

Summary
Addiction-associated behaviors such as drug craving and relapse are hypothesized to result from synaptic changes that persist long after withdrawal that are renormalized by drug reinstatement, although such chronic synaptic effects have not been identified. We report that exposure to the dopamine releaser methamphetamine for 10 days elicits a long-lasting (>4 month) depression at corticostriatal terminals which is reversed by methamphetamine readministration. Both methamphetamine-induced chronic presynaptic depression and the drug’s selective renormalization in drug-experienced animals are independent of corresponding long-term changes in synaptic dopamine release, but are due to alterations in D1 dopamine and cholinergic receptor systems. These mechanisms might provide a synaptic basis underlying addiction and habit learning and their long-term maintenance.
Introduction
Substance abuse is a chronic relapsing disorder in which drug reinstatement, even long after withdrawal, is thought to return the addict to a more stable, renormalized state (Ahmed and Koob, 2005; Koob, 1992; Redish, 2004). How drugs produce long-lasting neuroplastic changes, and how relapse provides compensation remains unknown, although a relationship between dopamine and corticostriatal synaptic activity is strongly implicated (Pessiglione et al., 2006; Vanderschuren and Kalivas, 2000). Most addictive drugs acutely increase synaptic dopamine, and in the case of the psychostimulants methamphetamine and amphetamine do so via stimulation-independent, non-vesicular reverse transport through the dopamine transporter and by inhibiting reuptake (Sulzer et al., 2005). The glutamatergic corticostriatal inputs are critical for the expression of behavioral and motoric responses (McFarland et al., 2003; Pessiglione et al., 2006; Pierce et al., 1996) and animals exposed to repeated psychostimulants exhibit enhanced behavioral responses to drug reinstatement long after withdrawal (Bickerdike and Abercrombie, 1997; Brady et al., 2005) with long-lasting reductions in basal extracellular glutamate and augmented glutamate release from corticostriatal inputs when the drugs are reinstated (McFarland et al., 2003; Pierce et al., 1996). Very long-lasting presynaptic effects of dopamine on the corticostriatal inputs that could contribute to habit formation, addiction, or allostatic renormalization, have not been reported, and we have taken advantage of new optical approaches to identify such changes.
Results
Repeated methamphetamine induces chronic presynaptic depression
To directly examine release from cortical terminals within the striatum (Figure 1A), we used the fluorescent tracer FM1-43 with multiphoton confocal microscopy in murine slice preparations. Stimulation of axons or cell bodies of projection neurons in layers 5–6 of the M1 motor cortex resulted in endocytosis of FM1-43 dye by recycling synaptic vesicles, revealing linear en passant arrays of fluorescent puncta characteristic of corticostriatal afferents (Bamford et al., 2004a; Bamford et al., 2004b). Following dye loading, cortical restimulation resulted in exocytosis of FM1-43 dye from the terminals, decreasing in a manner approximating first-order kinetics characteristic of synaptic vesicle fusion (Figure 1B). The kinetics of corticostriatal release were characterized by the halftime (t1/2), defined as the time required for terminal fluorescence to decay to half of its initial value.
Figure 1.
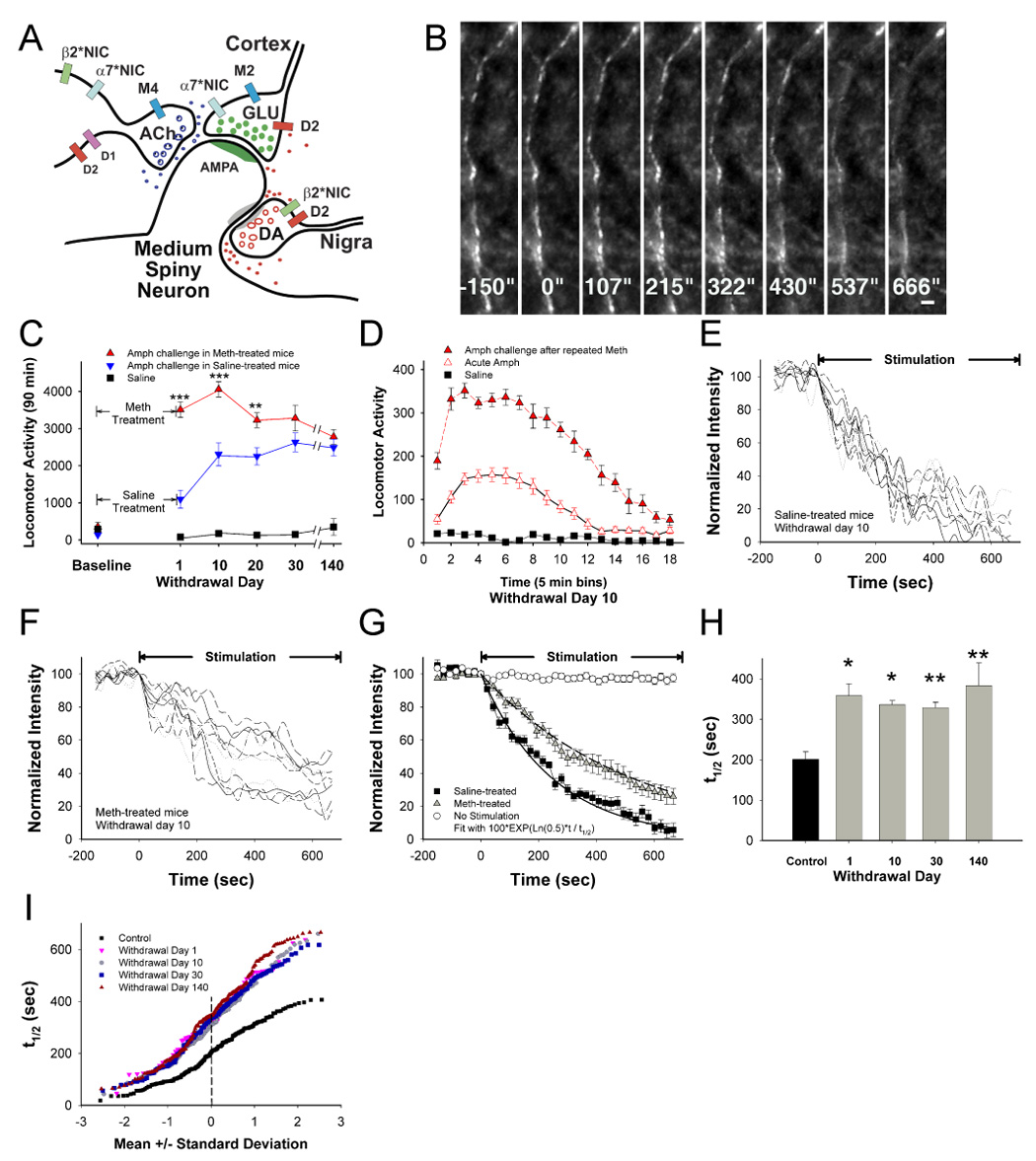
Chronic presynaptic depression (CPD). (A) In this simplified striatal microcircuit, dopaminergic (DA) nigrostriatal fibers and cholinergic (ACh) interneurons modulate excitatory glutamatergic (GLU) corticostriatal projections on medium spiny neurons. Neurotransmitter release is modified by D1 and D2 DA receptors, M2 and M4 muscarinic receptors and α7*- and β2*-nicotinic receptors. (B) Multiphoton images of corticostriatal terminals obtained from the forelimb motor striatum, located 1.0 – 1.5 mm from the site of cortical stimulation. Images captured every 21.5 seconds reveal en passant arrays of corticostriatal terminals. Restimulation at t=0 with 10 Hz pulses shows activity-dependent destaining of fluorescent puncta. Bar: 2 µm. (C) Amphetamine (AMPH; 2 mg/kg i.p.) -elicited locomotor activity measured by ambulation summed over 90 min was determined in mice following treatment with repeated saline or methamphetamine (METH) for 10 days. Repeated METH produced a 1370%–1970% increase in AMPH-elicited ambulation through 140 days of withdrawal (p<0.001, t-test with Bonferroni correction), significantly higher than saline-treated mice challenged with saline (F(5,70)=19; n=8 mice per condition; p<0.001). Repeated METH also produced a 12%–219% increase in ambulations compared to saline-treated mice also receiving AMPH challenges (F(5,70)=8.5; p<0.001, repeated measures ANOVA), although the difference between the two treatments narrowed after withdrawal day 20 (**p<0.01, ***p<0.001, ANOVA). All values are mean±SE. (D) AMPH-elicited locomotor activity 10 days following repeated METH was higher and of longer duration, when compared with responses from saline-treated mice challenged with AMPH (F(17,238)=9.1; n=8 mice per condition; p<0.001, repeated measures ANOVA). (E) Time-intensity analysis of FM1–43 destaining from individual puncta (n=8) in slices from saline-treated mice. Stimulation begins at t=0 sec. (F) FM1-43 destaining is depressed 10 days following repeated METH. (G) Mean±SE florescence intensity of puncta shown in panel E and F demonstrates preservation of 1st order release kinetics following repeated saline or METH. The plateau line represents fluorescence measurements in the absence of stimulation. (H) Repeated METH inhibits corticostriatal release halftimes (t1/2) over 140 days of withdrawal. n=4 mice per condition; *p<0.05, **p<0.01, t-test with Bonferroni correction. (I) Individual terminal responses from panel H are represented in a normal probability plot. All terminals were depressed during withdrawal.
We examined possible effects of repeated and intermittent methamphetamine administration on corticostriatal release. As the effects of methamphetamine and amphetamine on striatal dopamine transmission are identical and are not discriminated by humans, we chose methamphetamine, which is more widely available to drug abusers, to use for in vivo administration in mice. Mice were treated with saline (controls) or methamphetamine once per day (20 mg/kg/day i.p.) for 10 consecutive days. This dose of methamphetamine may mimic plasma levels reached with self-administration during “binges” (Davidson et al., 2005). Consistent with previous reports (Bickerdike and Abercrombie, 1997; Brady et al., 2005), repeated methamphetamine induced an enhanced locomotor response to an amphetamine challenge (2 mg/kg i.p.), 1–140 days following treatment (Figure 1C and 1D; p<0.001). In these animals, repeated methamphetamine inhibited corticostriatal release (Figure 1E – 1G), producing a highly prolonged state of corticostriatal depression in which the t1/2 for release increased by 63–90% during withdrawal (Figure 1H and 1I), an effect we term chronic presynaptic depression (CPD). When halftimes from individual terminals are presented relative to their standard deviation from the mean value, a straight line indicates a normally distributed (or single) population (Bamford et al., 2004b). Repeated methamphetamine produced CPD by inhibiting release from all terminals, shifting the population to a distribution that remained mostly normal (Figure 1I).
Drug reinstatement reverses CPD
We then examined corticostriatal activity during psychostimulant readministration. In saline-treated controls, we found a 33±12% depression of corticostriatal release in striatal slices prepared from mice challenged with a single dose of methamphetamine (20 mg/kg i.p., 30 min prior to sacrifice) in vivo (t1/2=273 vs. 203 sec for controls; Figure 2A; p<0.05). In striking contrast to controls, a methamphetamine challenge in vivo 10 days following repeated methamphetamine partially reversed CPD and potentiated release by 15%±2% (t1/2=335 vs. 285 sec following challenge; Figure 2A; p<0.05), an effect we term paradoxical presynaptic potentiation (PPP). Amphetamine also induced PPP in mice treated with a lower repeated dose of methamphetamine (t1/2=258 sec; 10 mg/kg/day, 10 days; Figure 2B) and did so by potentiating release from all terminals (Figure 2C and 2D).
Figure 2.
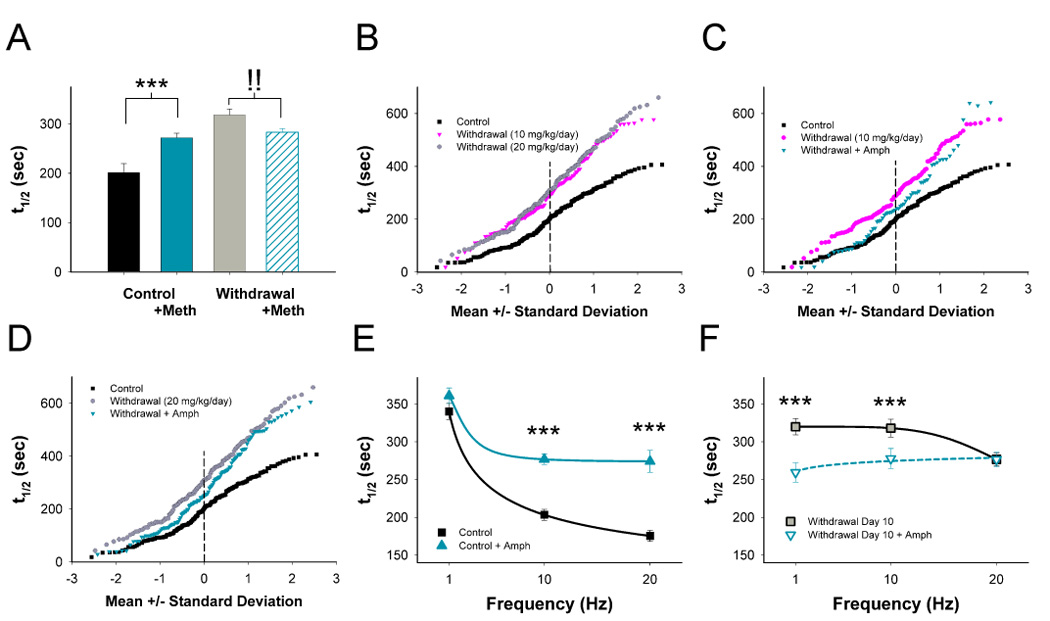
Paradoxical presynaptic potentiation (PPP). (A) A METH challenge in vivo decreases corticostriatal release in saline-treated controls (higher destaining halftime) but increases release on withdrawal day 10 following repeated METH. n=185–325 puncta per condition; ***p<0.01 compared to control without METH, !! p<0.01 compared to withdrawal without METH, Mann-Whitney. (B) Repeated METH at 10 and 20 mg/kg/day inhibits individual terminal responses on withdrawal day 10. An AMPH challenge 10 days following repeated METH at 10- (C) and 20 mg/kg/day (D) potentiated release from all terminals. Release half-times (t1/2) in slices from control (E) and METH–treated mice (F) on withdrawal day 10 following cortical stimulation at 1 Hz, 10 Hz and 20 Hz in the presence and absence of AMPH in vitro. ***p<0.001, Mann-Whitney.
Repeated methamphetamine abolishes frequency-dependent inhibition
Our previous studies demonstrate that the magnitude of dopamine’s inhibitory effect on corticostriatal activity is dependent on cortical stimulation frequency (Bamford et al., 2004b). We observed the effect of frequency-dependence by unloading corticostriatal terminals at 1 Hz, 10 Hz and 20 Hz before and after an amphetamine challenge (10 µM) in vitro. In saline-treated controls, amphetamine produced slower average unloading half-times at 10 Hz and 20 Hz (p<0.001) but not at 1 Hz (p>0.5; Figure 2E). The magnitude of dopamine inhibition became progressively greater at higher corticostriatal stimulation frequencies, with a 6% inhibition for the mean t1/2 values at 1 Hz (360/340 sec), a 26% inhibition at 10 Hz (276/203 sec), and a 36% inhibition at 20 Hz (275/175 sec; p<0.001 for interaction between amphetamine and stimulation frequency, F(2,1253)=7.6, two-way ANOVA). As such, dopamine provides low-pass frequency filtering at corticostriatal terminals.
On withdrawal day 10 following repeated methamphetamine (20 mg/kg/day, 10 days), terminal release was depressed at 10 and 20 Hz (p<0.001, repeated measures ANOVA; Figure 2F). Amphetamine in vitro accelerated release by 19% at 1 Hz (320/259 sec) and 13% at 10 Hz (318/277 sec) but had no effect at 20 Hz (276/276 sec; p<0.05 for interaction between amphetamine and stimulation frequency, F(2,1033)=5.3, two-way ANOVA). Thus, in contrast to controls where the greatest inhibitory effect of dopamine was seen at higher frequencies of stimulation, repeated methamphetamine produced the largest excitatory effect of dopamine at lower stimulation frequencies. Regardless of treatment or stimulation frequency, release closely approximated 1st order kinetics (r2>0.99; Supplemental Figure 1).
The depression in release following repeated methamphetamine was not due to inadequate FM1-43 loading of the recycling synaptic vesicle pool as loading stimulation frequencies of 1 Hz, 10 Hz, or 20 Hz (for 10 min) did not significantly affect unloading at 10 Hz either in saline-treated controls (t1/2=221 sec at 1 Hz, 203 sec at 10 Hz, and 234 sec at 20 Hz; not shown; n=82–391 puncta; p>0.5, Mann-Whitney) or following repeated methamphetamine (t1/2=300 sec at 1 Hz, 318 sec at 10 Hz, and 311 sec at 20 Hz; not shown; n=70–149 puncta; p>0.1, Mann-Whitney). Furthermore, the number of active terminals in each slice was similar following each loading frequency (not shown) and in both controls (38.1± 4puncta) and withdrawal (31.5±3 puncta; p=0.12, ANOVA). The reduced fractional release of label during exocytosis (Supplemental Figure 2) could be due to a reduced probability of recycling synaptic vesicles that undergo exocytic fusion per stimulus, a reduced amount of FM1-43 released per exocytic event, or a combination of these mechanisms.
Dopamine release is normal in methamphetamine-treated mice
We explored whether these repeated methamphetamine-induced changes in corticostriatal release relied on long-term changes in dopamine transmission. PPP could not depend on changes in dopamine neuronal firing, as it was measured in the striatal slice from which dopamine cell bodies were absent, but repeated methamphetamine might produce long-lasting changes in dopamine terminals. To test this possibility, we examined electrically evoked dopamine release and reuptake using cyclic voltammetry in the same preparation. Mice were treated with repeated saline or methamphetamine (20 mg/kg/day, 10 days). On withdrawal days 1, 10, 30 and 140, striatal slice preparations containing presynaptic dopamine terminals were stimulated by a single electrical pulse and the concentration and kinetics of dopamine release and reuptake were measured at sub-second resolution using fast-scan cyclic voltammetry as previously reported (Zhang and Sulzer, 2004). The only significant difference between saline and methamphetamine-treated mice in response to a single pulse stimulus was on withdrawal day 1, when evoked dopamine release was depressed by 57% (2.3 µM dopamine vs. 1.3 µM dopamine for controls and methamphetamine-treated mice, respectively; Supplemental Figure 3A; p<0.01). There was no change in evoked dopamine release on withdrawal day 10, 30 and 140.
We further examined mice for alterations in synaptic short-term presynaptic plasticity of the dopamine system. Dopamine release in response to train stimulus emulating phasic firing (4p and 10p at 100 Hz; Supplemental Figure 3B) was not altered on withdrawal day 1, 10, 30, or 140. The paired pulse ratio was not altered (Supplemental Figure 3C). The time constants for the fast component (τf) and the slow component (τs) were 4.9 sec and 16.7 sec respectively for withdrawal mice, and were no different than controls (6.6 sec and 16.5 sec respectively; p>0.5).
To confirm that we were not examining effects due to neurotoxicity in this protocol, mice were also treated with methamphetamine 10 mg/kg i.p. 4x at 2 hr intervals, an established neurotoxic regimen. As expected on withdrawal day 10, dopamine release was reduced to 39% of control values by this neurotoxic regimen (0.84 µM dopamine vs. 2.14 µM dopamine for controls and 4x methamphetamine-treated mice, respectively; Supplemental Figure 3D; p<0.001).
Finally, we examined amphetamine-induced dopamine release. The maximal level of striatal dopamine efflux reached ~8 µM within 6–20 min (Supplemental Figure 3E), similar to responses in untreated mice (Bamford et al., 2004b), confirming that a psychostimulant challenge elicits typical maximum levels of dopamine release in this preparation during withdrawal. Thus, although effects of methamphetamine on dopamine release apparently initiate CPD, the maintenance of CPD and PPP is apparently not due to changes in the ability of nigrostriatal terminals to release dopamine.
The lack of alterations in dopamine reuptake, short-term presynaptic plasticity, or the concentration of dopamine released by amphetamine detected during withdrawal indicates that repeated methamphetamine induces no long-lasting presynaptic alterations in dopamine neurotransmission. Thus, while increased dopamine transmission due to methamphetamine may have initiated long-term changes, the maintenance of CPD and the ability to produce PPP during withdrawal did not rely on an ongoing presynaptic alteration of dopamine transmission. The results further indicate that the protocols had no long-term neurotoxic effect on dopamine terminals.
Psychostimulants filter corticostriatal release via D2 receptors
Our previous results in the striatum of untreated mice showed that amphetamine inhibited exocytosis from less active corticostriatal terminals via activation of D2 receptors (D2R) (Bamford et al., 2004a; Bamford et al., 2004b). In saline-treated mice, a methamphetamine challenge in vivo depressed corticostriatal exocytosis (t1/2=272 vs. 201 sec for controls; Figure 3A and 3B; p<0.05). Similarly, acute amphetamine in vitro also decreased corticostriatal release (t1/2=263 vs. 203 sec for untreated slices; not shown; n=188–305 puncta; p<0.001, Mann-Whitney).
Figure 3.
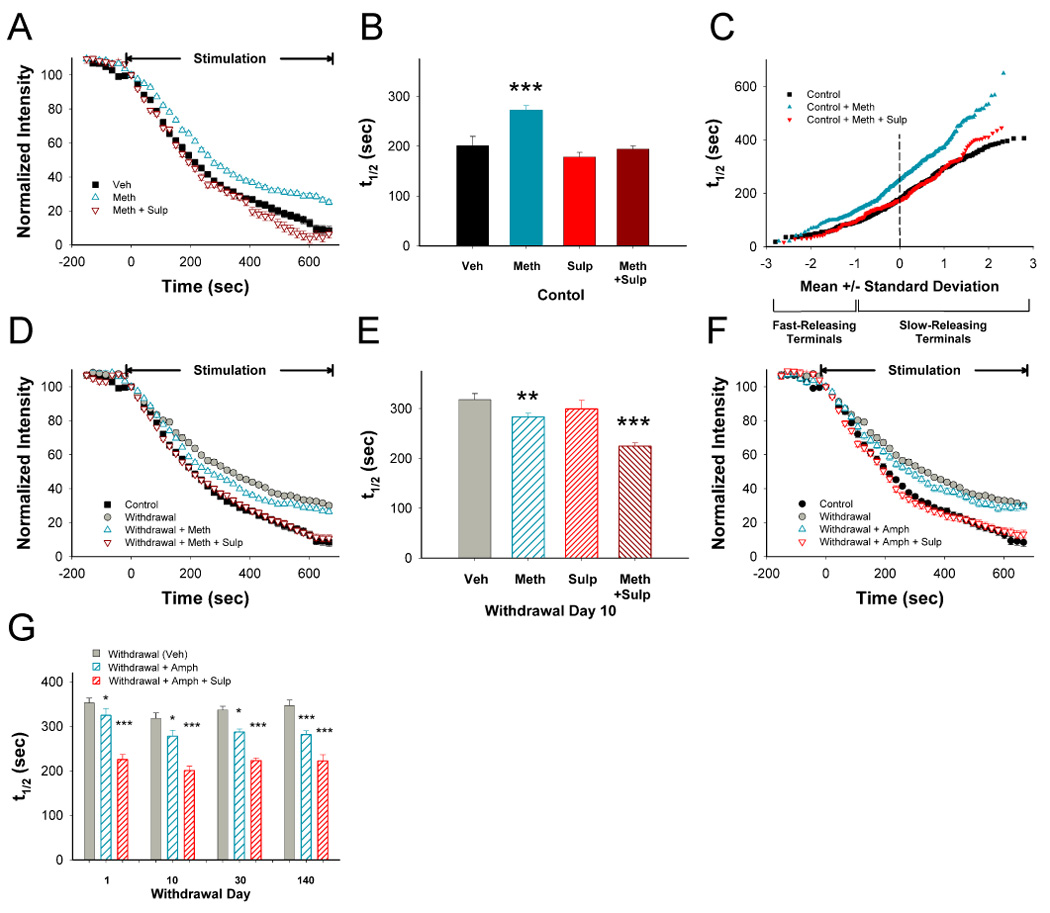
D2 receptors (D2R) remain inhibitory following repeated METH. (A) In slices prepared from mice treated with repeated saline, a METH challenge in vivo produced inhibition of FM1-43 destaining that was reversed by the D2R antagonist sulpiride (SULP) in vitro. (B) Distribution of mean t1/2 of release for FM1-43 destaining curves shown in panel A. n=188–325 puncta; ***p<0.001 compared to control (Veh), Mann-Whitney. (C) Individual terminal responses in saline-treated controls following a challenge with METH in vivo with and without SULP. Repeated methamphetamine produced more inhibition at the slowest-releasing terminals (greater t1/2). (D) On withdrawal day 10 following repeated METH, a METH challenge in vivo accelerated corticostriatal release. The addition of SULP in vitro further accelerated release to control halftimes. (E) Distribution of mean t1/2 for destaining curves shown in panel D. n=149–362 puncta; **p<0.01, ***p<0.001 compared to vehicle (Veh), Mann-Whitney. (F) On withdrawal day 10 following repeated METH, AMPH in vitro induced PPP while AMPH in combination with SULP normalized release. (G) Following repeated METH, AMPH in vitro induced PPP over 140 days of withdrawal while AMPH in combination with SULP normalized release. n=167–368 puncta for each condition; *p<0.05, **p<0.01 compared to Veh from the same withdrawal day, Mann-Whitney.
In controls, the D2R antagonist sulpiride (10 µM) in vitro slightly potentiated terminal release (t1/2=179 vs. 201 sec without sulpiride; Figure 3B; p>0.5), indicating some tonic activation of inhibitory D2R. However, sulpiride completely blocked inhibition by a methamphetamine challenge (t1/2=194 vs. 272 sec for methamphetamine in vivo with and without sulpiride in vitro; Figure 3A, 3B, and Supplemental Figure 4; p<0.001). A methamphetamine challenge in vivo created two reversible populations of terminals that diverged at −1 standard deviation below the mean, preferentially inhibiting slow-releasing terminals (~80%; Figure 3C). Thus, a methamphetamine challenge in vivo or amphetamine in vitro produced a D2R-dependent filter with filtering applied preferentially to terminals with the lowest probability of release.
D2 receptors remain inhibitory in methamphetamine withdrawal
We determined the effect of repeated methamphetamine on D2R-mediated corticostriatal filtering. On withdrawal day 10 following repeated methamphetamine, a methamphetamine challenge in vivo produced PPP (t1/2=335 vs. 285 sec following the challenge; Figure 3D and 3E; p<0.05). Similarly, an amphetamine challenge in vitro also potentiated release on withdrawal days 1–140 (Figure 3F and 3G).
On withdrawal day 10, sulpiride slightly potentiated terminal release (t1/2=299 vs. 335 sec without sulpiride; Figure 3E; p>0.3). However, it enhanced, rather than reversed PPP following a methamphetamine challenge in vivo, increasing corticostriatal release to control values (t1/2=227 sec; Figure 3D and 3E; p>0.5 compared to controls). Sulpiride also enhanced PPP due to amphetamine in vitro, potentiating release to control values (t1/2=203 sec; p>0.5) on withdrawal day 1–140 (Figure 3F, 3G and Supplemental Figure 4). Thus, in animals treated with repeated methamphetamine, a methamphetamine challenge in vivo or an amphetamine challenge in vitro induced PPP to partially normalize corticostriatal release, and PPP completely reversed CPD once D2R inhibition was blocked. The results demonstrate that PPP was not due to an activation of D2Rs since these receptors continued to be inhibitory during withdrawal.
CPD is reversed through D1 receptor actions
An alternate possibility is that psychostimulant activation of D1 receptors (D1R) might induce PPP. As in our previous studies (Bamford et al., 2004a; Bamford et al., 2004b), the D1R agonist SKF38393 (10 µM; t1/2=186 vs. 203 sec without SKF38393; p>0.5) or antagonist SCH23390 (1 µM; t1/2=193 sec; p>0.5) had little effect on corticostriatal release in saline-treated controls (Figure 4A and 4B). Furthermore, SCH23390 had no effect on corticostriatal release even when dopamine was released by amphetamine (t1/2=262 vs. 262 sec without SCH23390; Figure 4B; p>0.5). Thus, D1R stimulation did not significantly affect corticostriatal activity under control conditions.
Figure 4.
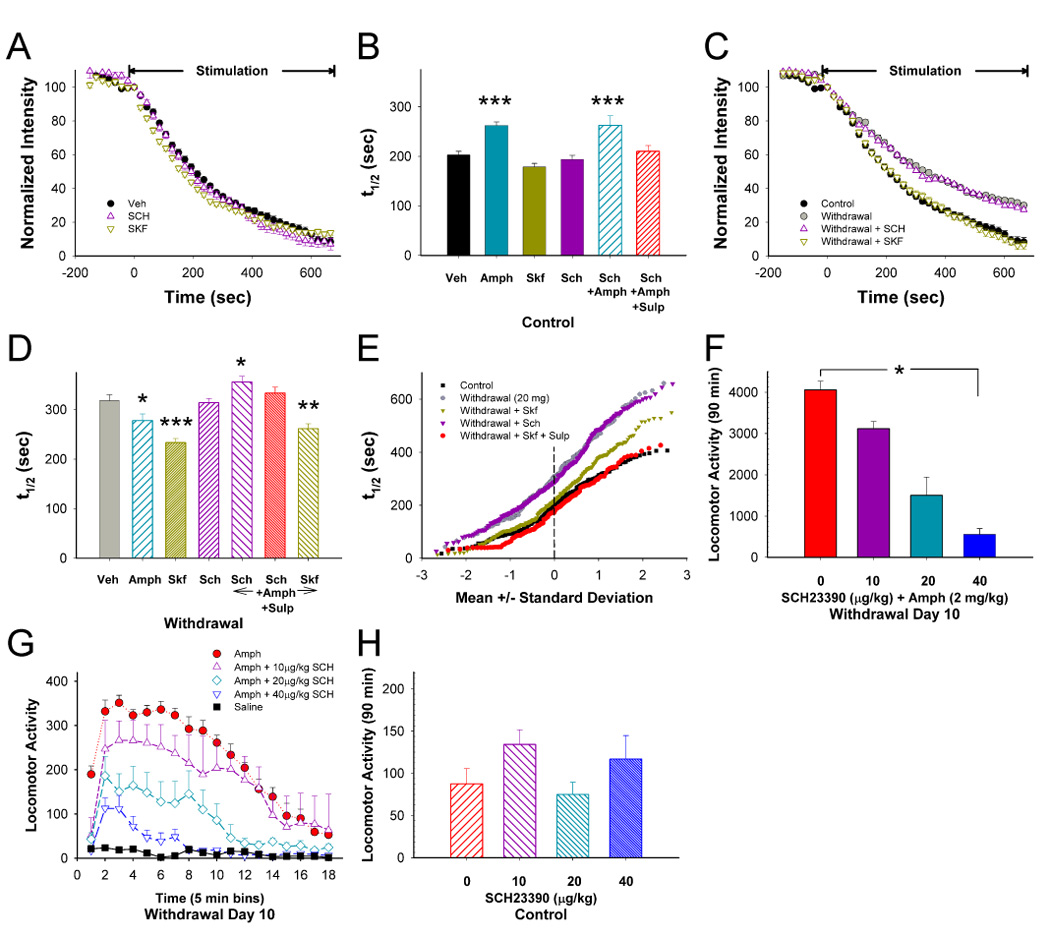
D1 receptor (D1R) stimulation reverses CPD. (A) Compared to untreated sections (Veh), the D1R agonist SKF38393 (SKF; n=169 puncta) and antagonist SCH23390 (SCH; n=386 puncta) in vitro had no effect on release in controls following repeated saline. (B) Distribution of mean t1/2 of release for destaining curves shown in panel A with additional experimental groups from controls. Compared to untreated sections (Veh; n=188 puncta), AMPH (n=305 puncta) inhibited release, but the D1R agonist SKF (n=169 puncta) and antagonist SCH (n=386 puncta) had no effect. In the presence of AMPH, SCH had no effect with (n=116 puncta) or without sulpiride (SULP; n=151 puncta). ***p<0.001 compared to Veh, Mann-Whitney. (C) 10 days following repeated METH (withdrawal), SKF accelerated release whereas SCH had no effect. (D) Distribution of mean t1/2 of release for destaining curves shown in panel C with additional experimental groups from withdrawal. AMPH in vitro (n=128 puncta) boosted release to elicit PPP. SKF (n=247 puncta) increased release to a greater extent than AMPH whereas SCH (n=266 puncta) had no effect. SCH (n=212 puncta) blocked the potentiating effect of AMPH. SCH in combination with SULP (n=161 puncta) also blocked accelerated release by AMPH whereas SKF (n=168 puncta) had little effect. *p<0.05, **p<0.01; ***p<0.001 compared to Veh (n=149 puncta), Mann-Whitney. (E) Individual terminal responses to D1 and D2R manipulation in withdrawal. (F) Mice were treated with METH (20 mg/kg/day i.p.) for 10 days. An AMPH challenge (2 mg/kg i.p.) on withdrawal day 10 induced sensitized locomotor ambulations summed over 90 min. The D1R antagonist SCH inhibited this locomotor response (*p<0.001; n=8 mice per treatment group) with a significant linear trend over dose levels (r2=0.97). (G) Interval locomotor responses for treatment groups in panel F. (H) Additional mice were treated with saline for 10 days. 10 days later, these mice were treated with the D1R antagonist SCH and challenged with saline. There were small variations in locomotor activity but at the doses used, SCH had no effect on locomotor activity (p=0.48; n=8 mice per treatment group; r2=0.01).
In marked contrast, on withdrawal day 10 following repeated methamphetamine, the D1R agonist SKF38393 strongly potentiated release and partially reversed CPD (t1/2=233 vs. 318 sec without SKF38393; Figure 4C and 4D; p<0.001) by renormalizing the activity of the faster releasing terminals (Figure 4E), whereas the D1R antagonist SCH23390 had no effect (t1/2=313 sec; Figure 4C – 4E; p>0.5). As expected, SCH23390 largely blocked the excitatory response produced with SKF38393 (t1/2=289 sec for SCH23390 and SKF38393; not shown; n=113 puncta; p>0.5 compared to SCH23390 alone, Mann-Whitney). The combination of sulpiride and SKF38393 further enhanced release and fully reversed CPD (t1/2=202 sec; p>0.5 compared untreated sections) by additionally accelerating exocytosis from slower terminals (Figure 4E). Combined SKF38393 and sulpiride also reversed CPD in mice treated with lower doses of methamphetamine (10 mg/kg/day; t1/2=225 vs. 307 sec without SKF38393 and sulpiride; not shown; n=250 puncta; p<0.001, Mann-Whitney). Amphetamine-induced D1R activation was responsible for PPP, as PPP was occluded by SCH23390 (t1/2=356 sec; p<0.001), even when sulpiride, which might be expected to enhance release by blocking any lingering D2R-mediated inhibition, was included with SCH23390 (t1/2=333 sec; Figure 4D; p<0.01). The excitatory effects of SKF38393 on amphetamine-induced PPP were not additive (t1/2=265 sec; p=0.04 compared to SKF38393 alone), and were identical to amphetamine alone (t1/2=263 sec; Figure 4D; p>0.5). Together, the results show that while D1R have no effect on corticostriatal release in controls, their actions become excitatory following repeated methamphetamine. Amphetamine has less excitatory effect than the D1R agonist as dopamine would also inhibit release through presynaptic D2R actions.
Locomotor activity is dependent on a new D1R effect
Since a psychostimulant challenge in withdrawal would produce striatal excitation and allow excessive locomotor responses through a D1R–mediated pathway, blockade of this receptor might prevent these ‘sensitized’ behavioral responses. Consistent with previous reports (Kuribara, 1995), we found that increasing concentrations of the D1R antagonist SCH23390 (10–40 µg/kg s.c.; 30 min prior to an amphetamine challenge) produced a dose-dependent reduction in locomotor responses to an amphetamine challenge (2 mg/kg) on withdrawal day 10 (Figure 4F and 4G; p<0.001), but had no effect on saline-treated controls (Figure 4H; p>0.5). Thus, both augmentation of corticostriatal release and enhanced locomotion are dependent on a new D1R effect that is seen only following repeated exposure to methamphetamine.
CPD and PPP are mediated through acetylcholine receptors
While D1R activation reversed CPD and mediated PPP, the results did not reveal where the responsible D1R was acting. We suspected that CPD and PPP might be mediated indirectly through cholinergic tonically active interneurons (TANs) that represent a small fraction of striatal neurons but provide the majority of striatal acetylcholine (ACh) transmission. Amphetamine exerts multiple state-dependent effects on striatal extracellular ACh efflux (DeBoer and Abercrombie, 1996). TANs possess D1R and D2R (DeBoer and Abercrombie, 1996; Yan et al., 1997), and their activity mediates corticostriatal responses including dopamine-dependent corticostriatal long-term depression (LTD) (Wang et al., 2006) via β2*- and α7*-type nicotinic receptors (nAChRs) on TANs (Azam et al., 2003), and α7* receptors found on corticostriatal terminals (Marchi et al., 2002; Pakkanen et al., 2005; Wang and Sun, 2005) that exert tonic excitation, and M2-type muscarinic ACh receptors (mAChRs) that are inhibitory (Calabresi et al., 2000; Hersch et al., 1994; Volpicelli-Daley et al., 2003; Zhang et al., 2002). nAChRs are rapidly desensitized at high agonist levels, in which case the agonists prevent tonic excitation and thus inhibit release (Wooltorton et al., 2003).
In slices from saline-treated mice, bath application of ACh (1–100 µM) potently inhibited release (Figure 5A), consistent with either mAChR-mediated depression and/or a desensitization of tonically activated nAChR (Quick and Lester, 2002). We determined that tonic ACh in controls was excitatory because vesamicol (5 µM), a potent inhibitor of vesicular ACh uptake, inhibited corticostriatal release in controls (t1/2=298 sec; n=135 puncta; not shown; p<0.001, Mann-Whitney) to a degree similar to CPD (318 sec; p>0.5, Mann-Whitney).
Figure 5.
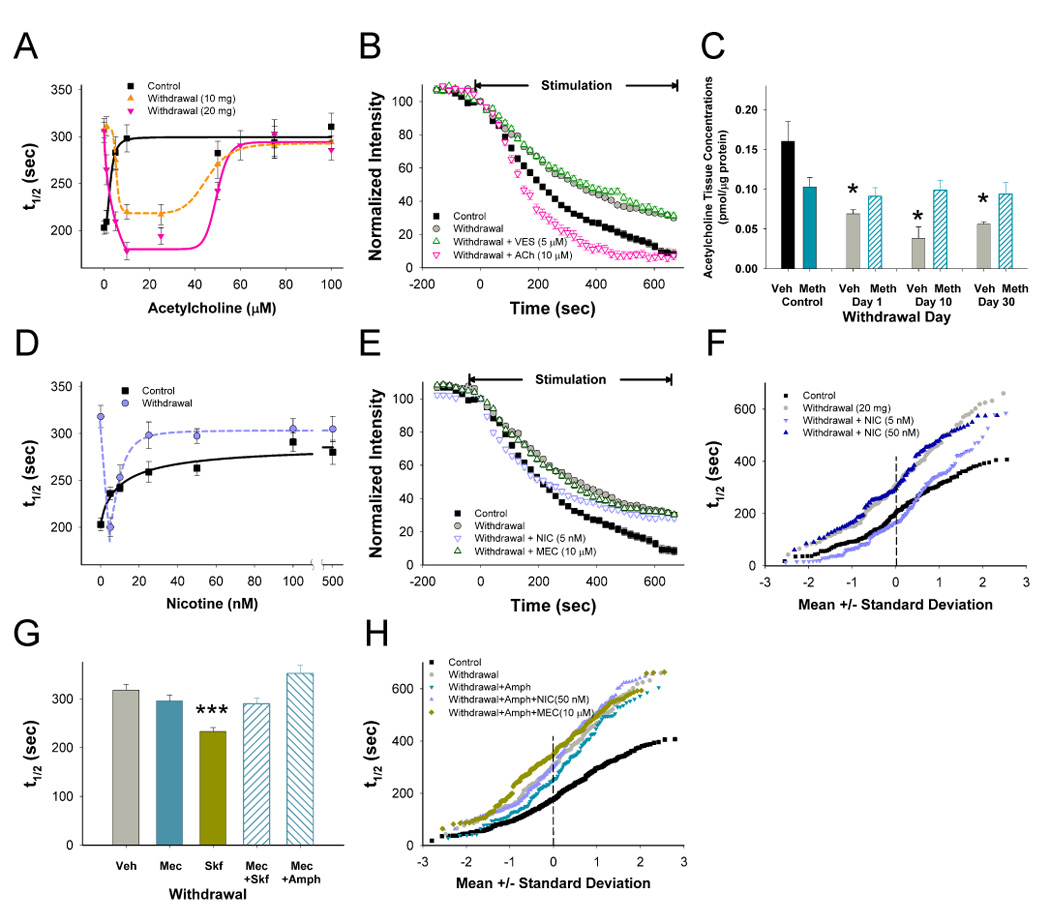
CPD and PPP are regulated through nAChRs. (A) Terminal release over a range of acetylcholine (ACh) concentrations 10 days following repeated saline- (control) and METH (withdrawal; 10 and 20 mg/kg/day, 10 days; n=30–381 puncta). (B) 10 days following repeated METH (20 mg/kg/day), vesamicol (VES) had little effect on CPD while ACh potentiated release to a greater extent than controls. (C) Striatal tissue concentrations of ACh, measured by HPLC, remained depressed during METH withdrawal. *p <0.01 compared to untreated control mice (Veh; n=8 slices from 4 mice; t-test). (D) In control slices, increasing concentrations of nicotine (NIC) inhibited release (t1/2=240 sec at IC50=3.52 nM; n=104–299 puncta). 10 days following repeated METH, release was accelerated at low concentrations of NIC (5 nM) but higher concentrations of NIC rapidly decreased release (IC50=12.5 nM; n=77–190 puncta). (E) On withdrawal day 10, low NIC concentrations accelerated release whereas the nAChR channel blocker mecamylamine (MEC) had little effect on CPD. (F) Individual terminal responses during withdrawal for low (5 nM) and high (50 nM) concentrations of NIC. (G) During withdrawal, MEC prevented potentiation of release by SKF and AMPH (n=149–247 puncta; ***p<0.001, Mann-Whitney). (H) Individual terminal responses during withdrawal demonstrate inhibition of AMPH-induced PPP by both NIC and MEC (n=60–188 puncta). Concentration dependence curves were fit with a Hill equation.
These cholinergic receptor responses were markedly altered by repeated methamphetamine. Low concentrations of bath-applied ACh reversed CPD in withdrawal and accelerated release beyond control halftimes (t1/2=178 sec at 10 µM ACh vs. 203 sec for controls; Figure 5A and 5B; p<0.05) suggesting a sensitized excitatory response to exogenous ACh. ACh also accelerated release on withdrawal day 10 following a lower daily dose of methamphetamine (10 mg/kg/day, 10 days; Figure 5A). Higher concentrations of ACh (>50 µM) expected to desensitize nAChR (Quick and Lester, 2002), inhibited release (Figure 5A). Whereas ACh depletion by vesamicol inhibited release in controls, it had no effect on CPD (t1/2=332 sec; n=126 puncta; Figure 5B; p>0.5, Mann-Whitney), confirming a loss of tonic excitatory ACh response in withdrawal.
As reductions in tonic ACh can rapidly enhance striatal nAChR (Pakkanen et al., 2005; Wooltorton et al., 2003; Zhou et al., 2001) and mAChR (Volpicelli-Daley et al., 2003) sensitivity, we examined the effects of repeated methamphetamine on striatal tissue ACh content. In saline treated mice, a methamphetamine challenge (20 mg/kg i.p., 30 min before sacrifice) decreased ACh content by 35% (p<0.05, t-test), while repeated methamphetamine decreased striatal ACh during withdrawal by 46%–76% (p<0.01), an effect partially reversed following methamphetamine reinstatement (Figure 5C; p<0.05, t-test).
Loss of nicotinic excitation results in CPD
This methamphetamine-induced reduction in ACh would likely perturb both nAChR and mAChR responses. In saline-treated controls, the classic nAChR agonist, nicotine (5–500 nM), inhibited corticostriatal release (Figure 5D), consistent with the compound’s ability to rapidly desensitize β2*-nAChR (Quick and Lester, 2002; Wooltorton et al., 2003) and prevent ongoing corticostriatal excitation by tonic ACh. Corticostriatal release is dependent on tonic excitation by nAChR as the nAChR antagonist mecamylamine reduced release (10 µM; t1/2=295 vs. 203 sec for controls; n=168 puncta; not shown; p<0.001, Mann-Whitney). Tonic nAChR excitation appeared due to actions at α7*-like nAChRs as the α7* antagonist methyllycaconitine (20 nM) inhibited corticostriatal release (t1/2=278 sec; n=186 puncta; p<0.001, Mann-Whitney). Likewise, choline (10 mM), an agonist that desensitizes α7*-nAChR (Turner, 2004), inhibited release in slices from saline-treated controls (t1/2=435 sec vs. 203 sec for controls; n=66 puncta; not shown; p<0.001, Mann-Whitney). In addition, the β2*- nAChR antagonist dihydro-β-erythroidine (DHβE; 300 nM) also reduced release (t1/2=279 sec; not shown; n=97 puncta; p<0.001, Mann-Whitney).
In contrast to controls, low concentrations of nicotine (5 nM) 10 days following repeated methamphetamine reversed CPD (t1/2=200 vs. 203 sec for controls; Figure 5D and 5E; p>0.5, Mann-Whitney) via a strong excitatory response that normalized release for all but the ~20% slowest terminals (Figure 5F). As expected, this effect was blocked by the β2*-nAChR antagonist DHβE (t1/2=317 sec; n=122 puncta; not shown; p>0.5, Mann-Whitney). Similar to bath-applied ACh, this potentiation was lost at higher nicotine levels (Figure 5D and 5F), consistent with β2*-nAChR desensitization (Wooltorton et al., 2003). Tonic nAChR excitation was not observed in methamphetamine withdrawal, as the nicotinic receptor blocker mecamylamine (t1/2=295 sec vs. 318 sec with and without mecamylamine; Figure 5E and 5G; p>0.5), the desensitizing α7*-nAChR agonist choline (t1/2=326 sec; n=127 puncta; not shown; p>0.5, Mann-Whitney) and the β2*-nAChR antagonist DHβE (t1/2=302 sec; n=99 puncta; not shown; p>0.5, Mann-Whitney) no longer inhibited release as they did in controls. AChR-induced PPP occurred downstream of D1R action, as mecamylamine blocked PPP elicited by the D1 agonist SKF38393 (t1/2=233 sec for SKF38393 vs. 290 sec for SKF38393 with mecamylamine; Figure 5G; p<0.001) and by amphetamine (t1/2=277 for amphetamine vs. 352 sec for amphetamine with mecamylamine; Figure 5G and 5H; p<0.001) as did desensitizing nicotine levels (50 nM; t1/2=330 sec for amphetamine and nicotine; Figure 5H; p<0.001).
Muscarinic receptors become sensitized during withdrawal
Next, we examined the effect of repeated methamphetamine on mAChR responses. In slices from saline-treated mice, the mAChR agonist muscarine (Figure 6A) inhibited release, while the antagonist, atropine (1–20 µM) had no effect (Figure 6B), indicating that tonic ACh did not inhibit corticostriatal activity via mAChR. Thus, in controls, tonic ACh exerts no inhibition at mAChR while providing ongoing excitation at nAChRs.
Figure 6.
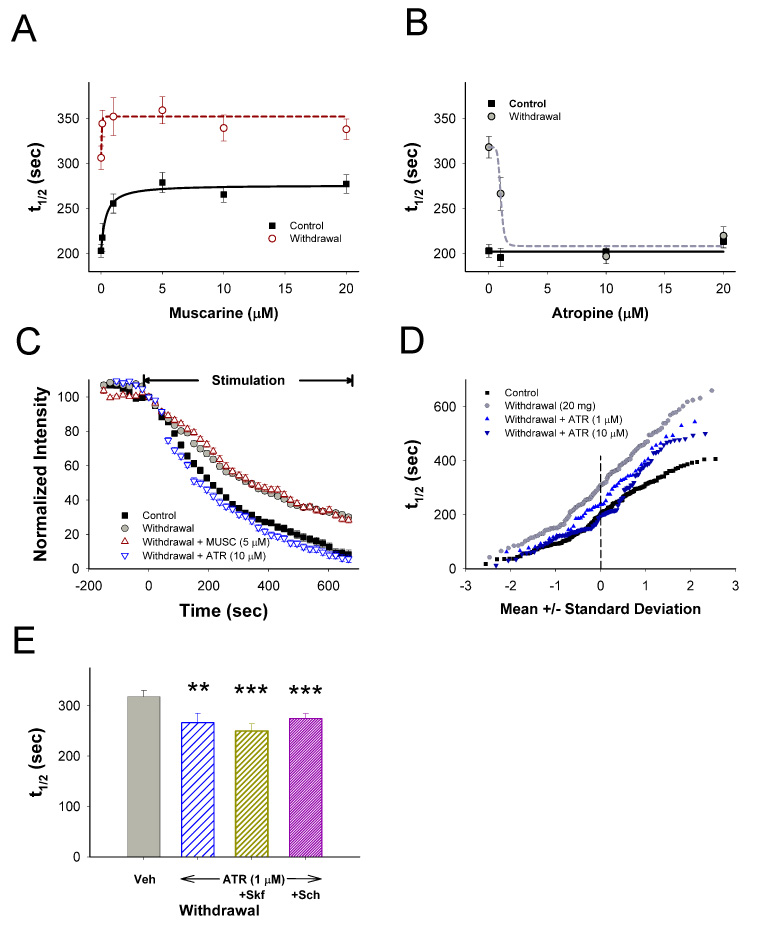
CPD develops through sensitized mAChRs. (A) Terminal release over a range of muscarinic (MUSC) concentrations from slices prepared from saline- (control) and METH-treated mice (withdrawal) on withdrawal day 10. MUSC inhibited release to a greater extent and at a lower dose in withdrawal (t1/2=342 sec at IC50=0.01 µM; n=57–176 puncta) than controls (t1/2=276 sec at IC50=0.38 µM; n=86–265 puncta). (B) Atropine (ATR) accelerated release (t1/2=263 sec at EC50=1.02 µM; n=55–254 puncta) in withdrawal but had no effect in controls (n=77–254 puncta). (C) MUSC inhibited release whereas ATR potentiated release in withdrawal. (D) Individual terminal responses from withdrawal mice with and without ATR (1 and 10 µM; n=55–381 puncta) are compared to controls. (E) In the presence of ATR (1 µM; n=155 puncta), SKF (n=94 puncta) and SCH (n=142 puncta) had little effect on corticostriatal release during METH withdrawal. **p<0.01, ***p<0.001 compared to Veh, Mann-Whitney.
Muscarine continued to be inhibitory in withdrawal (Figure 6A and 6C), but reached a maximum effect at a lower concentration (78% of maximal inhibition at 0.1 µM in controls vs. 98% of maximal inhibition in withdrawal; Figure 6A; p<0.001), consistent with hypersensitive mAChR responses. However, atropine reversed CPD (Figure 6B and 6C) at all varicosities except the slowest ~20% of the population (Figure 6D) a state nearly identical to that following the D1 agonist, SKF38393 (Figure 4E), or low concentrations of nicotine (Figure 5F) or ACh (10 µM; not shown).
Together, these data indicate that during withdrawal, low tonic ACh levels were associated with sensitized responses by both nAChR and mAChR. The sensitized mAChR response contributed to CPD and occurred downstream of D1R action, as atropine (1 µM) reversed CPD in the presence of either SKF38393 or SCH23390 (Figure 6E). The mAChR response was upstream of nAChR excitation, as both desensitizing concentrations of nicotine (50 nM; t1/2=310 vs. 196 sec for atropine (10 µM) alone; n=131 puncta; p<0.001, Mann-Whitney) and mecamylamine (t1/2=324 sec; not shown; n=101 puncta; p<0.001, Mann-Whitney) prevented atropine potentiation during withdrawal. mAChR activation, however, played no role in PPP, as atropine did not block amphetamine excitation in withdrawal (t1/2=278 sec for amphetamine vs. 248 sec with amphetamine and atropine (10 µM); not shown; n=128 puncta; p>0.5, Mann-Whitney).
Thus, withdrawal mice selectively exhibited two, long-lasting forms of methamphetamine-induced presynaptic corticostriatal plasticity. CPD is due to a tonic inhibition mediated by reduced tonic nAChR excitation combined with a tonic mAChR inhibition, whereas PPP is due to psychostimulant-induced D1 activation that boosts corticostriatal release by activating nAChRs. These results are consistent with evidence that both nAChR and mAChR sensitivity are strongly regulated by ACh input, with low ACh levels generally promoting supersensitivity (Overstreet and Djuric, 2001). This balance between opposing ACh effects is altered by methamphetamine-induced sensitized nAChR and mAChR responses. As was observed following simulation of PPP by low nicotine levels, withdrawal mice are very sensitive to nAChR excitation, although higher nicotine or ACh levels cause desensitization and eliminate PPP.
CPD and PPP in post-synaptic medium spiny neurons
We expected that changes in glutamate release from cortical afferents during CPD and PPP would be reflected in post-synaptic medium spiny neurons. Mice were treated with saline (n=8) or methamphetamine (20 mg/kg/day i.p.; n=9) for 10 days. Recordings from medium spiny neurons in voltage-clamp mode (n=28 from saline- and n=31 from methamphetamine-treated mice), obtained 10 days after the last injection, revealed no differences in passive membrane properties between groups (membrane capacitance 97.5±3.3 and 93.8±2.4 pF, input resistance 87.0±4.4 and 92.9±8.4 MΩ, time constant 1.5±0.1 and 1.6±0.1 ms, respectively). The average frequency of spontaneous excitatory post-synaptic currents (sEPSCs; Figure 7A left and 7B) was higher in cells from saline- compared to methamphetamine-treated mice (1.17±0.11 Hz and 0.94±0.07 Hz, p=0.036), providing electrophysiological evidence supporting CPD. In a subset of cells (n=6 from saline- and n=7 from methamphetamine-treated mice) tetrodotoxin (TTX; 1 μM) was used to isolate miniature (m) EPSCs (Figure 7A right and 7C). In this group the frequency of sEPSCs also was significantly higher in saline- than in methamphetamine-treated animals (p=0.033). After TTX the average frequency of mEPSCs (Figure 7C) remained significantly higher (p=0.047) in cells from control (1.2±0.2 Hz) compared to methamphetamine-treated (0.7±0.1 Hz) mice. Differences in frequency were more dramatic after TTX, indicating that in the absence of this blocker cortical pyramidal neuron firing may be increased in methamphetamine-treated mice compared to controls, possibly as a compensatory mechanism. In contrast, average mEPSCs amplitudes were similar between groups (10.4±0.9 pA in cells from saline-and 8.8±0.8 pA in cells from methamphetamine-treated animals). This indicates that in methamphetamine-treated mice there is a depression of synaptic transmission in the corticostriatal pathway, and this depression is independent of action potentials as it persists in the presence of TTX. Evidence for reduced glutamate transmission was also obtained from evoked EPSCs. The current required to evoke EPSCs (Figure 7D) was significantly higher in cells from methamphetamine-treated (0.46±0.05 mA) than in cells from saline-treated (0.32±0.04 mA) animals (p=0.021). The average evoked EPSC amplitude was determined at threshold intensity + 0.1 mA in cells from saline and methamphetamine-treated mice. At 0.42 mA the average EPSC amplitude in control cells was −104.3±11.7 pA (n=18) and at 0.56 mA the amplitude in methamphetaminetreated cells was −93.3±10.8 pA (n=23). Thus, in order to obtain comparable responses, higher intensities need to be used in methamphetamine-treated than in control mice, providing further evidence of CPD.
Figure 7.
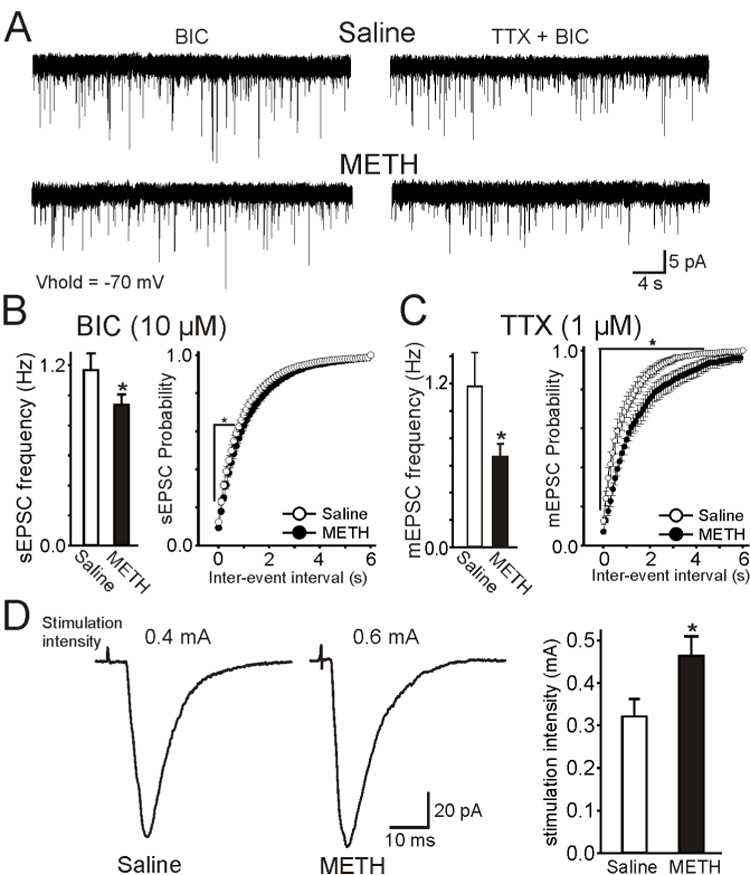
Response to CPD in medium spiny neurons (MSNs). (A) Traces represent spontaneous (s) EPSCs in the presence of bicuculline (BIC, 10 µM, a GABAA receptor blocker) alone (left) or BIC and tetrodotoxin (TTX; right) in MSNs from saline- and METH-treated animals at a holding potential of −70 mV. (B) In the presence of BIC only, there was a small but significant reduction of sEPSCs in cells from METH- compared to saline-treated mice. Histogram on the right is a cumulative inter-event interval distribution of sEPSCs. Intervals were significantly different (p<0.05). (C) In a subset of cells TTX was added to isolate mEPSCs. After TTX, there was a significant decrease in mEPSC frequency in cells from METH- compared to saline-treated mice. Histogram on the right is a cumulative inter-event interval distribution of mEPSCs. (D) Responses evoked in MSNs by stimulation of the cortical layers in saline- and METH-treated animals. More stimulation intensity was needed to induce responses of similar amplitude in cells from METH- than in cells from salinetreated mice. Traces represent the average of 3 responses. The graph on the right indicates that the threshold current required to induce responses was significantly higher in cells from METH- compared to saline-treated mice. Student’s t-tests or ANOVAs were used for group comparisons. Asterisks indicate differences were statistically significant (p<0.05).
To determine if PPP also could be demonstrated in postsynaptic neurons, amphetamine (10 µM) was bath applied to examine its effects on sEPSCs. Amphetamine produced a small reduction (3%) in average frequency of sEPSCs in cells (n=5) from saline-treated mice while it significantly increased the frequency (34%) in cells (n=8) from methamphetamine-treated mice (p=0.02, Supplemental Figure 5A). Further, PPP was likely mediated by D1 receptors as bath application of the D1R agonist SKF38393 (10 µM) produced no significant change (7% increase) in the frequency of sEPSCs in cells (n=6) from saline-treated mice but significantly increased (34% increase) the frequency in cells (n=7) from methamphetamine-treated mice (p=0.015; Supplemental Figure 5B). As expected, the D1R antagonist SCH23390 (1 µM) had no effect on the frequency of sEPSCs (n=5 cells from saline- and n=6 cells from methamphetamine-treated animals; Supplemental Figure 5C). In contrast, bath application of the D2R antagonist sulpiride (10 µM) significantly increased the frequency of sEPSCs in both groups (58% in saline- and 28% in methamphetamine-treated animals, p=0.007 and p=0.015 respectively; Supplemental Figure 5D). However, addition of amphetamine produced a further increase (12%) in cells from methamphetamine-treated mice whereas it reduced (10%) the frequency in cells from control mice (not shown). Overall, these electrophysiological data support the optical recordings of presynaptic release and demonstrate that CPD and PPP produce alterations in the excitation of post-synaptic neurons.
Discussion
We report that repeated methamphetamine treatment causes long-lasting synaptic changes in the corticostriatal pathway that were previously suggested by theoretical models to underlie drug dependence. The CPD induced by the drug occurs at corticostriatal terminals and is independent of long-term changes in striatal dopamine terminals. PPP by drug reinstatement occurs both in vivo and in vitro exclusively in animals that have undergone withdrawal and acts to partially renormalize synaptic activity. While the precise mechanisms underlying CPD and PPP require elucidation, the data indicate that D1 dopamine and cholinergic responses are required for these long-term adaptations to drug administration.
CPD was indicated by a decreased rate of exocytosis of the recycling synaptic vesicle pool in motor corticostriatal terminals in animals exposed to repeated methamphetamine, together with a reduction in spontaneous and mEPSCs, as well as by the increased threshold required to evoke EPSCs in methamphetamine-treated animals. The optical recordings indicate that the changes were presynaptic, while the electrophysiological results confirm a presynaptic locus, as they occurred in the presence of TTX, and as the amplitude of mEPSCs were not different in cells from saline- or methamphetamine-treated animals. PPP was clearly observed by the increased rate of exocytosis of the recycling vesicle pool with psychostimulant reinstatement which only occurred in animals previously exposed to repeated methamphetamine, as well as by the paradoxical increase in sEPSCs after amphetamine and a D1R agonist, an effect never observed in control conditions.
How might dopamine release during repeated methamphetamine exert longlasting changes in ACh transmission and initiate CPD and PPP without a concomitant long-lasting change in dopamine release? Opposing D1R-excitatory and D2R-inhibitory mechanisms regulate cholinergic efflux in the striatum (Bertorelli and Consolo, 1990; DeBoer and Abercrombie, 1996), as TANs possess D2R that inhibit ACh release (Yan et al., 1997), and D1R that enhance ACh efflux (Figure 8) (Abercrombie and DeBoer, 1997; DeBoer and Abercrombie, 1996; Le Moine et al., 1991; Yan et al., 1997). Under control conditions, responses to dopamine favor D2R-mediated inhibition of ACh efflux (DeBoer and Abercrombie, 1996). ACh accelerates corticostriatal release through α7*-nAChR (Marchi et al., 2002; Pakkanen et al., 2005; Wang and Sun, 2005) and inhibits corticostriatal release through M2 mAChRs (Calabresi et al., 2000; Hersch et al., 1994) with mAChR responses submissive to alterations in nAChR sensitivity (Wang and Sun, 2005). Our data are consistent with dominant regulation by tonic nAChR in control mice, as mAChR blockade by atropine does not affect release, while nAChR blockade with mecamylamine, ACh depletion with vesamicol, or desensitization of nAChR by nicotine and choline are each inhibitory. The lack of tonic ACh influence via mAChR on control corticostriatal activity is in agreement with previous literature (Malenka and Kocsis, 1988). It may be that the tonic levels of ACh are normally too low to desensitize nAChR, but that when higher levels are reached, there is an allosteric regulation of mAChRs which provides enhanced affinity to ACh (Wang and Sun, 2005).
Figure 8.
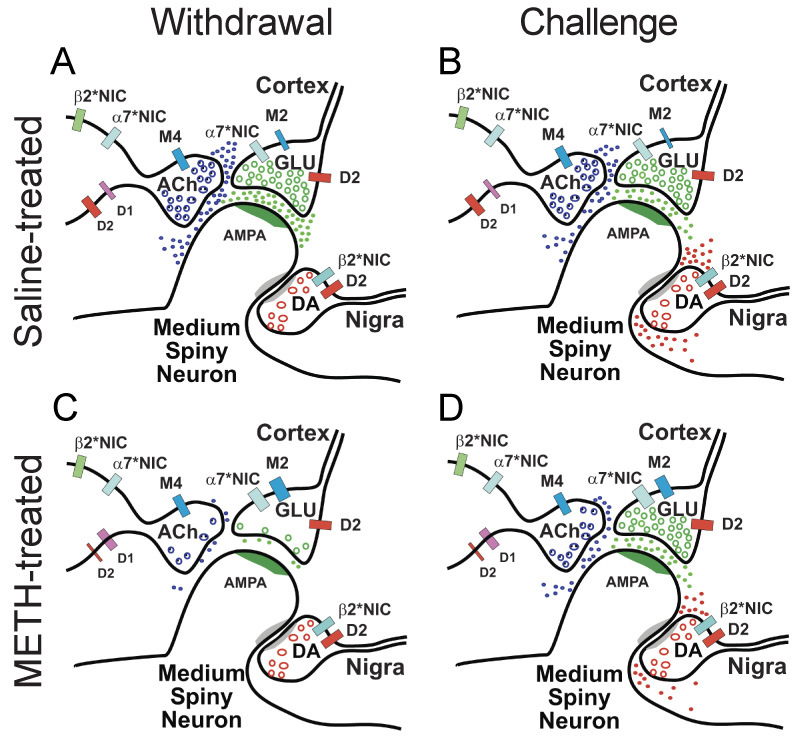
Proposed mechanism for METH-induced synaptic plasticity. (A) The simplified striatal circuit is composed of medium spiny neurons that receive excitatory glutamatergic (GLU) corticostriatal projections, modulatory dopaminergic (DA) nigrostriatal fibers, and tonically active acetylcholine (ACh) – releasing interneurons (TANs). ACh modulates GLU release (Malenka and Kocsis, 1988) through excitatory α7*-nicotinic (NIC) (Marchi et al., 2002; Pakkanen et al., 2005) and inhibitory M2 mAChRs (Calabresi et al., 2000) located on corticostriatal terminals (Hersch et al., 1994) and regulates its own release through M4 muscarinic (Zhang et al., 2002) and both α7*-NIC and β2*-NIC autoreceptors (Azam et al., 2003). (B) Under control conditions, DA released by a psychostimulant inhibits GLU release from a subset of cortical terminals via D2R (Bamford et al., 2004b). Although TANs possess both inhibitory D2R (Yan et al., 1997) and excitatory D1R (Le Moine et al., 1991; Yan et al., 1997), D2R responses predominate so that DA reduces ACh efflux from striatal cholinergic interneurons (DeBoer and Abercrombie, 1996). (C) Following repeated METH, a reduction in ACh availability sensitizes muscarinic and nicotinic receptors. Enhanced muscarinic inhibition and reduced nicotinic excitation promotes CPD. (D) During withdrawal, DA released by a psychostimulant challenge induces PPP. DA increases ACh efflux (Bickerdike and Abercrombie, 1997) through TAN D1R responses (Berlanga et al., 2003) to excite GLU release through α7*-nAChRs.
The situation in drug-naïve animals is markedly altered in withdrawal, possibly because repeated methamphetamine reduces ACh levels, limiting corticostriatal nAChR excitation and sensitizing both mACh and nACh receptors (Siegal et al., 2004). Persistent dopamine release during repeated methamphetamine may additionally uncouple D1R/D2R synergisms (Hu and White, 1994; Kashihara et al., 1999) on TAN neurons, favoring D1R excitation (Berlanga et al., 2003) so that methamphetamine challenge during withdrawal activates TAN D1R and enhances ACh release (Bickerdike and Abercrombie, 1997) to activate PPP. The dependence of PPP on D1R and nAChR activation could contribute to the ability of D1 antagonists to block sensitized locomotor responses or drug self-administration in rodents (Ciccocioppo et al., 2001).
Our data do not directly indicate the locus of AChRs responsible for methamphetamine-induced corticostriatal plasticity. The nAChRs that mediate PPP may be on corticostriatal terminals (Marchi et al., 2002; Pakkanen et al., 2005; Wang and Sun, 2005) or TANs (Azam et al., 2003). Likewise, the mAChRs responsible for CPD may also be at presynaptic sites (Calabresi et al., 2000; Hersch et al., 1994), on TANs (Zhang et al., 2002), or elsewhere. The mAChR may be an inhibitory TAN autoreceptor (Zhang et al., 2002), since nAChR stimulation is required to reverse CPD.
An advantage of presynaptic optical measurements is that variability between individual presynaptic terminals can be analyzed. Our FM1-43 loading protocol is fairly extensive (10 min, 10 Hz), and saturates those terminals capable of dye uptake, i.e., additional stimulation results in no additional labelled terminals. As CPD in withdrawal is reversed by pharmacological treatment following loading, it is not due to a decreased number of active terminals or a smaller pool of recycling synaptic vesicles, but rather a decreased probability of fusion of recycling vesicles. A decreased probability of synaptic vesicle fusion is consistent with the decreased mEPSC frequency in the presence of TTX following withdrawal.
The distribution of individual cortical terminal halftimes in controls demonstrated that stimulation of D2R during periods of high cortical activity depresses release from the majority of cortical terminals, preferentially inhibiting the activity of the terminals with the lowest probability of release, an effect that occurs in the dynamic and kinetic range of dopamine input associated with both salient behavioral stimuli and psychostimulants (Bamford et al., 2004b). Thus, dopamine release associated with salience during learning would reinforce specific corticostriatal connections by filtering out less effective cortical terminal inputs (Bamford et al., 2004b). Repeated methamphetamine would disrupt this filtering mechanism by inducing CPD. The induction of CPD is dopamine-dependent, but CPD continues to be expressed even when dopamine release returns to normal. This indicates that long-lasting plasticity, once initiated, does not require a corresponding long-lasting change in the dopamine system. Subsequent psychostimulant readministration, however, would enhance striatal ACh release by activating D1R, and thus induce PPP by accelerating exocytosis from corticostriatal terminals. PPP provides a mechanism by which drug readministration renormalizes synaptic function following withdrawal, a feature long suggested to be required for addiction, and may favor the conversion of LTD to LTP (Nishioku et al., 1999). Since striatal LTD and LTP are implicated in memory for habitual behaviors (Jog et al., 1999; Packard and Knowlton, 2002), these findings support the idea that the striatum is likely to be the site for storage of information related to locomotor sensitization and drug addiction (Gerdeman et al., 2003; Koob, 1992).
Experimental Procedures
Animals and statistics
Experimental procedures were carried out in accordance with the USPHS Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of Washington, Columbia University and UCLA. C57BL/6 mice aged 12–16 weeks were obtained from Jackson Labs (Bar Harbor, Maine). Mice were treated with methamphetamine (10 or 20 mg/kg/day, i.p.) or with an equal volume of 0.9% saline by daily injection for 10 days. In some studies, mice were challenged by a single dose of methamphetamine (20 mg/kg, i.p.) or amphetamine (2 mg/kg i.p.) in vivo. Mice were anesthetized with Nembutal or ketamine/xylazine prior to sacrifice. Mice for electrochemical recordings were treated in University of Washington and shipped to Columbia University. Some mice were treated at Columbia University to exclude possible effects of stress. For in vivo studies, mice were sacrificed 30 min following methamphetamine when dopamine efflux is expected to reach peak concentrations (McFarland et al., 2003). To ensure equilibrium, sections were exposed to pharmacological agents for 10 min before stimulation-mediated unloading. All drugs were obtained from Sigma (St. Louis, MO).
Values given in the text and in the figures are mean±SE. To establish differences in FM1–43 release between groups of mice exposed to saline or methamphetamine, release halftimes from each mouse were averaged and significance was determined using t-test with Bonferroni correction with n=number of mice. Differences between non-parametric release halftimes (t1/2) following receptor perturbation were determined using the Mann-Whitney test with n=number of puncta. Comparisons between groups of puncta represent data collected from 4–6 mice and comparisons between groups of mice represent the average of 149–439 puncta from 6–12 slices per mouse. Differences were considered significant at levels of p<0.05. Changes in terminal subpopulations were determined graphically using normal probability plots by comparing individual terminal release to normally-distributed data.
Behavioral protocol
Locomotor responses were determined using animal activity monitor cages as described in the Supplemental Data.
Optical imaging with FM1-43
Optical recordings of cortical afferents in the motor striatum were obtained as previously described (Bamford et al., 2004a) and are further detailed in the Supplemental Data.
Electrochemical recordings with cyclic voltammetry
Striatal dopamine release was studied in 3–5 pairs of methamphetamine treated mice and their saline controls for each withdrawal day, i.e., day 1, day 10, day 30, and day 140, using fast-scan cyclic voltammetry. Electrochemical recordings and electrical stimulation were adapted from previous studies (Schmitz et al., 2001) and the procedures are described further in the Supplemental Data.
Detection of striatal ACh concentrations
ACh tissue concentrations were determined by high performance liquid chromatography, based on a reaction with acetylcholinesterase and choline oxidase (Vanderbilt Kennedy Center, Vanderbilt, TN) according to previous publications (Bertrand et al., 1994; Damsma et al., 1985) as further described in the Supplemental Data.
Electrophysiology
Electrophysiological recordings in medium spiny neurons were obtained as previously described (Cepeda et al., 1998) and are further detailed in the Supplemental Data.
Supplementary Material
Supplemental Data The Supplemental Data for this article can be found online at:
Acknowledgements
We thank Drs Richard Palmiter, Patricio O’Donnell, Robert H. Edwards, Paul Philips, Larry Zweifel, Dennis Dever and Lisa H. Zimberg, and also Ian J. Bamford. Supported by NINDS K02NS052536, R01NS060803, P30 HD02274, Child Neurology Society, Colleen Giblin Charitable Foundation for Pediatric Neurology, and by Royalty Research Award, Vision Research Center and Children’s Hospital and Regional Medical Center, University of Washington, Seattle, WA (all to NB), NIDA DA07418 and the Picower and Parkinson’s Disease Foundations (DS), NS33538 (MSL) and NARSAD (HZ)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abercrombie ED, DeBoer P. Substantia nigra D1 receptors and stimulation of striatal cholinergic interneurons by dopamine: a proposed circuit mechanism. J Neurosci. 1997;17:8498–8505. doi: 10.1523/JNEUROSCI.17-21-08498.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed SH, Koob GF. Transition to drug addiction: a negative reinforcement model based on an allostatic decrease in reward function. Psychopharmacology (Berl) 2005;180:473–490. doi: 10.1007/s00213-005-2180-z. [DOI] [PubMed] [Google Scholar]
- Azam L, Winzer-Serhan U, Leslie FM. Co-expression of alpha7 and beta2 nicotinic acetylcholine receptor subunit mRNAs within rat brain cholinergic neurons. Neuroscience. 2003;119:965–977. doi: 10.1016/s0306-4522(03)00220-3. [DOI] [PubMed] [Google Scholar]
- Bamford NS, Robinson S, Palmiter RD, Joyce JA, Moore C, Meshul CK. Dopamine modulates release from corticostriatal terminals. J Neurosci. 2004a;24:9541–9552. doi: 10.1523/JNEUROSCI.2891-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford NS, Zhang H, Schmitz Y, Wu NP, Cepeda C, Levine MS, Schmauss C, Zakharenko SS, Zablow L, Sulzer D. Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron. 2004b;42:653–663. doi: 10.1016/s0896-6273(04)00265-x. [DOI] [PubMed] [Google Scholar]
- Berlanga ML, Olsen CM, Chen V, Ikegami A, Herring BE, Duvauchelle CL, Alcantara AA. Cholinergic interneurons of the nucleus accumbens and dorsal striatum are activated by the self-administration of cocaine. Neuroscience. 2003;120:1149–1156. doi: 10.1016/s0306-4522(03)00378-6. [DOI] [PubMed] [Google Scholar]
- Bertorelli R, Consolo S. D1 and D2 dopaminergic regulation of acetylcholine release from striata of freely moving rats. J Neurochem. 1990;54:2145–2148. doi: 10.1111/j.1471-4159.1990.tb04922.x. [DOI] [PubMed] [Google Scholar]
- Bertrand N, Beley P, Beley A. Brain fixation for acetylcholine measurements. J Neurosci Methods. 1994;53:81–85. doi: 10.1016/0165-0270(94)90147-3. [DOI] [PubMed] [Google Scholar]
- Bickerdike MJ, Abercrombie ED. Striatal acetylcholine release correlates with behavioral sensitization in rats withdrawn from chronic amphetamine. J Pharmacol Exp Ther. 1997;282:818–826. [PubMed] [Google Scholar]
- Brady AM, Glick SD, O'Donnell P. Selective disruption of nucleus accumbens gating mechanisms in rats behaviorally sensitized to methamphetamine. J Neurosci. 2005;25:6687–6695. doi: 10.1523/JNEUROSCI.0643-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Pisani A, Bernardi G. Acetylcholine-mediated modulation of striatal function. Trends Neurosci. 2000;23:120–126. doi: 10.1016/s0166-2236(99)01501-5. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Colwell CS, Itri JN, Chandler SH, Levine MS. Dopaminergic modulation of NMDA-induced whole cell currents in neostriatal neurons in slices: contribution of calcium conductances. J Neurophysiol. 1998;79:82–94. doi: 10.1152/jn.1998.79.1.82. [DOI] [PubMed] [Google Scholar]
- Ciccocioppo R, Sanna PP, Weiss F. Cocaine-predictive stimulus induces drug-seeking behavior and neural activation in limbic brain regions after multiple months of abstinence: reversal by D(1) antagonists. Proc Natl Acad Sci U S A. 2001;98:1976–1981. doi: 10.1073/pnas.98.4.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsma G, Westerink BH, Horn AS. A simple, sensitive, and economic assay for choline and acetylcholine using HPLC, an enzyme reactor, and an electrochemical detector. J Neurochem. 1985;45:1649–1652. doi: 10.1111/j.1471-4159.1985.tb07238.x. [DOI] [PubMed] [Google Scholar]
- Davidson C, Lee TH, Ellinwood EH. Acute and chronic continuous methamphetamine have different long-term behavioral and neurochemical consequences. Neurochem Int. 2005;46:189–203. doi: 10.1016/j.neuint.2004.11.004. [DOI] [PubMed] [Google Scholar]
- DeBoer P, Abercrombie ED. Physiological release of striatal acetylcholine in vivo: modulation by D1 and D2 dopamine receptor subtypes. J Pharmacol Exp Ther. 1996;277:775–783. [PubMed] [Google Scholar]
- Gerdeman GL, Partridge JG, Lupica CR, Lovinger DM. It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci. 2003;26:184–192. doi: 10.1016/S0166-2236(03)00065-1. [DOI] [PubMed] [Google Scholar]
- Hersch SM, Gutekunst CA, Rees HD, Heilman CJ, Levey AI. Distribution of m1-m4 muscarinic receptor proteins in the rat striatum: light and electron microscopic immunocytochemistry using subtype-specific antibodies. J Neurosci. 1994;14:3351–3363. doi: 10.1523/JNEUROSCI.14-05-03351.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XT, White FJ. Loss of D1/D2 dopamine receptor synergisms following repeated administration of D1 or D2 receptor selective antagonists: electrophysiological and behavioral studies. Synapse. 1994;17:43–61. doi: 10.1002/syn.890170106. [DOI] [PubMed] [Google Scholar]
- Jog MS, Kubota Y, Connolly CI, Hillegaart V, Graybiel AM. Building neural representations of habits. Science. 1999;286:1745–1749. doi: 10.1126/science.286.5445.1745. [DOI] [PubMed] [Google Scholar]
- Kashihara K, Ishihara T, Akiyama K, Abe K. D1/D2 receptor synergism on CREB DNA-binding activities in the caudate-putamen of rat. Neurol Res. 1999;21:781–784. doi: 10.1080/01616412.1999.11741014. [DOI] [PubMed] [Google Scholar]
- Koob GF. Drugs of abuse: anatomy, pharmacology and function of reward pathways. Trends Pharmacol Sci. 1992;13:177–184. doi: 10.1016/0165-6147(92)90060-j. [DOI] [PubMed] [Google Scholar]
- Kuribara H. Dopamine D1 receptor antagonist SCH 23390 retards methamphetamine sensitization in both combined administration and early posttreatment schedules in mice. Pharmacol Biochem Behav. 1995;52:759–763. doi: 10.1016/0091-3057(95)00173-t. [DOI] [PubMed] [Google Scholar]
- Le Moine C, Normand E, Bloch B. Phenotypical characterization of the rat striatal neurons expressing the D1 dopamine receptor gene. Proc Natl Acad Sci U S A. 1991;88:4205–4209. doi: 10.1073/pnas.88.10.4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Kocsis JD. Presynaptic actions of carbachol and adenosine on corticostriatal synaptic transmission studied in vitro. J Neurosci. 1988;8:3750–3756. doi: 10.1523/JNEUROSCI.08-10-03750.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi M, Risso F, Viola C, Cavazzani P, Raiteri M. Direct evidence that release-stimulating alpha7* nicotinic cholinergic receptors are localized on human and rat brain glutamatergic axon terminals. J Neurochem. 2002;80:1071–1078. doi: 10.1046/j.0022-3042.2002.00805.x. [DOI] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23:3531–3537. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioku T, Shimazoe T, Yamamoto Y, Nakanishi H, Watanabe S. Expression of long-term potentiation of the striatum in methamphetamine-sensitized rats. Neurosci Lett. 1999;268:81–84. doi: 10.1016/s0304-3940(99)00386-9. [DOI] [PubMed] [Google Scholar]
- Overstreet DH, Djuric V. A genetic rat model of cholinergic hypersensitivity: implications for chemical intolerance, chronic fatigue, and asthma. Ann N Y Acad Sci. 2001;933:92–102. doi: 10.1111/j.1749-6632.2001.tb05816.x. [DOI] [PubMed] [Google Scholar]
- Packard MG, Knowlton BJ. Learning and memory functions of the Basal Ganglia. Annu Rev Neurosci. 2002;25:563–593. doi: 10.1146/annurev.neuro.25.112701.142937. [DOI] [PubMed] [Google Scholar]
- Pakkanen JS, Jokitalo E, Tuominen RK. Up-regulation of beta2 and alpha7 subunit containing nicotinic acetylcholine receptors in mouse striatum at cellular level. Eur J Neurosci. 2005;21:2681–2691. doi: 10.1111/j.1460-9568.2005.04105.x. [DOI] [PubMed] [Google Scholar]
- Pessiglione M, Seymour B, Flandin G, Dolan RJ, Frith CD. Dopamine-dependent prediction errors underpin reward-seeking behaviour in humans. Nature. 2006;442:1042–1045. doi: 10.1038/nature05051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce RC, Bell K, Duffy P, Kalivas PW. Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci. 1996;16:1550–1560. doi: 10.1523/JNEUROSCI.16-04-01550.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick MW, Lester RA. Desensitization of neuronal nicotinic receptors. J Neurobiol. 2002;53:457–478. doi: 10.1002/neu.10109. [DOI] [PubMed] [Google Scholar]
- Redish AD. Addiction as a computational process gone awry. Science. 2004;306:1944–1947. doi: 10.1126/science.1102384. [DOI] [PubMed] [Google Scholar]
- Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D. Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci. 2001;21:5916–5924. doi: 10.1523/JNEUROSCI.21-16-05916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegal D, Erickson J, Varoqui H, Ang L, Kalasinsky KS, Peretti FJ, Aiken SS, Wickham DJ, Kish SJ. Brain vesicular acetylcholine transporter in human users of drugs of abuse. Synapse. 2004;52:223–232. doi: 10.1002/syn.20020. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Turner TJ. Nicotine enhancement of dopamine release by a calcium-dependent increase in the size of the readily releasable pool of synaptic vesicles. J Neurosci. 2004;24:11328–11336. doi: 10.1523/JNEUROSCI.1559-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderschuren LJ, Kalivas PW. Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology (Berl) 2000;151:99–120. doi: 10.1007/s002130000493. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Hrabovska A, Duysen EG, Ferguson SM, Blakely RD, Lockridge O, Levey AI. Altered striatal function and muscarinic cholinergic receptors in acetylcholinesterase knockout mice. Mol Pharmacol. 2003;64:1309–1316. doi: 10.1124/mol.64.6.1309. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun X. Desensitized nicotinic receptors in brain. Brain Res Brain Res Rev. 2005;48:420–437. doi: 10.1016/j.brainresrev.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, Tkatch T, Lovinger DM, Surmeier DJ. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron. 2006;50:443–452. doi: 10.1016/j.neuron.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Wooltorton JR, Pidoplichko VI, Broide RS, Dani JA. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci. 2003;23:3176–3185. doi: 10.1523/JNEUROSCI.23-08-03176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Song WJ, Surmeier J. D2 dopamine receptors reduce N-type Ca2+ currents in rat neostriatal cholinergic interneurons through a membrane-delimited, protein-kinase-C-insensitive pathway. J Neurophysiol. 1997;77:1003–1015. doi: 10.1152/jn.1997.77.2.1003. [DOI] [PubMed] [Google Scholar]
- Zhang H, Sulzer D. Frequency-dependent modulation of dopamine release by nicotine. Nat Neurosci. 2004;7:581–582. doi: 10.1038/nn1243. [DOI] [PubMed] [Google Scholar]
- Zhang W, Basile AS, Gomeza J, Volpicelli LA, Levey AI, Wess J. Characterization of central inhibitory muscarinic autoreceptors by the use of muscarinic acetylcholine receptor knock-out mice. J Neurosci. 2002;22:1709–1717. doi: 10.1523/JNEUROSCI.22-05-01709.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4:1224–1229. doi: 10.1038/nn769. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data The Supplemental Data for this article can be found online at: