

Abstract
We developed a cell division–activated Cre-lox system for stochastic recombination of loxP-flanked loci in mice. Cre activation by frameshift reversion is modulated by DNA mismatch-repair status and occurs in individual cells surrounded by normal tissue, mimicking spontaneous cancer-causing mutations. This system should be particularly useful for delineating pathways of neoplasia, and determining the developmental and aging consequences of specific gene alterations.
Valuable mouse cancer models exist that combine conditional expression of the Cre recombinase with various loxP-flanked tumor suppressor or oncogene alleles1. In these model systems, ‘cancer’ is induced by gene alteration ubiquitously throughout the target tissue or in a selected cell type. But this does not accurately mimic the natural process of sporadic cancer initiation and progression. Two Cre-lox models that facilitate stochastic cancer gene alterations in isolated cells rely on homologous recombination to induce activation of an oncogene2, or to induce inactivation of one or more tumor suppressor genes on the same chromosome3. The former system is restricted in that it is only applicable to activation of an oncogenic K-ras allele2. The latter more flexible system uses engineered loxP-FRT sites to induce mitotic recombination of individual chromosomes containing modified genes3. Here we report a highly versatile system that features Cre-mediated stochastic genetic changes in single cells or cell lineages in normal tissue. The system can be applied to any loxP-flanked allele, is dependent on cell division and can be modulated by DNA mismatch-repair status.
To construct an inactive but revertible Cre allele, we first engineered an 11-bp A·T run in a modified version of Cre4 without altering the nuclear localization signal (Supplementary Methods online). We added an extra A·T base pair, creating a +1 bp out-of-frame Cre allele, which we termed 12A-Cre. To ensure efficient expression at the intended target locus, we added a splice acceptor and internal ribosome entry site to 12A-Cre. We cloned the modified 12A-Cre plus a neo module between homology arms of the DNA mismatch repair gene Pms2 (ref. 5) to generate a Pms2-Cre targeting vector. Targeting resulted in an out-of-frame Cre gene under the control of the Pms2 promoter, which is expressed in several cell types, including stem cells of the mouse intestine6. Targeting to Pms2 removed exon 2, creating a null allele, which we refer to as Pms2cre (Fig. 1a). Because of mismatch-repair deficiency, Pms2cre/cre mice should have increased frequency of −1 bp frameshifts7 and hence increased Cre reversion relative to Pms2cre/+ mice. Therefore, Cre activation frequency can be modulated appropriately for a particular study by breeding Pms2cre/+ or Pms2cre/cre mice. Notably, this system should better mimic sporadic carcinogenesis because Cre activation is stochastic, limited to individual cells and linked to cell division.
Figure 1. Generation and characterization of Pms2cre mice.
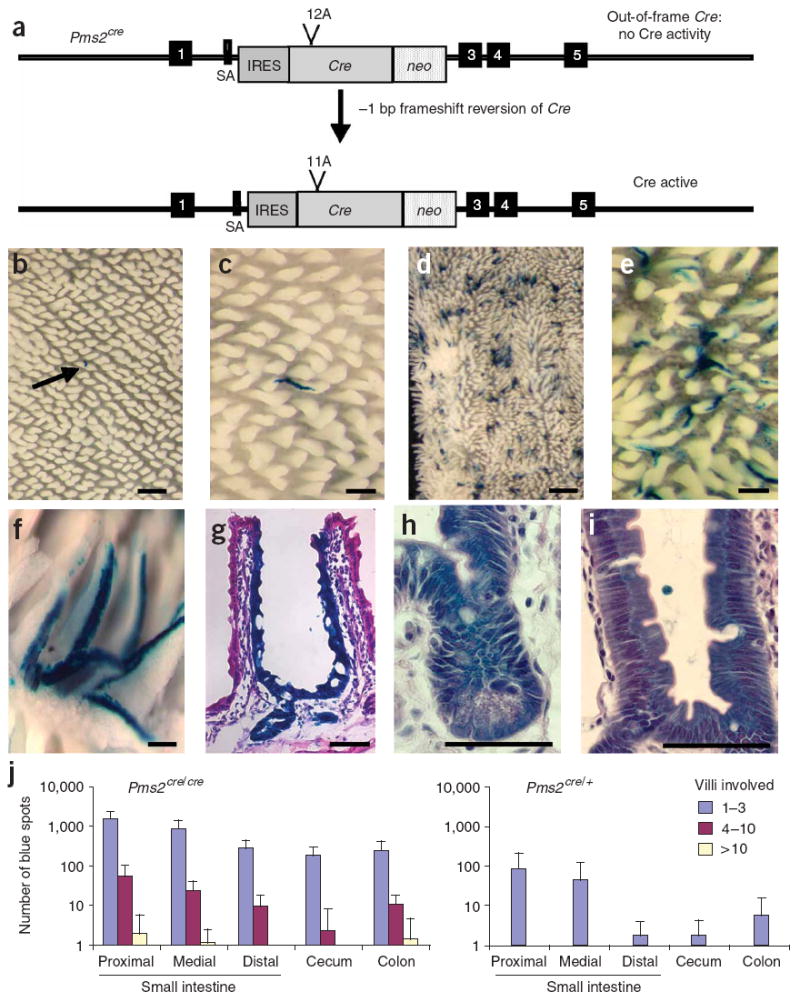
(a) A schematic of the Pms2cre allele activation by a −1-bp frameshift in the 12A run. SA, splice acceptor; IRES, internal ribosome entry site. Boxes 1 and 3–5 represent exons.
(b–f) β-galactosidase staining in whole-mount mouse intestinal sections. Different magnification images of Pms2cre/+ (b,c) and Pms2cre/cre (d–f) small intestine. Scale bars, 1 mm (d), 0.5 mm (b), 0.25 mm (c,e) and 0.1 mm (f). Arrow in b indicates the single spot in the field.
(g–i) Hematoxylin and eosin–stained 5-μm paraffin sections of Pms2cre/cre small intestine crypt. Scale bars, 50 μm (g) and 25 μm (h,i).
(j) Distribution of average number of blue spots in the gastrointestinal tract of age-matched Pms2cre/cre (n = 10) and Pms2cre/+ (n = 4) mice. Error bars, s.d.
Because of our interest in intestinal cancers, we focused on Cre activation in the gastrointestinal tract. To estimate relative Cre reversion in Pms2cre/+ and Pms2cre/cre mice, we bred them to Cre-inducible Gt(rosa)26tm1Sor lacZ reporter mice (hereafter referred to as Rosa26r)8. We examined progeny over a 12-month period and visualized Cre activation in the intestine by β-galactosidase staining as ‘ribbons’ of blue staining up villus sides, or ‘spots’9. As expected, Cre reversion in Pms2cre/cre (mismatch repair–deficient) mice was elevated ~100-fold relative to Pms2cre/+ (mismatch repair–proficient) mice (Fig. 1). The average total numbers of spots in the Pms2cre/+ and Pms2cre/cre mice were 26 and 3,300, respectively. We frequently observed a ribbon-like pattern of β-galactosidase staining from crypt base to the villus tip, with a greater number of blue spots in the proximal small intestine and trending toward fewer spots in the distal small intestine (Fig. 1). The total number of blue spots appeared to increase with age (Supplementary Fig. 1a online), consistent with mutations accumulating with age in the epithelium. Patch size was typically small, consistent with the progeny of a single stem or progenitor cell contributing to 1–3 villi (Fig. 1). This small patch size suggests stem cells with reactivated Cre usually remain confined to a single crypt with a single crypt supplying cells to several adjacent villi10. Typically, each of the four differentiated cell types in a particular crypt and associated villus appeared to stain positive for β-galactosidase (Fig. 1), consistent with Cre reversion having occurred in a stem cell. Microdissection of individual villi followed by a PCR assay for Cre-mediated recombination at the Rosa26r locus was consistently positive in blue-stained villi but negative in unstained villi (Supplementary Fig. 1b). Additionally, we observed patches of β-galactosidase–expressing (Cre-activated) cells in all tissues examined: intestine, pancreas, kidney, liver and muscle (data not shown), as expected based on the distribution of Pms2 expression6.
Numbers and sizes of β-galactosidase–positive patches provide a baseline indicator of intestinal stem cell fates after stochastic Cre reactivation. Each crypt contains multiple stem cells within a niche10, and the inactivation or activation of a tumor suppressor or oncogene, respectively, may change the number of blue-staining cells by providing a selective advantage (more or larger patches) or disadvantage (fewer or smaller patches) to that stem cell, even in the absence of visible histological changes. In turn, clonal evolution, that is, the replacement of cell populations by the progeny of a single altered cell, within a normal appearing tissue can be visualized. As a next step, we tested the oncogenic allele, LSL-K-rasG12D (ref. 11) activated by Cre-mediated excision of a stop codon, in our system. Pms2cre/cre, Rosa26r, LSL-K-rasG12D mice became moribund at 5 weeks of age because of high lung tumor burden, a phenotype previously associated with activation of K-rasG12D (ref. 11). Lung tumors stained blue, consistent with Cre activation and subsequent K-rasG12D expression (data not shown). We scored intestines from Pms2cre/cre, Rosa26r, LSL-K-rasG12D mice for the number and size of blue-staining regions (Fig. 2a). The numbers of blue ‘spots’ in the Pms2cre/cre, Rosa26r, LSL-K-rasG12D mice did not change, but the number of stained villi per patch, or blue patch size, increased in the small intestine and cecum (Fig. 2a,b). The morphology of most blue-staining villi was normal, with a few irregularly sized villi (Fig. 2c). The increase in patch size implies that sporadic activation of K-rasG12D can confer a selective advantage at some stage, apparently by facilitating dominance of that stem cell over wild-type stem cells12, including the ability to spread over larger areas, presumably through crypt fission13. This apparent clonal expansion and crypt fission of K-rasG12D–expressing cells would facilitate the subsequent acquisition of the additional alterations needed to confer a visible neoplastic phenotype.
Figure 2. Activation of K-ras results in larger regions of β-galactosidase staining.
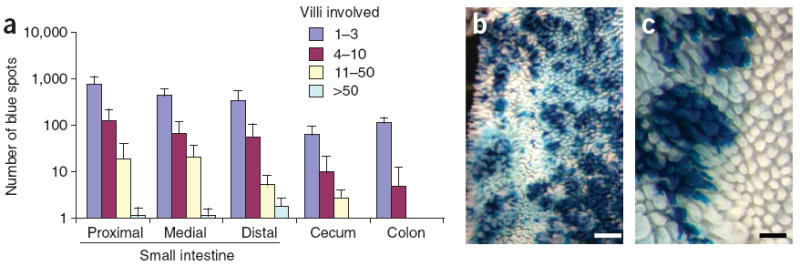
(a) Distribution of average spots per region in Pms2cre/cre, LSL-K-rasG12D mice (n = 5). Error bars, s.d. The size spot per area increases compared to Pms2cre/cre, Rosa26r mice. (b,c) β-galactosidase staining in whole-mount small intestine from Pms2cre/cre, LSL-K-rasG12D mice shows expanded (large) regions of staining. Scale bars, 1 mm (b) and 0.5 mm (c).
We developed a Cre-lox system that facilitates monitoring the short- and longer-term consequences of genetic changes in single cells in normal tissue in the mouse. This system relies on a stochastic reversion event, which activates Cre recombinase expression and can be modulated by mismatch-repair status. Visualization of cell lineages experiencing Cre activation is facilitated by staining for Cre-inducible β-galactosidase, which also allows monitoring the relative survival and developmental capacity of mutant cells even in the absence of visible neoplasia. In this way, clonal evolution in normal-appearing tissues may be visualized. Patterns of β-galactosidase staining revealed that Cre activation can occur in stem cells and/or proliferating progenitors. Normally the progeny of a single stem cell are confined to a single crypt, but sporadic mutations may alter a stem cell’s survival or ability to spread. Some mutations may be initially neutral, whereas others may be lethal or reduce fitness relative to wild-type stem cells within a niche. Alternately some mutations, as illustrated with LSL-K-rasG12D, may confer a selective advantage, leading to expanded β-galactosidase–positive regions.
Because Pms2cre/cre mice are defective in mismatch repair, random mutations in the genome could complicate interpretations. However, controls can be studied in parallel, for example, mice that do not harbor tumor suppressor or oncogene Cre targets. Furthermore, although Pms2cre/cre mice have a reduced lifespan with a half-life of 10 months (data not shown), they can be useful for determining ‘short–term’ consequences of stochastic knockout of specific tumor suppressor loci individually, or of several loci in combination (unpublished data). Additionally, this system has inherent flexibility because either mismatch repair–proficient Pms2cre/+ mice, which are not prone to cancer, or mismatch repair–deficient Pms2cre/cre mice can be used to modulate the activation rate of Cre. Judicious use of Pms2cre/+ or Pms2cre/cre mice will be directed by the nature and number of Cre targets (loxP-flanked alleles) in a particular study.
Because Pms2 is expressed in multiple tissues and mismatch repair is likely to be active in a variety of stem cell types, the Pms2cre system described here offers flexibility in terms of probing both normal and abnormal development in a variety of tissues. In addition, having a widely expressed promoter driving Cre offers the potential to determine for any given tumor whether Cre activation occurring in the tissue or cell type that gave rise to the tumor and/or an underlying or surrounding tissue type was critical. Nevertheless, it may be advantageous in certain situations to use a tissue-specific promoter, such as the villin promoter, which would target Cre activation to the epithelium of the mouse gut14.
Supplementary Material
Note: Supplementary information is available on the Nature Methods website.
Acknowledgments
We thank M. Wong, N. Erdeniz, J. Johnson, O. Reilly and K. MacDonald for critically reading the manuscript. This work was supported by US National Institutes of Health grants R37 GM32741 to R.M.L. and 2 RO1 CA 80077 to D.S.
Footnotes
Published online at http://www.nature.com/naturemethods
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Jonkers J, Berns A. Nat Rev Cancer. 2002;2:251–265. doi: 10.1038/nrc777. [DOI] [PubMed] [Google Scholar]
- 2.Johnson L, et al. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 3.Wang W, Warren M, Bradley A. Proc Natl Acad Sci USA. 2007;104:4501–4505. doi: 10.1073/pnas.0607953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewandoski M, Martin GR. Nat Genet. 1997;17:223–225. doi: 10.1038/ng1097-223. [DOI] [PubMed] [Google Scholar]
- 5.Baker SM, et al. Cell. 1995;82:309–319. doi: 10.1016/0092-8674(95)90318-6. [DOI] [PubMed] [Google Scholar]
- 6.Narayanan L, et al. Proc Natl Acad Sci USA. 1997;94:3122–3127. doi: 10.1073/pnas.94.7.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harfe BD, Jinks-Robertson S. Annu Rev Genet. 2000;34:359–399. doi: 10.1146/annurev.genet.34.1.359. [DOI] [PubMed] [Google Scholar]
- 8.Soriano P. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 9.Wong MH, Hermiston ML, Syder AJ, Gordon JI. Proc Natl Acad Sci USA. 1996;93:9588–9593. doi: 10.1073/pnas.93.18.9588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marshman E, Booth C, Potten CS. Bioessays. 2002;24:91–98. doi: 10.1002/bies.10028. [DOI] [PubMed] [Google Scholar]
- 11.Jackson EL, et al. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calabrese P, Tavare S, Shibata D. Am J Pathol. 2004;164:1337–1346. doi: 10.1016/S0002-9440(10)63220-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDonald SA, et al. Cell Cycle. 2006;5:808–811. doi: 10.4161/cc.5.8.2641. [DOI] [PubMed] [Google Scholar]
- 14.Robine S, et al. Proc Natl Acad Sci USA. 1985;82:8488–8492. doi: 10.1073/pnas.82.24.8488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary information is available on the Nature Methods website.