and diluted further in PBS or tissue culture medium immediately before use. 1-NPZ was prepared in sterile water and further diluted in PBS or tissue culture medium. Tissue culture medium was obtained from Gibco BRL.
Radiation source
A 1000 W xenon UV solar simulator equipped with a Schott WG-320 atmospheric attenuation filter (1 mm thick), a visible/infrared band pass blocking filter (Schott UG-11; 1 mm thick), and a diachronic mirror to further reduce visible and infrared energy (Oriel) was used to provide solar-simulated UV radiation (UVA + UVB, 290 to 400 nm). The intensity and spectral output of the WG-320 equipped solar simulator were measured with an Optronics model OL 754 scanning spectrophotometer (Optronics Laboratories), as described previously (15). Approximately 10% of the incident light was in the UVB (290 to 320 nm) portion of the spectrum. During irradiation, the mice were held individually in a specially constructed Plexiglas container with a quartz glass top, to prevent cage mates from climbing on top of each other and interfering with the UV dose applied. Spectrophotometer readings were taken through the quartz glass top. During the irradiation period the mice were conscious and had full range of movement.
Effect of PAF and 5-HT2A receptor antagonists on skin cancer induction and progression
Two protocols were used. In the skin cancer induction experiments, the mice received an intraperitoneal (ip) injection of 500 nmol of a PAF or a 5-HT2A receptor antagonist or received an ip injection of the vehicle (1:5000 dilution of DMSO in PBS), 1h before irradiation. The dose was chosen based on previous findings that 500 nmol was optimal at blocking UV-induced immune suppression (11, 13). The mice were then exposed to 1.25 kJ/m2 (approximately one half a minimal erythemal dose) (16) of solar simulated UVB radiation. The mice were irradiated on Mondays, Wednesdays and Fridays. The experiment was terminated when approximately 100% of the animals in the UV + vehicle control developed a skin cancer. The percentage of mice with tumors (number of tumor positive animals ÷ total number of mice in the group) × 100 was calculated. All positive skin tumors were confirmed by histopathology. In the tumor progression experiments, the mice were exposed to 1.25 kJ/m2 of solar simulated UVB radiation (three times a week, as above) until at least one papilloma was evident by visual examination. The UV exposure was then stopped and the animals were randomly assigned to one of three groups. One group was injected three times a week (Monday, Wednesday, Friday) with 500 nmol of the vehicle, the second was injected three times a week with 1-NPZ, a 5-HT2A receptor antagonist, and the third group was injected three times a week with PCA-4248, a PAF receptor antagonist. The mice were examined daily for skin cancer induction. The number of tumors per animal was counted. All tumors were excised and tumor incidence (squamous cell carcinoma) was confirmed by histopathology. Statistically significant differences in tumor number and tumor incidence between the vehicle treated control and the experimental groups were determined by use of the Fisher’s exact test; probabilities less than 0.05 were considered significant. There were 8 to 10 mice per group. Each tumor experiment was repeated at least two times to ensure reproducibility of the results.
Immunohistochemistry
SKH-hr1 mice were exposed to 1.5 kJ/m2 solar simulated UVB radiation, with or without the PAF or 5-HT2A receptor antagonists. Skin samples were harvested at various time points (3, 6, 9, 12, 24, 48 and 72 hours) post irradiation. They were embedded in Tissue-Tek OCT medium (Miles Laboratories) and snap frozen in liquid nitrogen. Five-μm sections were cut with a cryostat. IL-10 was detected using anti-mouse IL-10 antibody (JES5-2A5.11, rat IgG; BD-Pharmingen) and COX2 was detected using rabbit polyclonal to COX2 (ab15191, rabbit IgG; Abcam). Active caspase 3 was detected using monoclonal antibody (C92-605, rabbit IgG; BD Pharmingen). Isotype-matched control rat and mouse antibodies were acquired from Sigma. Terminal dUTP Nick-End Labeling (TUNEL) positive cells were detected using a commercial kit according to the manufacturer’s instructions (Promega).
Sunburn cells
Approximately 1 cm2 of dorsal skin was excised, fixed immediately in 4% buffered formaldehyde, and sectioned at 5 μM for hematoxylin and eosin (H&E) staining. Sunburn cells were counted in the interfollicular epidermis; at least 10 random high-power fields per section (20X magnification) were counted from tissues after 24h exposure to UV-irradiation. Counts were expressed as the number of sunburn cells per centimeter length of epidermis, as determined with a calibrated eyepiece micrometer (Nikon Inc). Statistically significant differences (p < 0.05) between the controls (UV only) and the experimental groups (UV + receptor antagonist) were determined with the Student’s t-test.
Real-Time PCR Amplification
Real-time PCR amplification was performed on an ABI Prism 7500 thermo cycler (Applied Biosystems), using default settings, in a final volume of 20 μL, including 0.5, 1, or 2.5 μL of DNA template. The TaqMan Universal PCR Master Mix 1X (Applied Biosystems) was used with 500 nM of each primer and 200 nM of TaqMan MGB probe. Real-time PCR was performed using TaqMan Universal PCR mix, primers and fluorescent probes specific for COX-2 (Ptgs2), IL-10 and GAPDH (Taqman Gene Expression Assay Regents, Applied Biosystems). Outputs of real-time amplifications were analyzed using SDS 7500 1.1 software (Applied Biosystems). Threshold cycle (CT) values for COX-2 were normalized to glyceraldhyde-3-phosphate dehydrogenase (GAPDH) using the following equation: 1.8(GAPDH-COX-2) X 10,000), where GAPDH is the CT of the GAPDH control, COX-2 is the CT of COX-2, and 10,000 is an arbitrary factor to bring all values above one. Similar calculations were done to quantitative IL-10 expression. There were 10 mice in each treatment group and RNA was isolated from each individual mouse. The means and the standard deviation for each treatment group were calculated and statistical differences between each experimental group were determined by using a one way analysis of variance (ANOVA) followed by the Student-Newman-Keuls Multiple comparison test (GraphPad Prism Software V4).
Results
Suppression of skin cancer induction by PAF and Serotonin receptor antagonists
The immune suppression induced by UV exposure is a major risk factor for skin cancer induction (6, 7). Because we previously demonstrated that PAF and 5-HT2A receptor antagonists block UV-induced immune suppression (11, 13, 14), we wanted to test the hypothesis that blocking 5-HT and PAF receptor binding would block skin cancer induction. Injecting PCA-4248, a selective PAF receptor antagonist into UV-irradiated mice significantly suppressed skin cancer induction (Figure 1A; p < 0.02, vs. UV + Vehicle). As a positive control in this experiment, another group of mice were injected with 0.2 μg/mouse of SC58236, a selective COX-2 inhibitor (17), because others have shown that neutralizing cyclooxygenase-2 function blocks UV-induced carcinogenesis (18, 19). Neutralizing COX-2 activity suppressed skin cancer induction (p < 0.003 vs. UV + vehicle). When the mice were injected with 1-NPZ, a selective 5-HT2A receptor antagonist (Figure 1B), skin cancer induction was also significantly suppressed (p < 0.03, vs. UV + Vehicle). These data indicate that in addition to blocking UV-induced immune suppression, PAF and 5-HT2A receptor antagonists block UV-induced carcinogenesis.
Figure 1.
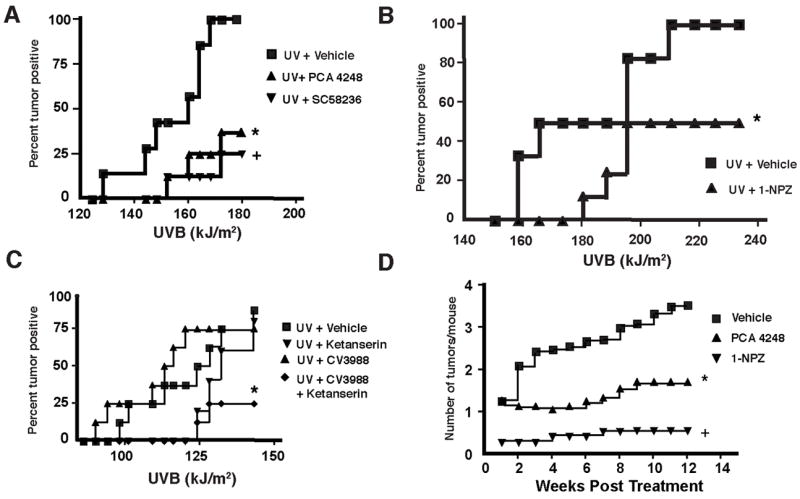
Suppression of skin cancer induction and progression by PAF and serotonin receptor antagonists. A. PAF receptor antagonist suppresses skin cancer induction. Hairless mice were exposed to 1.25 kJ/m2 of UVB radiation on Mondays, Wednesday and Fridays. 1 hr prior to irradiation, the mice received an ip injection of PCA-4248 or a selective COX-2 inhibitor (SC58236). * Indicates a significant suppression of tumor incidence vs. the UV + Vehicle control; p <0.02. + Indicates a significant suppression of tumor incidence vs. the UV + Vehicle control; p <0.003. B. 5-HT2A-receptor antagonist suppresses skin cancer induction. Hairless mice were exposed to 1.25 kJ/m2 of UVB radiation on Mondays, Wednesday and Fridays. 1 hr before irradiation, the mice were injected with 1-NPZ. * Indicates a significant suppression of tumor incidence vs. the UV + Vehicle control; p < 0.03. C. PAF and 5-HT2A-receptor antagonists act in concert to block skin cancer induction. Mice were injected (ip) with sub-optimal doses (500 pmol) of CV-3899, a PAF receptor antagonist or ketanserin, a 5-HT2A receptor antagonist, or with a mixture of the two (500 pmol of each). * Indicates a significant suppression of tumor incidence vs. the UV + Vehicle control; p < 0.04. D. PAF and 5-HT2A receptor antagonists block the progression of skin cancer. Mice were exposed to sufficient UV radiation to induce a papilloma, and then they were removed from protocol, and injected with a PAF (PCA-4248) or a 5-HT2A (1-NPZ) receptor antagonist. * Indicates a significant suppression of tumor number vs. mice injected with the vehicle only; p < 0.01. + Indicates a significant suppression of tumor number vs. mice injected with the vehicle only.
Next we wanted to determine if PAF and 5-HT2A receptor antagonists can act in a synergistic manner to block skin cancer induction. In previous work, dealing with UV-induced immune suppression, we noted that injecting 500 nmol of either a PAF or 5-HT2A receptor antagonist into a UV-irradiated mouse blocked immune suppression, but lower doses (500 pmol) were ineffective (11, 13, 14). To determine if these agents could synergize to prevent photocarcinogenesis, we injected a series of mice with sub-optimal doses (500 pmol) of ketanserin, a selective 5-HT2A receptor antagonist, or CV-3988, a selective PAF receptor antagonist, or a mixture of ketanserin and CV-3988 (Figure 1C). Injecting a sub-optimal dose of the PAF or 5-HT2A receptor antagonist alone did not prevent skin cancer induction, as there was no difference in tumor incidence from the positive control. When however, the mice received a mixture of 500 pmol of CV-3988 and 500 pmol of ketanserin, significant suppression of tumor induction occurred (p < 0.04, UV + Vehicle vs. UV + CV3988 + ketanserin). These data indicate that the PAF and serotonin receptor antagonists act synergistically to prevent skin cancer induction.
We also asked if blocking the binding of PAF and/or 5-HT2A to their receptors could prevent the progression of skin cancer. This experiment was modeled after those originally described by Pentland and colleagues (19). In these experiments, hairless mice are exposed to UV radiation until the animal develops a tumor. The mouse is then removed from the UV protocol, and the chemo-preventative regimen is started. Mice that do not receive any further treatment generally go on to develop multiple skin tumors. In our experiments, we removed the mice from the UV when they developed obvious skin damage and at least one visible papilloma. The mice were then injected with 500 nmol of a selective PAF (PCA-4248) or 5-HT2A (1-NPZ) receptor antagonist (Monday, Wednesday, and Friday) and monitored for skin cancer development. Results from this experiment are shown in Figure 1D. As expected, multiple tumors developed in the group of mice that were injected with the vehicle after being removed from the UV protocol. However, in mice receiving PCA-4248, the selective PAF receptor antagonist, we noted a significant reduction in skin cancer number (p < 0.01 vs. the Vehicle control). Similarly, injecting the 5-HT2A receptor antagonist, 1-NPZ significantly suppressed the development tumors in the treated mice (p < 0.001 vs. the vehicle control). These findings indicate that blocking the binding of PAF and/or serotonin to their receptors, will block the progression of UV-induced skin cancers.
Inhibition of epidermal damage by PAF and 5-HT2A receptor antagonists
A single exposure to UV radiation initiates several physiological and cellular changes including cytokine induction (20), prostaglandin E2 (PGE2) production (21), and dermal damage leading to apoptosis (22, 23). We wanted to determine if treating the mice with the receptor antagonists also affects UV-induced skin damage. PAF and serotonin receptor antagonists (1000 nmol of each) were injected ip into SKH-hr1 mice 1h prior to irradiation with 1.5 kJ/m2 of UVB irradiation. We first measured sunburn cell formation (Figure 2A). UV-irradiated mice showed a high number of sunburn cells with characteristic morphology of condensed pyknotic nuclei and diffused cytoplasm. These dysmorphic keratinocytes were scattered throughout the epidermis. Treating the mice with the receptor antagonists statistically suppressed sunburn cell formation (80–90% fewer sunburn cells; p < 0.001 vs. the UV only control) (Figure 2B).
Figure 2.
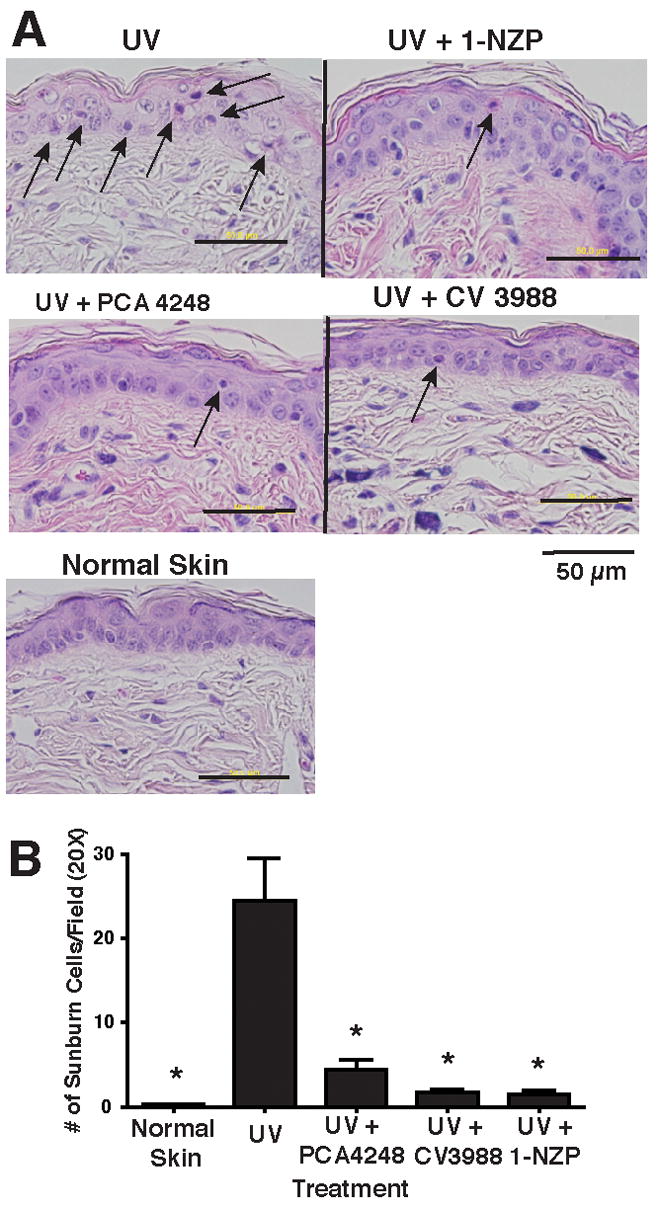
Suppression of skin damage by PAF and 5-HT2A receptor antagonists. A. Hairless mice were exposed to 1.25 kJ/m2 of UVB radiation and 24 hour later, sunburn cell formation was measured. Treatment groups include: Normal skin (No UV); UV only and UV+ PAF receptor antagonists PCA-4248 and CV3988 and UV+ serotonin receptor antagonist, 1-NPZ. Arrows indicate sunburn cells. B. Sunburn cells were counted in the interfollicular epidermis (10 random fields per section @ 20X magnification; 5 sections from 5 different mice). Counts were expressed as the number of sunburn cells per/20X field, as determined with a calibrated eyepiece micrometer. * Indicates a significant (p < 0.003) reduction in the sunburn cell number vs. UV control.
Sunburn cells develop after irreparable UV-induced DNA damage and eventually they are removed by apoptosis. Next we employed TUNEL analysis in situ to measure apoptosis (Figure 3A). Because we noted maximal numbers of apoptotic cells 24h after UV-irradiation in initial experiments, we measured the effect that injecting the PAF and 5-HT2A receptor antagonists had on apoptosis 24h after UV exposure (Figure 3A). Both the PAF and 5-HT2A receptor antagonists inhibited apoptosis. We also measured apoptotic cell formation 48 h and 72 h after UV exposure to rule out the possibility of delayed apoptosis by antagonists. Delayed apoptosis was not evident (data not shown).
Figure 3.
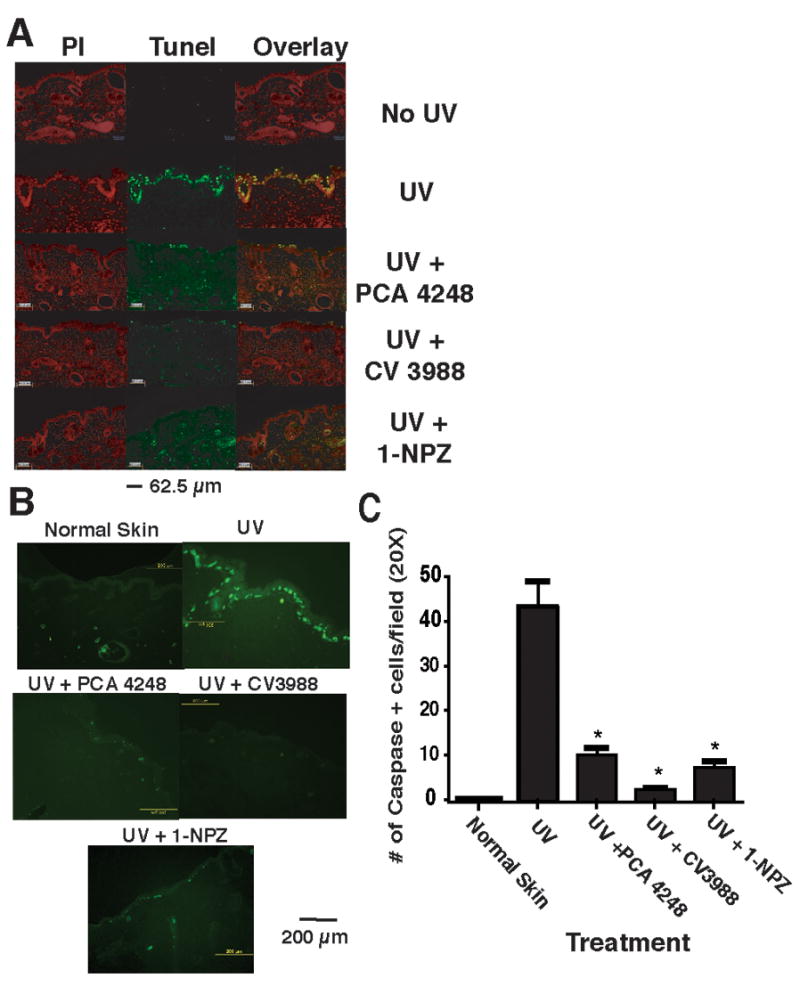
PAF and 5-HT2A receptor antagonists block UV-induced apoptosis. A: Apoptosis was determined by TUNEL staining. Skin sections taken after 24 h after exposing the mice to 1.5 kJ/m2 UVB radiation. Treatment groups include: Normal skin (No UV); UV only; mice injected with the PAF receptor antagonist PCA-4248 and exposed to UV; mice injected with the PAF receptor antagonist CV-3988 and exposed to UV; mice injected with the serotonin receptor antagonist 1-NPZ and exposed to UV. The left panel (red nuclear staining) shows propidium iodide staining. The middle panel shows TUNEL positive (green staining) cells. The overlay shows the merging of propidium iodide staining with TUNEL positive cells to confirm nuclear (yellow) staining. B: Immunofluorescence staining for active caspase-3. Skin sections were incubated with cleaved active caspase-3 antibody followed by a secondary antibody conjugated to fluorescein isothiocyanate (FITC) and visualized under a fluorescent microscope. Treatment groups include: Normal skin (No UV); UV only; mice injected with the PAF receptor antagonist PCA-4248 and exposed to UV; mice injected with the PAF receptor antagonist CV-3988 and exposed to UV; mice injected with the serotonin receptor antagonist 1-NPZ and exposed to UV. C: Enumeration of caspase-3 positive cells. * Indicates a significant reduction in caspase-3 positive cells in PAF and 5-HT2A receptor antagonists treated mice vs. UV control; p < 0.001.
Caspase-3 plays an important role in mediating UV-induced apoptosis (24). Next, we used an immunofluorescence assay to determine if cells undergoing apoptosis express activated caspase-3 (Figure 3B). No caspase-3 positive cells were found in the epidermis or dermis of un-irradiated control mice. At 12h post irradiation, caspase-3 positive cells were present in the basal and upper epidermis of UV-irradiated mice. Injecting either the PAF or 5-HT2A receptor antagonists significantly (p < 0.001) decreased the number of active caspase-3 positive cells found 12h post irradiation (Figure 3C). Our results indicate that PAF and 5-HT2A receptor antagonist treatment prevents apoptosis after a single exposure to UV radiation.
Inhibition of cytokines and inflammatory mediator production by PAF and 5-HT2A receptor antagonists
Another consequence of acute UV damage is the up-regulation of epidermal cytokine production. Real-time PCR was used to determine if prior injection of PAF and/or 5-HT2A receptor antagonists would block UV-induced cytokine production (Figure 4A). We noted optimal IL-10 mRNA expression 24h after UV exposure. The UV-induced up-regulation of IL-10 mRNA expression was significantly suppressed in the receptor antagonist treated mice (p < 0.001 vs. UV-only control). The inhibition of UV-induced IL-10 secretion in the skin by the receptor antagonists was confirmed by immunohistochemical analysis (Figure 4B). These data indicate that blocking the binding of PAF or 5-HT2A to its receptor suppresses UV-induced cytokine production. Similar results were observed when COX-2 expression was measured. Nine hours post UV-irradiation, COX2 mRNA was up regulated, and treating the UV-irradiated mice with either a PAF or 5-HT2A receptor antagonist reduced COX2 mRNA expression to a degree similar to that found in the non-irradiated controls (Figure 4C). The effect of the receptor antagonists on UV-induced COX2 protein expression in the skin was confirmed by immunohistochemical analysis. (Figure 4D).
Figure 4.
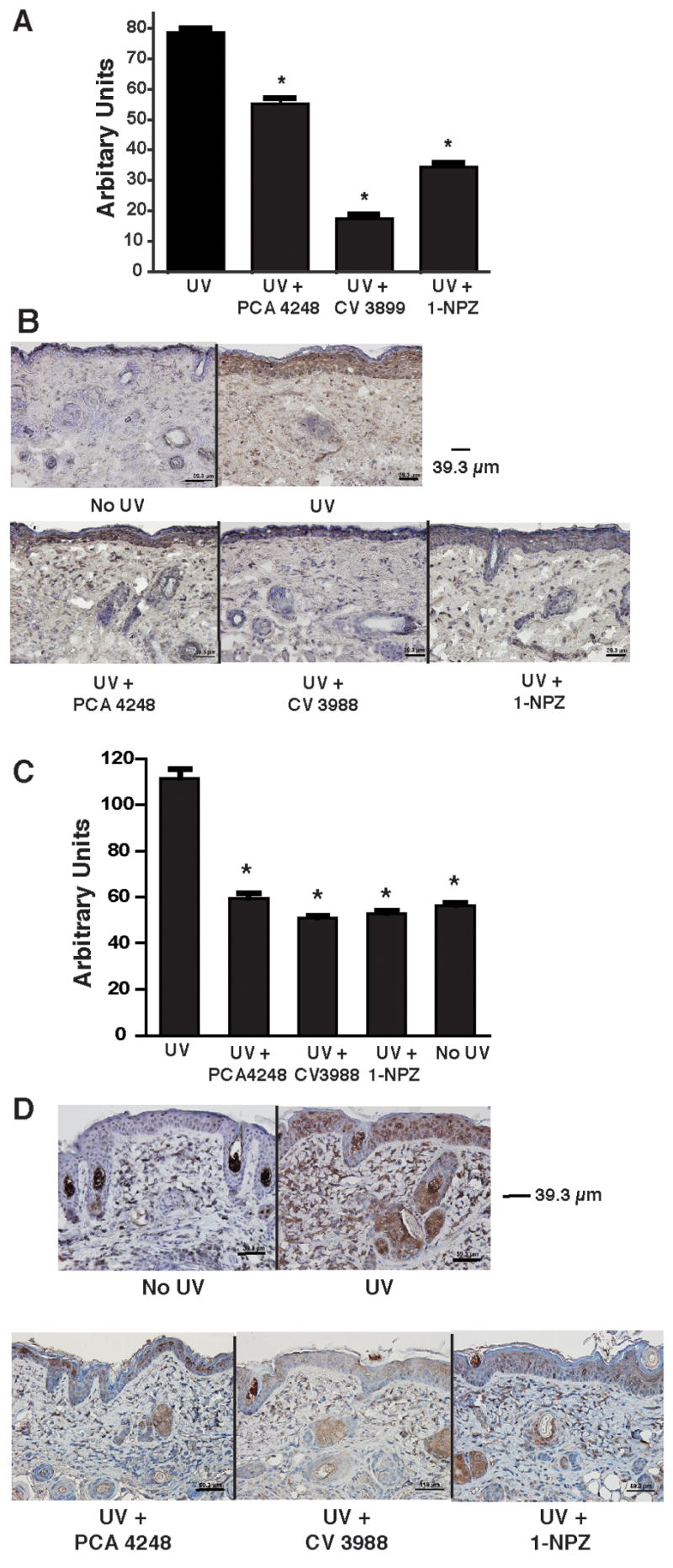
Inhibition of UV-induced IL-10, and COX2 by PAF and 5-HT2A receptor antagonists. A: Expression of IL-10 mRNA by real time PCR. Samples were taken 24h post UV exposure from mice exposed to UV only, or from mice exposed to UV and treated with PAF and/or 5-HT receptor antagonists. The data is expressed as arbitrary units relative to GAPDH mRNA expression. * indicates a significant difference (p < 0.001) from the UV only control. B. Immunohistochemistry was used to measure UV-induced IL-10 protein production. IL-10 in normal skin (No UV), mice only exposed to UV and mice exposed to UV and injected with PAF and/or 5-HT receptor antagonists was measured 48h post UV exposure. C: Expression of COX-2 mRNA by real time PCR. Samples were taken 9h post UV exposure from mice exposed to UV only, or from mice exposed to UV and treated with PAF and/or 5-HT receptor antagonists. The data is expressed as arbitrary units relative to GAPDH mRNA expression. * indicates a significant difference (p < 0.001) from the UV only control. D. Immunohistochemistry was used to measure UV-induced COX2 protein production. COX-2 in normal skin (No UV), mice only exposed to UV and mice exposed to UV and injected with a PAF or a 5-HT receptor antagonist was measured 48h post UV exposure.
Discussion
The UV radiation in sunlight is the primary cause of skin cancer (1). In addition, UV radiation is immunosuppressive, and the immune suppression induced by UV radiation is a major risk factor for skin cancer induction (7). Because we previously demonstrated that PAF and 5-HT2A receptor binding plays an essential role in UV-induced immune suppression (11, 13), we tested the hypothesis that PAF and 5-HT2A receptor antagonists could block UV-induced photocarcinogenesis. Here we show that PAF and 5-HT2A receptor antagonists block skin cancer induction and progression. In addition, we demonstrate that injecting PAF and 5-HT2A receptor antagonists into mice treated with a single dose of UV radiation, blocked UV-induced skin damage, including, sunburn cell formation, apoptosis, and cytokine secretion. These data, and our previous findings, indicating that PAF and 5-HT2A receptor antagonists block UV-induced immune suppression (11, 13), indicate that these reagents are working at multiple levels to block photocarcinogenesis.
In the carcinogenesis studies, we tested the effect of PAF and 5-HT2A receptor antagonists on both tumor induction and tumor progression. In both cases, the use of multiple, structurally unrelated PAF and serotonin receptor antagonists blocked carcinogenesis. The data presented in Figure 1D is particularly important because it more closely mimics the human model of skin cancer treatment. Generally, a patient comes into the skin cancer clinic with a obvious lesion, it is removed, and the patient is advised to stay out of the sun, apply sunscreen when he/she does go outside, and if available, use some type of chemopreventive agent to prevent the development of a second skin cancer. Our experiment was designed to mimic this situation. Once a papilloma was evident, the UV exposure was halted, and the mice were injected with the PAF or the 5-HT2A receptor antagonists. We noted significant suppression in the development of second tumors. The number of tumors generated in this experiment is somewhat lower than that reported by others (19). Two reasons may explain this fact. First, we used a sub-erythemal UV dose, one-half the minimal erythemal dose, in these experiments. Here again, this was done to more closely model human exposure to sunlight, which generally does not include multiple successive sun burning episodes (i.e., Monday, Wednesday, Friday) over a span of 25 to 35 weeks. Second, in our analysis we only counted skin cancers (squamous cell carcinoma) and not papillomas, as is often done in other experiments of this type. Because not every papilloma progresses into a skin cancer, and it is common to find regression of papillomas, even in the presence of continual UV exposure, we only counted tumors in our experiments.
The data presented in Figure 1C indicates that the PAF and 5-HT2A receptor antagonists work synergistically to block skin cancer induction. In this experiment, the mice were injected with 500 pmol of the receptor antagonists, rather than the normal 500 to 1000 nmol dose. Dose response experiments carried out in the past indicated that injecting 500 pmol of either PAF or 5-HT2A receptor antagonists had no effect in UV-induced immune suppression (11, 13). We noted the same here, injecting the mice with 500 pmol of ketanserin or CV-3988 did not prevent skin cancer induction. When however, the mice were injected with a cocktail of both drugs (500 pmol of each), we noted substantial suppression of skin cancer induction, indicating that the drugs are working synergistically. The mechanism is not known, but we suspect that UV-induced cis-UCA is binding to a cell in the skin, perhaps a dermal mast cell, causing it to secrete PAF, which induces COX-2 up-regulation and PGE2 secretion, leading to immune suppression (25) and photocarcinogenesis (18, 19). Studies are in progress to test this hypothesis.
Our data suggests that one mechanism by which 5-HT2A and PAF receptor antagonists block UV-induced skin cancer induction is by preventing the initial damage to the skin. We used an acute exposure to UV radiation in hairless mice, with a relatively low dose of UVB radiation, to examine the effects of the receptor antagonists on sunburn cell formation, apoptosis, and cytokine production. In all cases treating the mice with the 5-HT2A and/or the PAF receptor antagonists prevented UV-induced skin damage. Our finding that PAF receptor binding plays a role in UV-induced apoptosis confirms and extends previous reports (26, 27). Similarly, studies with cardiac and neuronal tissues have documented a role for 5-HT2A receptor binding in apoptosis (28, 29). Our data is the first to implicate 5-HT2A receptor binding in apoptosis induction in the skin after exposure to the common environmental carcinogen, UV radiation.
The reversal of apoptosis in our system may at first appear to be counter-intuitive, in that drugs that prevent apoptosis also block skin cancer induction. Note however, that we measured the effect these drugs have on UV-induced skin damage after a single acute exposure to a subcarcinogenic dose of UV radiation. We suggest that the effect here may be similar to what has been reported regarding the up-regulation of the tumor suppressor gene, p53 after UV exposure (22, 23). Initially, UV-damage up-regulates normal p53 protein, which serves to suppress tumor induction by controlling apoptosis. After continual UV exposure, the gene is mutated and the tumor suppressing function is aborted. We have never looked at the effect of PAF and 5-HT receptor antagonists on apoptosis after multiple UV exposures, so it is impossible to project what would happen. We have only used these findings to conclude that these drugs can block initial UV-induced skin damage. Any other conclusion would be an over interpretation of the data.
In summary, we show that PAF and 5-HT2A receptor antagonists inhibit UV-induced skin cancer induction and progression. In addition, we show that PAF and 5-HT2A receptor antagonists inhibit UV-induced skin damage, in that they prevent cytokine release, and prevent UV-induced apoptosis after a single exposure to UV radiation. These findings suggest that PAF and 5-HT2A receptor antagonists affect UV-induced carcinogenesis at two distinct levels: First by preventing UV-induced damage in the skin. Second, by preventing UV-induced immune suppression. Our findings indicate that PAF and 5-HT2A receptor antagonists maybe considered as novel chemopreventive agents for sunlight-induced skin cancer.
Acknowledgments
The authors thank Professor H.N. Ananthaswamy for his comments and critical review of the manuscript.
Footnotes
This work was supported by grants from the National Cancer Institute, (CA112660, CA088943, CA75575; SEU). The animal facility and the histology core at MD Anderson are supported in part by a NCI Cancer Center Support Grant (CA 16672). Dr Sreevidya was supported by an NCI training grant (T32-CA-09598-15).
References
- 1.Urbach F. Ultraviolet radiation and skin cancer of humans. J Photochem Photobiol B: Biol. 1997;40:3–7. doi: 10.1016/s1011-1344(97)00029-8. [DOI] [PubMed] [Google Scholar]
- 2.Elwood JM, Jopson J. Melanoma and sun exposure: An overview of published studies. Int J Cancer. 1997;73:198–203. doi: 10.1002/(sici)1097-0215(19971009)73:2<198::aid-ijc6>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 3.Chen JG, Fleischer AB, Smith ED, et al. Cost of Nonmelanoma Skin Cancer Treatment in the United States. Dermatol Surg. 2001;27:1035–8. doi: 10.1046/j.1524-4725.2001.01004.x. [DOI] [PubMed] [Google Scholar]
- 4.Housman TS, Feldman SR, Williford PM, et al. Skin cancer is among the most costly of all cancers to treat for the Medicare population. J Am Acad Dermatol. 2003;48:425–9. doi: 10.1067/mjd.2003.186. [DOI] [PubMed] [Google Scholar]
- 5.Kripke ML. Antigenicity of murine skin tumors induced by ultraviolet light. J Natl Cancer Inst. 1974;53:1333–6. doi: 10.1093/jnci/53.5.1333. [DOI] [PubMed] [Google Scholar]
- 6.Fisher MS, Kripke ML. Suppressor T lymphocytes control the development of primary skin cancers in ultraviolet-irradiated mice. Science. 1982;216:1133–4. doi: 10.1126/science.6210958. [DOI] [PubMed] [Google Scholar]
- 7.Yoshikawa T, Rae V, Bruins-Slot W, Van den Berg JW, Taylor JR, Streilein JW. Susceptibility to effects of UVB radiation on induction of contact hypersensitivity as a risk factor for skin cancer in humans. J Invest Dermatol. 1990;95:530–6. doi: 10.1111/1523-1747.ep12504877. [DOI] [PubMed] [Google Scholar]
- 8.Penn I. Post-transplant malignancy: the role of immunosuppression. Drug Saf. 2000;23:101–13. doi: 10.2165/00002018-200023020-00002. [DOI] [PubMed] [Google Scholar]
- 9.De Fabo EC, Noonan FP. Mechanism of immune suppression by ultraviolet irradiation in vivo. I Evidence for the existence of a unique photoreceptor in skin and its role in photoimmunology. J Exp Med. 1983;157:84–98. doi: 10.1084/jem.158.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Norval M, El-Ghorr AA. Studies to determine the immunomodulating effects of cis-urocanic acid. Methods. 2002;28:63–70. doi: 10.1016/s1046-2023(02)00210-4. [DOI] [PubMed] [Google Scholar]
- 11.Walterscheid JP, Nghiem DX, Kazimi N, et al. Cis-urocanic acid, a sunlight-induced immunosuppressive factor, activates immune suppression via the 5-HT2A receptor. Proc Natl Acad Sci U S A. 2006;103:17420–5. doi: 10.1073/pnas.0603119103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pei Y, Barber LA, Murphy RC, et al. Activation of the epidermal platelet-activating factor receptor results in cytokine and cyclooxygenase-2 biosynthesis. J Immunol. 1998;161:1954–61. [PubMed] [Google Scholar]
- 13.Walterscheid JP, Ullrich SE, Nghiem DX. Platelet-activating factor, a molecular sensor for cellular damage, activates systemic immune suppression. J Exp Med. 2002;195:171–9. doi: 10.1084/jem.20011450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolf P, Nghiem DX, Walterscheid JP, et al. Platelet-activating factor is crucial in psoralen and ultraviolet A-induced immune suppression, inflammation, and apoptosis. Am J Pathol. 2006;169:795–805. doi: 10.2353/ajpath.2006.060079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim TH, Ullrich SE, Ananthaswamy HN, Zimmerman S, Kripke ML. Suppression of delayed and contact hypersensitivity responses in mice have different UV dose responses. Photochem Photobiol. 1998;68:738–44. [PubMed] [Google Scholar]
- 16.Kim TH, Ananthaswamy HN, Kripke ML, Ullrich SE. Advantages of using hairless mice versus haired mice to test sunscreen efficacy against photoimmune suppressions. Photochem Photobiol. 2003;78:37–42. doi: 10.1562/0031-8655(2003)078<0037:aouhmv>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 17.Seibert K, Zhang Y, Leahy K, et al. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc Natl Acad Sci USA. 1994;91:12013–7. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fischer SM, Lo HH, Gordon GB, et al. Chemopreventive activity of celecoxib, a specific cycloogenase-2 inhibitor, and indomethacin against ultraviolet-light induced skin carcinogenesis. Mol Carcinogenesis. 1999;25:231–40. [PubMed] [Google Scholar]
- 19.Pentland AP, Schoggins JW, Scott GA, Khan KNM, Han RJ. Reduction of UV-induced skin tumors in hairless mice by selective COX-2 inhibition. Carcinogenesis. 1999;20:1939–44. doi: 10.1093/carcin/20.10.1939. [DOI] [PubMed] [Google Scholar]
- 20.Ullrich SE. Mechanisms underlying UV-induced immune suppression. Mutat Res. 2005;571:185–205. doi: 10.1016/j.mrfmmm.2004.06.059. [DOI] [PubMed] [Google Scholar]
- 21.Grewe M, Trefzer U, Ballhorn A, Gyufko K, Henninger H, Krutmann J. Analysis of the mechanism of ultraviolet (UV) B radiation-induced prostaglandin E2 synthesis by human epidermoid carcinoma cells. J Invest Dermatol. 1993;101:528–31. doi: 10.1111/1523-1747.ep12365904. [DOI] [PubMed] [Google Scholar]
- 22.Soehnge H, Ouhtit A, Ananthaswamy HN. Mechanisms of induction of skin cancer by UV radiation. Front Biosci. 1997;2:d538–51. doi: 10.2741/a211. [DOI] [PubMed] [Google Scholar]
- 23.Melnikova VO, Ananthaswamy HN. Cellular and molecular events leading to the development of skin cancer. Mutat Res. 2005;571:91–106. doi: 10.1016/j.mrfmmm.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 24.Svobodova A, Zdarilova A, Maliskova J, Mikulkova H, Walterova D, Vostalova J. Attenuation of UVA-induced damage to human keratinocytes by silymarin. J Dermatol Sci. 2007;46:21–30. doi: 10.1016/j.jdermsci.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 25.Shreedhar V, Giese T, Sung VW, Ullrich SE. A cytokine cascade including prostaglandin E2, IL-4, and IL-10 is responsible for UV-induced systemic immune suppression. J Immunol. 1998;160:3783–9. [PubMed] [Google Scholar]
- 26.Barber LA, Spandau DF, Rathman SC, et al. Expression of the platelet-activating factor receptor results in enhanced ultraviolet B radiation-induced apoptosis in a human epidermal cell line. J Biol Chem. 1998;273:18891–7. doi: 10.1074/jbc.273.30.18891. [DOI] [PubMed] [Google Scholar]
- 27.Ma X, Bazan HE. Platelet-activating factor (PAF) enhances apoptosis induced by ultraviolet radiation in corneal epithelial cells through cytochrome c-caspase activation. Curr Eye Res. 2001;23:326–35. doi: 10.1076/ceyr.23.5.326.5445. [DOI] [PubMed] [Google Scholar]
- 28.Rajesh KG, Suzuki R, Maeda H, Murio Y, Sasaguri S. 5-HT2 receptor blocker sarpogrelate prevents downregulation of antiapoptotic protein Bcl-2 and protects the heart against ischemia-reperfusion injury. Life Sci. 2006;79:1749–55. doi: 10.1016/j.lfs.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 29.Capela JP, Ruscher K, Lautenschlager M, et al. Ecstasy-induced cell death in cortical neuronal cultures is serotonin 2A-receptor-dependent and potentiated under hyperthermia. Neuroscience. 2006;139:1069–81. doi: 10.1016/j.neuroscience.2006.01.007. [DOI] [PubMed] [Google Scholar]