

Abstract
The study tested the hypothesis that estrogen controls epithelial paracellular resistance through modulation of myosin. The objective was to understand how estrogen modulates non-muscle myosin-II-B (NMM-II-B), the main component of the cortical actomyosin in human epithelial cervical cells. Experiments used human cervical epithelial cells CaSki as a model, and end points were NMM-II-B phosphorylation, filamentation, and MgATPase activity. The results were as follows: 1) treatment with estrogen increased phosphorylation and MgATPase activity and decreased NMM-II-B filamentation; 2) estrogen effects could be blocked by antisense nucleotides for the estrogen receptor-α and by ICI-182,780, tamoxifen, and the casein kinase-II (CK2) inhibitor, 5,6-dichloro-1-β-(D)-ribofuranosylbenzimidazole and attenuated by AG1478 and PD98059 (inhibitors of epithelial growth factor receptor and ERK/MAPK) but not staurosporine [blocker of protein kinase C (PKC)]; 3) treatments with the PKC activator sn-1,2-di-octanoyl diglyceride induced biphasic effect on NMM-II-B MgATPase activity: an increase at 1 nM to 1 μM and a decrease in activity at more than 1 μM; 4) sn-1,2-dioctanoyl diglyceride also decreased NMM-II-B filamentation in a monophasic and saturable dose dependence (EC50 1–10 μM); 5) when coincubated directly with purified NMM-II-B filaments, both CK2 and PKC decreased filamentation and increased MgATPase activity; 6) assays done on disassembled NMM-II-B filaments showed MgATPase activity in filaments obtained from estrogen-treated cells but not estrogen-depleted cells; and 7) incubations in vitro with CK2, but not PKC, facilitated MgATPase activity, even in disassembled NMM-II-B filaments. The results suggest that estrogen, in an effect mediated by estrogen receptor-α and CK2 and involving the epithelial growth factor receptor and ERK/MAPK cascades, increases NMM-II-B MgATPase activity independent of NMM-II-B filamentation status.
An important role of estrogen in females is the regulation of transport across reproductive tract epithelia. The major route of transport in secretory epithelia, such as the female reproductive tract, is the paracellular (intercellular) pathway. Free movement of water and solutes in the paracellular pathway is restricted by the resistance of the tight junctions (RTJ) and the resistance of the lateral intercellular space (RLIS) in series (1). The RTJ determines the overall paracellular resistance; however, in leaky epithelia such as the vaginal and cervical epithelia, the contributions of the RLIS to the total resistance can be significant (2). The RLIS is determined by the geometry of the intercellular space and therefore reciprocally by the width and shape of the epithelial cells that define this space. Formation of a dynamic, flexible cytoskeleton would allow cells facilitated changes in cell width in response to stimuli. For instance, capillary hydrostatic gradients in the subluminal to luminal direction expand the intercellular space and decrease the RLIS (2). Therefore, epithelia composed of cells with dynamic cytoskeleton would expand the intercellular space and decrease the RLIS more readily than epithelia composed of cells with a rigid cytoskeleton.
Estrogen increases the permeability of vaginal and cervical epithelia by two known mechanisms: modulation of the junctional protein occludin, thereby decreasing the RTJ (3, 4), and stimulation of actin depolymerization, thereby facilitating epithelial cell deformability and decreasing the RLIS (5, 6). A recent study has suggested that in addition to actin depolymerization, estrogen can also decrease the RLIS through modulation of actin-myosin interaction (7). The latter mechanism is relatively little understood and was the focus of the present study.
Myosins are protein molecular motors that bind to actin in an ATP-dependent manner. Upon ATP binding, the myosin head detaches from actin and hydrolyzes ATP. In muscle cells the main function of myosin is generation of force for contractility and movement. In nonmuscle cells, myosin interactions with cytoskeletal actin involve regulation of cytokinesis, cell motility, and cell polarity (8). In epithelial cells, nonmuscle myosin-II interactions with actin regulate exocytosis (9). Point mutations in nonmuscle myosin II heavy chains can result in phenotypes affecting kidneys, platelets, leukocytes, heart and brain, and the inner ear (10). Abrogation of myosin-II function can disrupt actin polymerization and lead to cell detachment, and has been reported to be associated with kidney and bowel disease (8, 11).
Nonmuscle myosins are composed of the ubiquitous conventional class II myosins, which are hexameric proteins consisting of two heavy chains and two pairs of light chains. The carboxyl-terminal half of the two heavy chains dimerize via α-helical coiled-coil to form a long rod (tail) through which myosin molecules can self-associate to form filaments. The amino-terminal halves of the two heavy chains (heads) form two separate motor (catalytic) domains, which bind actin and hydrolyze ATP. Of the three different forms of nonmuscle myosin-II (NMM-II) (8, 12), NMM-II-B is the most abundant cortical myosin in epithelial vaginal-cervical cells (7). NMM-II-B has adapted for maintaining tension in a static manner; it displays a low steady-state MgATPase activity and a positive thermodynamic coupling between actin and ADP binding (13, 14). These characteristics confer an advantage of energetic economy within the cell and make its working cycle more suited for sustained maintenance of tension, as opposed to short duration force generation that occurs with muscle myosins (14) and the NMM-II-A (15).
Myosin-II functions can be regulated by phosphorylation (8), and a recent study suggested that estrogen can regulate NMM-II-B interaction with actin through phosphorylation of NMM-II-B heavy chains (7). The specific objective of the present study was to better understand the molecular mechanisms involved in estrogen modulation of NMM-II-B with emphasis on estrogen modulation of NMM-II-B MgATPase activity. The background hypothesis was the current model of myosin-II regulation of cytoskeletal actin (8). The model predicts that in resting conditions myosin is bound to actin in a rigor modus that confers cytoskeletal stability. Detachment of myosin from actin and relaxation of the cytoskeleton in preparation for contraction (muscle cells) or increased state of deformability (epithelial cells) requires binding of ATP to the motor head domain and MgATPase-dependent hydrolysis of ATP (15). The model also predicts that efficient myosin motors can hydrolyze ATP at a greater rate, and conditions that render the myosin motor less efficient would tend to diminish ATP hydrolysis. Accordingly, optimal activity of the MgATPase depends on homodimerization of NMM-II-B filaments.
The results in the human epithelial cervical CaSki cells challenge this hypothesis. The results show that estrogen, in an effect mediated by the estrogen receptor (ER)-α, the epithelial growth factor receptor (EGFR) and ERK/MAPK cascades and casein kinase-II (CK2), increases NMM-II-B phosphorylation and decreases filamentation of the myosin. Estrogen also increases NMM-II-B MgATPase activity by a mechanism that is apparently unrelated to the modulation of filamentation. These findings are novel and suggest that estrogen-induced CK2-mediated phosphorylation of NMM-II-B can up-regulate NMMHC-II-B MgATPase activity independent of NMM-II-B filamentation status.
Materials and Methods
Chemicals, supplies, and antibodies
All chemicals were obtained from Sigma Chemical (St. Louis, MO) unless specified otherwise. 5,6-dichloro-1-β-(D)-ribofuranosylbenzimidazole (DRB) was obtained from Calbiochem (La Jolla, CA). Rabbit polyclonal antihuman NMM-II heavy chain (NMMHC-II)-A antibody (PRB-440P) and its antigenic peptide, and the antihuman NMM-II-B heavy chain (NMMHC-II-B) antibody (PRB-445P) were from Covance Research Products (Berkeley, CA). Mouse monoclonal antihuman protein kinase C (PKC)-α antibody, rabbit polyclonal antiphosphopan-PKC (Ser660), and the Src inhibitor PP1 were from Alexis Corp. (Nottingham, UK). Mouse anti-ERα antibody (catalog CBL425) was from Chemicon (currently Millipore, Billerica, MA). Mouse polyclonal anti-ERβ antibody was from Acris (Hiddenhausen, Germany). The antitubulin antibody (hybridoma supernatant, clone E7, generated with the antigen of β-tubulin-galactosidase/ftz fusion protein) was from the Developmental Studies Hybridoma Bank, University of Iowa (Iowa City, IA). Mouse monoclonal antibody recognizing tyrosine phosphorylated ERK1–2 and rabbit polyclonal antibody recognizing total ERK1–2 were from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell cultures
Experiments used CaSki cells (obtained from the American Type Culture Collection, Manassas, VA), a line of estrogen-responsive transformed cells that retain phenotypic characteristics of human endocervical epithelial cells (16). The methodology of cell culturing was described (5, 7, 16, 17). For assays, cultures were shifted to modified Ringer solution, composed of (millimoles) NaCl (120), CaCl2 (1.2), MgSO4 (1.2), KCl (5), NaHCO3 (10 mM, before equilibration with 95% O2-5% CO2), HEPES (10), glucose (5), and 0.1% BSA plus 0.2 nM epithelial growth factor (EGF).
Depletion of estrogens
The methodology of steroid-hormones depletion (including estrogens) was described (5, 7).
Protein methods
Phosphorylation, immunoprecipitation, and immunoblotting assays, as well as cell fractionation by the freeze/thaw method were described (17). Extraction and purification of NMM-II-B filaments were described (7, 18). The method of purification yields a single 200-kDa band that is immunodetected by the anti NMMHC-II-B antibody (7). Purified NMM-II-B filaments were stored in 6 M urea until assayed. Cellular total protein was measured as described (19). Densitometry was done using Arcus II scanner (AGFA, New York, NY) and Un-Scan-It gel automated digital software (Silk Scientific, Orem, UT).
NMM-II-B phosphorylation in vitro
In vitro phosphorylation of NMM-II-B by CK2 was carried out in 50 μ l solution, containing 0.1–15 μg per 10 μl NMM-II-B filaments plus 40 μl of buffer composed of 10 mM 3[N-morholino]propanesulfonic acid (pH 7.2), 5 mM MgCl2, 30 mM NaCl, 2 mM EGTA, 0.4 μM protein kinase A inhibitor, 10 mM β-glycerol phosphate, 0.4 mM Na orthovanadate, 0.5 mM dithiothreitol, 0.1 mM ATP, 50 μM (2.5 μCi) of [γ-32P]ATP (Upstate, Charlottesville, VA) plus 0.2 μg/tube of purified CK2 from rat liver (Sigma) dissolved in PBS. In vitro phosphorylation of NMM-II-B by PKC was carried out in 50 μ l solution, containing 0.1–15 μg per 10 μl NMM-II-B filaments plus 40 μl of buffer composed of 40 mM Tris-HCl (pH 7.5), 0.15 M NaCl, 0.25 mM CaCl2, 5 mM MgCl2, 0.1 mM dithiothreitol, 5% glycerol, 2 mM phosphatidylserine, 5 μg/ml leupeptin, 0.1 mM ATP, 50 μM (2.5 μCi) [γ-32P]ATP, plus 45 ng (0.075 U) of purified rat brain PKC (Calbiochem) dissolved in PBS. Both in vitro phosphorylation assays were started by the addition of 0.1 mM ATP plus 50 μM (2.5 μCi) [γ-32P]ATP (Upstate). Reactions were carried out at 30 C for 15 min (CK2) and 25 min (PKC) to reach maximal velocity (Vmax) (not shown). The reactions were terminated by the addition of 3 N H2SO4-4% silicotungstate. Mixtures were transferred to nitrocellulose squares, washed several times with 0.75% phosphoric acid, followed by a final wash with 100% acetone. Bound radioactivity was measured using liquid scintillation counting. Kinases activities were determined in terms of ATP hydrolysis as [γ-32P]Pi release and expressed as mole Pi per mole NMM-II-B per minute.
NMM-II-B assembly/disassembly assays
NMM-II-B filament assembly assays were done as follows. Purified NMM-II-B filaments at concentrations of 1–500 μg/ml (about 20 nM to 10 μM) were incubated with 2.5 mM MgSO4 and 10 mM NaCl at 4 C in the presence of 10 mM imidazole-HCl (pH 7.5) and 0.1 mM Ca2+ to induce filament assembly. At the completion of incubations, samples were centrifuged at 25,000 × g for 30 min at 4 C, and the protein content in the supernatant (i.e. nondimerized NMM-II-B) was measured and expressed per total protein measured before centrifugation as the percent nondimerized NMM-II-B. To ascertain complete recovery of non-dimerized filaments, aliquots from the supernatant were centrifuged at 90,000 × g for 20 min. Assays in which sediment was found, indicating inappropriate separation of the monomers from the oligomers, were omitted from analysis (less than 3% of assays). NMM-II-B filament assembly status was expressed in terms of the critical concentration of NMM-II-B necessary to induce dimerization from the curve of percent nondimerized NMM-II-B vs. added protein. The critical concentration of NMM-II-B necessary for dimerization was defined as the concentration that induced maximal oligomerization.
NMM-II-B disassembly assay was done using an existing method (20) with some modifications. NMM-II-B filaments stored in 6 M urea were dialyzed against 50 mM NaCl, 10 mM imidazole hydrochloride (pH 7.0), 10 mM MgCl2, and 0.1 mM EGTA to induce fragment assembly. Aliquots were obtained to determine protein concentration, the assembled protein was precipitated by centrifugation at 25,000 × g for 10 min, and the protein concentration of the resultant supernatant was adjusted to 0.5 mg/ml heavy chain. Fifty micrograms of the protein (about 1.05 μM heavy chain dimer) was incubated in 10 mM imidazole hydrochloride (pH 7.5), 0.1 mM EGTA, 5 mM MgCl2, and various concentrations of NaCl, in a final volume of 0.5 ml in microcentrifuge tubes. The reaction mixtures were incubated on ice for 4 h and then centrifuged at 15,000 × g for 30 min. An aliquot (100 μl) of the supernatant was removed for measurement of the protein remaining in the supernatant to determine nonassembled NMM-II-B (expressed as percent NMM-II-B recovered in the supernatant).
NMM-II-B MgATPase assays
Determinations of NMM-II-B steady-state MgATPase activity in terms of ATP hydrolysis were done using a well-described method (21) with some modifications. Purified NMM-II-B filaments were suspended in 0.1 ml to concentrations of 1–500 μg/ml in a solution composed of 0.5 M KCI, 10 mM imidazole chloride (pH 7.0), 10 mM MgCl2 (for MgATPase activity) at 30 C, plus various concentrations of F-actin (0–200 μM). F-actin was prepared by dissolving purified G-actin (Sigma) in 2 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 0.2 mM CaCl2, 0.2 mM ATP, 100 mM KCl, and 0.5 mM bimercaptoethanol. Under these conditions the added F-actin remains in the polymerized form for the duration of the experiment. In preliminary experiments the involvement of KATPase and CaATPase activities was ruled out, using 2 mM EDTA or 10 mM CaCl2, respectively.
Nonradioactive assays were started by the addition of ATP (0–7.5 mM) and incubated at 37 C for 30 min. Rates were obtained from the average of three time points at the steep increase of Pi release. The reaction was stopped by adding a mixture of 0.2 ml of 1:1 2-butanol-benzene with 0.5 ml of 3 N H2SO4-4% silicotungstate, and the protein was precipitated by repeated vortexing for 10–15 sec after adding 50 μl of 10% ammonium molybdate. After separation of the phases, 0.1 ml of the upper (organic) phase was added to 0.2 ml of 0.73 N H2SO4 in ethanol and the phosphomolybdate complex was then reduced by adding 15 μl of a solution of stannous chloride (prepared by diluting 1 ml of 10% stannous chloride in 12 N HCl with 25 ml of 1 N H2S04). The blue color was read at 720 nm (OD = 4.1/μ mo Pi).
Radioactive assays were started by adding ATP (0–7.5 mM) plus 0.1 mM (5 μCi) of [γ-32P]ATP (Upstate) and incubated at 30 C for 30 min. Rates were obtained from the average of three time points at the steep increase of [32P]Pi release. The reaction was stopped and the inorganic/organic phases were separated as above. An aliquot (0.1 ml) of the organic phase was used to determine bound radioactivity by liquid scintillation counting. MgATPase activity was determined in terms of ATP hydrolysis as [32P]Pi release and expressed either as mole Pi per (mole NMM-II-B per minute) or in terms of Vmax as rate per second. The positive control was adult rat muscle myosin, which under these conditions yielded a maximum rate of 2.5/sec (not shown).
Molecular biology techniques
ER-specific antisense oligonucleotides (ASO) and random control oligonucleotide (CLO) were designed from the published sequences of the human ERα(22) (NM_012389); the human ERβ(23) (AJ002602); and the G protein-coupled receptor GPR30 [a membrane receptor responsive to the classical ligands of the ERα and ERβ(24), (AF027956)] by using HYBsimulator (Advanced Gene Computing Technologies, Irvine, CA) and choosing the melting temperature of the oligonucleotides as 34 C. Possible sequences that would have the given melting temperature were identified from the target mRNA, and the cross-hybridization against the whole sequence in the GenBank database of each oligonucleotide was calculated. The oligomers ASO that would hybridize to the coding region of the ERα, ERβ, and GPR30 mRNAs were selected as 5′-TCA TGG TCA TGG TCC GT-3′, 5′-CAT CAC AGC AGG GCT ATA-3′, and 5′-TTG GGA AGT CAC ATC CAT-3′, respectively. The CLO was composed of a random 18-mer sequence AGA ACG TTA CTT ACA CTG with GC content (32%) similar to the antisense oligonucleotides. The CLO was designed such that no cross-hybridization against the ER genes would occur.
To assess the effects of the ASOs and CLO on ERα, ERβ, or GPR30 mRNA expression, CaSki cells were treated with or without ASO or CLO at concentrations of 10 μM. RT-PCR assays were carried out after 2 d and assay experiments after 3–4 d. RT-PCR assays used the following primers and conditions: ERα forward, 5′-TTA CGA AGT GGG CAT GAT GA-3′, reverse, 5′-ATC TTG TCC AGG ACT CGG TG-3′(product size, 711 bp, 35 cycles); ERβ forward, 5′-GAG GCC TCC ATG ATG ATG TC-3′, reverse 5′-TCT CCA GCA GCA GGT CAT AC-3′(product size 610 bp, 35 cycles); GPR30 forward, 5′-TAA TAA GTC GAC GGG TCT CTT CCT-3′, reverse, 5′-ATT ATT GGA TCC TAC ACG GCA CTG C-3′(product size, 652 bp, 35 cycles) (AF027956); and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (XM_227696) forward 5′-CCA TGT TCG TCA TGG GTG TGA ACC A-3′, revers, 5′-GCC AGT AGA GGC AGG GAT GAT GTT C-3′(product size 229 bp, cycles 25). End points for successful transfections would have been 50% or greater reduction in ERα, ERβ, or GPR30 mRNA, compared with GAPDH mRNA [using real-time PCR as we described (3)]; no significant effects on GAPDH mRNA; and no significant effects of the CLO.
Determinations of CK2 activity and ERK1–2 activation
CK2 activity in cell lysates was determined by the ability to phosphorylate the decapeptide RRREEETEEE and expressed in terms of the accumulated Pi in nanomoles Pi per minute per milligram protein as we described (7). ERK1–2 activation status was determined in terms of ERK1–2 phosphorylation by Western blots and analysis by densitometry of the densities of the phosphorylated ERK1–2 relative to total ERK1–2 as we described (7).
Data presentation and analysis
Data are presented as means (± SD). Data of trends were fitted by using GB-STAT (Dynamic Microsystems Inc., Silver Spring, MD), and significance of differences was estimated by ANOVA.
Results
Modulation of NMM-II-B phosphorylation
In vivo (intact cells)
To understand whether estrogen modulates phosphorylation of NMM-II-B, cells were shifted to steroid-free medium and treated with one of the hormones or agents shown in Fig. 1, A and D. Drugs were used at doses and durations of time that produced submaximal-maximal effects based on data from preliminary experiments (not shown). The intracellular ATP pool of CaSki cells was labeled by incubation of cells with [32P]orthophosphate, and cell lysates were immunoprecipitated with the anti NMMHC-II-B antibody. Autoradiography revealed phosphorylation of NMM-II-B at baseline conditions in estrogen-depleted cells that increased after treatment with a physiological concentration of 17β-estradiol in the woman (10 nM) (Fig. 1, A and D). Treatments with ASOs for the ERβ and GPR30 or progesterone (1 μM, 48 h) had no effects on NMM-II-B phosphorylation, and cotreatments of these agents with 17β-estradiol did not significantly affect estrogen actions (not shown). Treatments with the ASO-ERα, ICI-182,780, or tamoxifen had no significant effect on NMM-II-B phosphorylation (Fig. 1D). However, in estrogen-treated cells, cotreatments with ASO-ERαICI-182,780, or tamoxifen blocked 17β-estradiol increase in NMM-II-B phosphorylation (Fig. 1, A and D). These data suggest the involvement of ERαin estrogen increase in NMM-II-B phosphorylation.
FIG. 1.

Phosphorylation of NMM-II-B filaments. A–D, Experiments in vivo (intact cells). CaSki cells were shifted for 3 d to steroid-free medium (SFM) and treated before assays with the vehicle (C, control) or 10 nM 17β-estradiol (E, 48 h), 10 μM ICI-182,780 (ICI, 24 h), DRB (6 h), or 10 μM staurosporin (STA, 8 h), alone or in combination. Cells were labeled with [32P]orthophosphate; lysates were immunoprecipitated (IP) with the anti-NMMHC-II-B antibody, fractionated on 6% PAGE, and autoradiographed (upper panels) or were immunoblotted only with the antibody (IB) (lower panels). The experiments were repeated three times with similar trends. D shows the densitometry analysis [means ±SD, arbitrary units (A.U.)] of the data in A–C as well as experiments using cells treated with 10 μM ASO-ERα(48 h) or tamoxifen (TMX, 24 h), alone or in combination with estrogen (Est). Data were analyzed in terms of phosphorylated NMM-II-B per total NMM-II-B and normalized to densitometry of the control band (10 U). a, P < 0.01–0.05, compared with SFM, C; b, P < 0.05–0.01, compared with SFM, C, and + Est. E–I, Phosphorylation by CK2 and PKC in vitro, determined in terms of ATP hydrolysis and [32P]Pi accumulation. E and F, Effects in vitro of CK2 (E) and PKC (F) using RRREEETEEE or the MARCKS-PSD, respectively, as substrates (added at concentrations of 1 and 5 μM, respectively). C, Control (no added kinase); HI, heat-inactivated kinases (5 min at 60 C). Experiments also involved coincubations with 50 μM DRB or 50 μM staurosporin (STA), respectively. Shown are means (± SD) of three experiments. a, P < 0.01. G–I, Effects of CK2 and PKC on phosphorylation of purified NMM-II-B filaments in vitro. G and H show dependence of CK2 (G) and PKC (H) on NMM-II-B concentration, in the absence (filled circles) and presence of inhibitors (empty circles) (DRB or staurosporin, respectively, both added at a concentration of 50 μM). Data were normalized to zero in control samples (C, no added kinase). I, Purified NMM-II-B filaments were coincubated with CK2 or PKC, in the absence or presence of 50 μM DRB or 50 μM staurosporin, respectively. Shown are means (±SD) of three experiments. a, P < 0.01.
Treatment of estrogen-depleted cells with the CK2 inhibitor DRB had no significant effect on NMM-II-B phosphorylation, but when coadministered with estrogen, it blocked estrogen increase in NMM-II-B phosphorylation (Fig. 1, B and D). These data suggest that CK2 is not significantly involved in phosphorylation of the NMM-II-B under baseline conditions, but it mediates estrogen increase in NMM-II-B phosphorylation.
Treatment of estrogen-depleted cells with the PKC inhibitor staurosporine decreased NMM-II-B phosphorylation (Fig. 1D), suggesting PKC is involved in the phosphorylation of NMM-II-B under baseline conditions. The effect of stau-rosporin was dose related, beginning already at 100 nM and reaching saturation at about 10 μM without causing toxic effects to cells (not shown). In cells cotreated with estrogen plus staurosporine, NMM-II-B phosphorylation was higher than in estrogen-depleted cells but lower than in cells treated only with estrogen (Fig. 1, C and D). These data suggest additive effects of estrogen and staurosporine.
In vitro (NMM-II-B filaments)
The data in Fig. 1, A–D, suggested involvement of PKC and CK2 in the baseline and the estrogen-induced phosphorylation of NMM-II-B, respectively. To test more directly whether CK2 and PKC can induce phosphorylation of NMM-II-B, purified NMM-II-B filaments were exposed in vitro to CK2 and PKC. The control experiments are shown in Fig. 1, E–I. Positive control experiments for CK2 included transfer of the [γ-32P] to the synthetic CK2-specific peptide RRREEETEEE (Sigma) (Fig. 1E), and for PKC the transfer of [γ-32P] to the phosphorylation site domain (PSD) of myristoylated Ala-rich C kinase substrate (MARCKS-PSD) (BIOMOL, Plymouth Meeting, PA). Negative control experiments used heat-inactivated CK2 or PKC. There were no kinase activities in the absence of added kinases, and in reactions done in the presence of heat-inactivated kinases, the kinase activities were significantly inhibited (Fig. 1, E and F). Also, [γ-32P]Pi release by CK2 was inhibited by more than 97% in the presence of DRB, and [γ-32P]Pi release by PKC was inhibited by more than 95% in the presence of staurosporine.
Fig. 1, G–I, shows that in experiments using purified NMM-II-B filaments as substrate, CK2 and PKC induced ATP hydrolysis in vitro. The effects of CK2 and PKC depended on the concentration of added NMM-II-B (i.e. the substrate), and reached saturation at about 300 ng/μl added NMM-II-B (Fig. 1, G and H). The effect of CK2 was inhibited by DRB (Fig. 1G) and that of PKC by staurosporine (Fig. 1H). Figure 1I summarizes three experiments, showing that both kinases induced significant ATP hydrolysis and that the effects could be inhibited by coincubations with DRB and staurosporine, respectively. Collectively, these data suggest that CK2 and PKC can directly phosphorylate NMM-II-B filaments.
Estrogen modulation of NMM-II-B filamentation
Phosphorylation of NMM-II-B filaments can lead to decreased dimerization (25) and abrogate filamentation (8). To understand whether the effects shown in Fig. 1 involve modulation of homodimerization of NMM-II-B filaments, NMM-II-B filaments were extracted, and filaments assembly status was determined in terms of the critical concentration of filaments necessary to induce homodimerization in vitro. The NMM-II-B critical concentration of filaments extracted from estrogen-treated cells was higher than of filaments extracted from estrogen-depleted cells (Fig. 2A). ICI-182,780 and DRB alone had no significant effect, but both drugs blocked estrogen effect (Fig. 2A). Treatment with staurosporine alone had no significant effect, and it did not modulate the effect of estrogen (Fig. 2A). Cotreatments with DRB plus staurosporine had no additional effect to that of staurosporine alone (not shown).
FIG. 2.
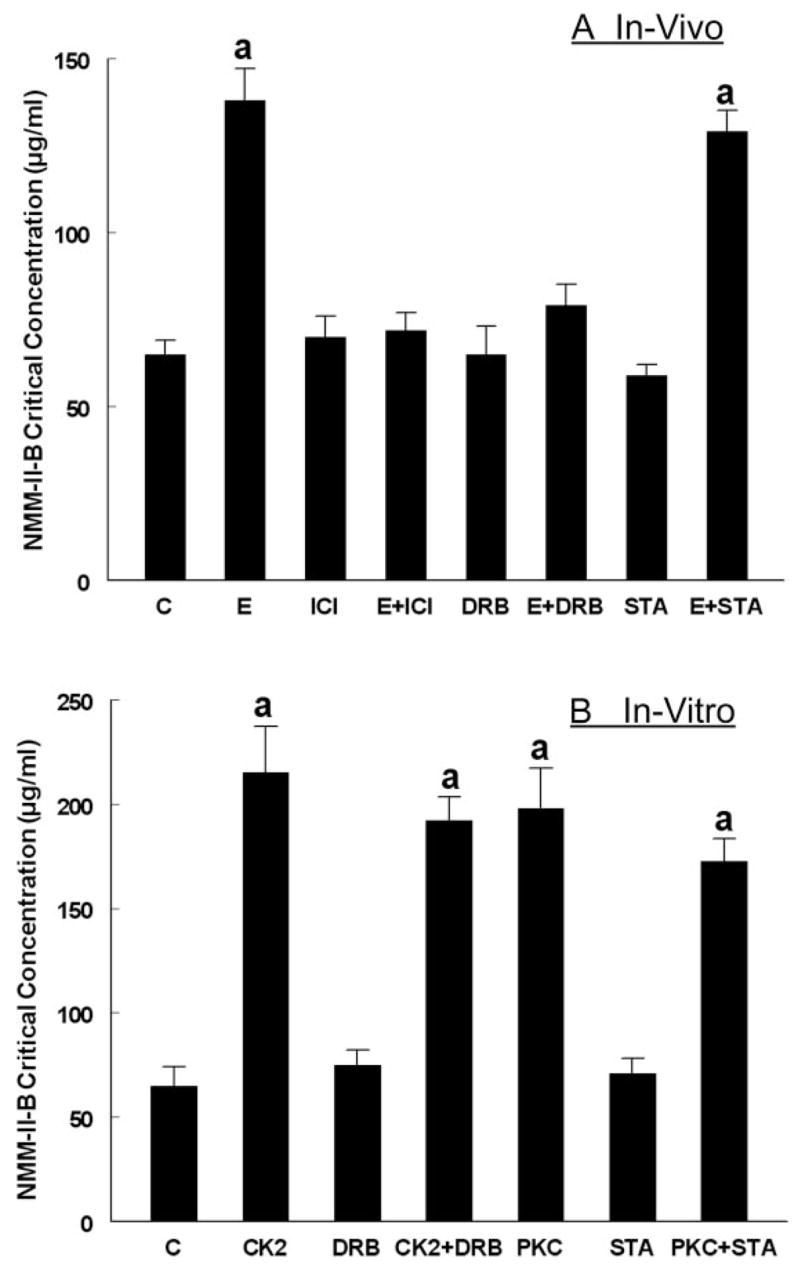
Assembly of purified NMM-II-B filaments determined in terms of the NMM-II-B critical concentration (CC). A, Experiments in vivo (intact CaSki cells, as in Fig. 1, A–C). B, Experiments in vitro, using purified NMM-II-B filaments (described in Fig. 1I). a, P < 0.02–0.01, compared with control (C).
The direct effects of CK2 and PKC on NMM-II-B filamentation are shown in Fig. 2B. When coincubated with purified NMM-II-B filaments, both kinases induced significant increases in the NMM-II-B critical concentration. DRB and staurosporine alone had no significant effects, but coincubations with DRB and staurosporine inhibited CK2 and PKC effects, respectively (Fig. 2B). These data suggest that CK2 and PKC directly decrease NMM-II-B filamentation.
Collectively, the data in Fig. 2 indicate that treatment with estrogen decreases NMM-II-B filamentation and suggest the involvement of ERα and CK2 in the estrogen inhibition of filamentation.
Estrogen modulation of NMM-II-B MgATPase activity
Phosphorylation and decreased filamentation of NMM-II-B could abrogate NMM-II-B MgATPase activity (8). To assay modulation of NMM-II-B MgATPase activity in CaSki cells, preliminary experiments were done to determine baseline conditions. The results shown in Fig. 3, A and B, used radioactive assays and the experiments in Figs. 4–6 used nonradioactive methods with similar trends. In the absence of actin, addition of ATP to purified NMM-II-B filaments induced a biphasic increase in ATP hydrolysis (insert in Fig. 3A): a fast increase that was too rapid to be accurately evaluated, followed by a slower time- and dose-related increase (Fig. 3A). The calculated Vmax [1.89 mmol Pi/(mole NMM-II-B per minute)] and the KATPase (67 μM ATP) were in the range reported for NMM-II-B from other types of nonmuscle nonepithelial cells (8). ATP hydrolysis was 10-fold higher when the assays were conducted in the presence of F-actin (Fig. 3B), and the calculated Vmax (0.115/sec) and KATPase (37 μM F-actin) were also in the range reported for other non-muscle nonepithelial cells (8).
FIG. 3.
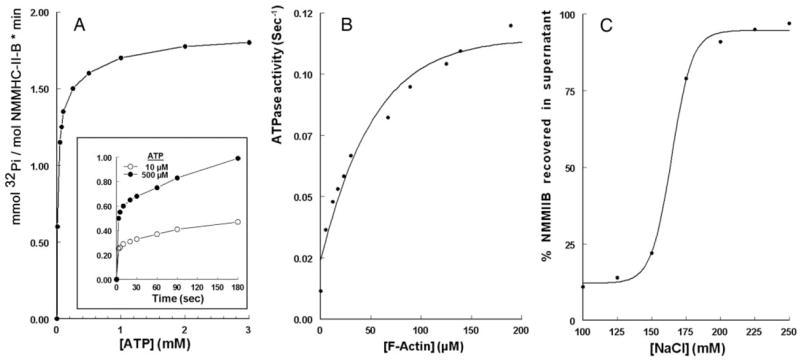
A, Steady-state ATP-dependence of ATP hydrolysis by purified NMM-II-B filaments. Insert, Time course of ATP hydrolysis. The ATP dose-related increase in [32P]Pi release was calculated from the slower steady-state increases in [32P]Pi release (>5 sec, insert). Data were fitted to the Michaelis-Menten equation: V = (ATP) · Vmax/KATPase + (ATP), P < 0.01 for two experiments. In the example, the calculated Vmax was 1.89 mmol Pi per (mole NMM-II-B per minute) and KATPase 67 μM ATP. B, Steady-state F-actin-dependent ATP hydrolysis by purified NMM-II-B filaments. Data were fit to the Michaelis-Menten equation: V = (actin) · Vmax/KATPase + (actin), P < 0.01 for two experiments. In the specific example, the calculated Vmax was 0.115/sec and the KATPase 37 μM F-actin. Experiments in A and B were done in low ionic (Na+ and Ca2+) strength to maintain NMM-II-B filaments in homodimerized state. C, Steady-state NaCl-dependent disassembly of NMM-II-B filaments, determined in terms of percent NMM-II-B recovered in the supernatant after incubation at 0–4 C with increasing concentrations of NaCl. The curve is means of two experiments.
FIG. 4.
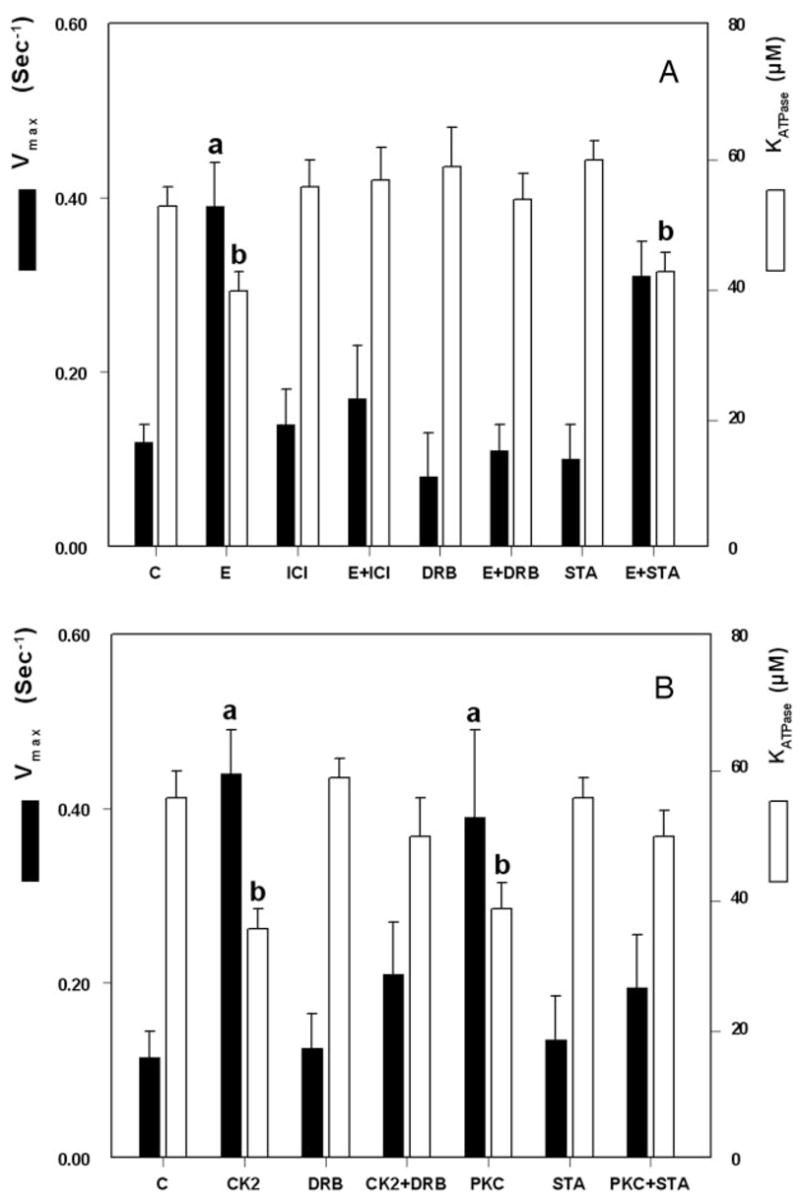
Steady-state F-actin-dependent ATP hydrolysis by purified NMM-II-B filaments. Vmax (filled bars) and KATPase (empty bars) were determined from fitting the results in each category to the Michaelis-Menten equation. A, NMM-II-B filaments were obtained from cells treated in vivo with one or more of the indicated drugs as in Fig. 1, A–C. B, Purified NMM-II-B filaments were coincubated with one or more of the indicated kinases and agents in vitro as described in Fig. 1I. Shown are means (±SD) of three to four experiments. a and b, P < 0.05–0.01, compared with control (C).
FIG. 6.
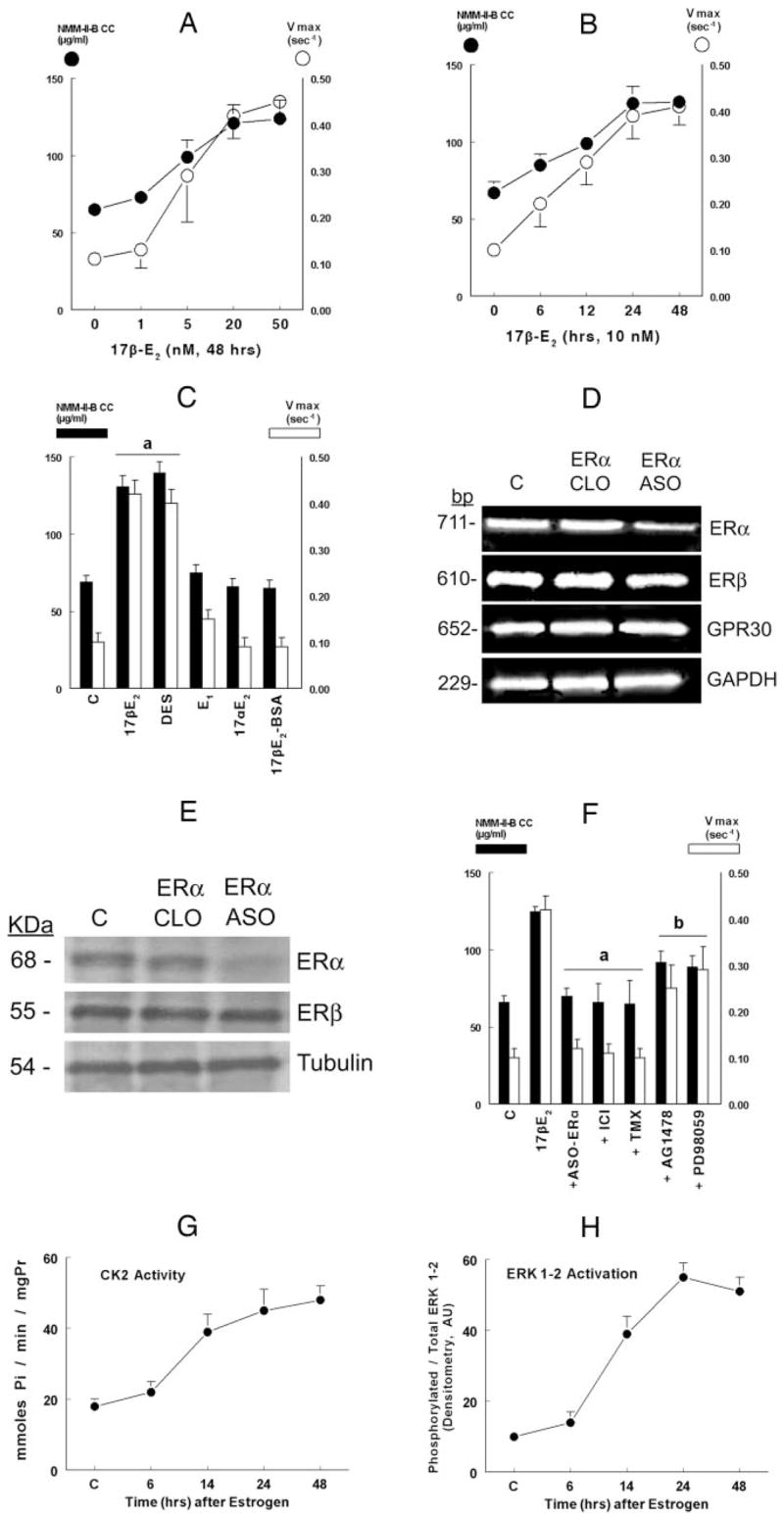
Pharmacology and mechanisms of estrogen effects. CaSki cells were shifted for 3 d to steroid-free medium and treated with the vehicle (C, control) or one of the indicated hormones/drugs/agents, alone or in combination, and purified NMM-II-B filaments were used for the experiments. Estrogen effects on NMM-II-B filamentation were determined in terms of NMM-II-B critical concentration (CC, filled circles or bars). Estrogen effects on F-actin-dependent ATP hydrolysis were determined in terms of Vmax (empty circles or bars). A, Dose-response reactions. Cells were treated with one of the indicated concentrations of 17β-estradiol (17β-E2) for 48 h. B, Time-response reactions. Cells were treated with 10 nM 17β-E2 for the indicated durations of time. C, Specificity of estrogen effect. Cells were treated with 10 nM 17β-E2 or one of the following estrogens (all for 48 h): diethylstilbestrol (DES, 10 nM); estrone (E1, 100 nM); 17β-estradiol (17βE2, 10 nM); and 17β-estradiol-6-[o-carboxymethyl] oxime-BSA (17β-E2-BSA, 1 μM). D and E, Effects of ASO-ERα on ERα, ERβ, GPR30, and GAPDH steady-state mRNA levels (determined by real-time PCR) (D) and ERα, ERβ, and tubulin protein steady-state levels (determined by Western blotting) (E). Cells were treated for 48 h with 10 nM 17β-E2 alone (C, control) or in combination with the CLO-ERα or ASO-ERα(both used at 10 μM for 48 h). In E, after blotting with the anti-ERα antibody, gels were reprobed and blotted with the antitubulin antibody. The experiments were repeated twice with similar trends. F, Effects of inhibitors on NMM-II-B filamentation and F-actin-dependent ATP hydrolysis. Cells were treated for 48 h with 10 nM 17β-E2 alone, or plus one of the following: ASO-ERα(10 μM, 48 h); ICI-182,780 (ICI, 10 μM, 24 h); tamoxifen (TMX, 10 μM, 24 h), AG1478 (5 μM, 12 h), or PD98059 (10 μM, 6 h). Shown are means ±SD of three to six repeats. a, P < 0.01, compared with control, C (serum-free conditions); b, P < 0.05–0.01, compared with control and 17β-E2. G and H, Estrogen effects on CK2 activity (G) and ERK1–2 activation (H). Estrogen-depleted cells were treated with 10 nM 17β-E2. At the indicated times after start of treatment, dishes with cells were used for the assays. Shown are means ±SD of three repeats; trends were significant at P < 0.01 in both cases. mgPr, Milligram (total cellular) protein (G). In H, data were normalized to densitometry of the control band [10 arbitrary units (AU)].
A similar methodology was used to determine effects of estrogen on NMM-II-B MgATPase activity. F-actin-MgATPase-Vmax was higher (Fig. 4A, filled bars), and F-actin-KATPase lower (Fig. 4A, empty bars) in NMM-II-B filaments extracted from estrogen-treated CaSki cells than from estrogen-depleted cells. ICI-182,780 and DRB alone had no significant effect on the Vmax or KATPase levels, but both drugs blocked estrogen effects (Fig. 4A). Staurosporine alone had no significant effect on Vmax or on KATPase, and it did not affect estrogen modulation of these rates (Fig. 4A).
The direct effects of CK2 and PKC on NMM-II-B MgATPase activity are shown in Fig. 4B. Both CK2 and PKC induced significant increases in MgATPase Vmax rates, and coincubations with DRB and staurosporine inhibited CK2 and PKC effects, respectively (Fig. 4B, filled bars). CK2 and PKC also decreased KATPase; DRB and staurosporine alone had no significant effects, but when coadministered with the kinases, respectively, they blocked the decreases in KATPase induced by the kinases (Fig. 4B, empty bars). These data suggest that CK2 and PKC directly increase NMMHC-II-B MgATPase activity.
Collectively, the data in Fig. 4 indicate that treatment with estrogen increases NMM-II-B MgATPase activity and suggest the involvement of the ERα and CK2 in estrogen effect.
Estrogen effects on NMMHC-II-B MgATPase are independent of effects on filamentation
The results in Figs. 2 and 4 suggested, for the first time, that estrogen can increase NMM-II-B MgATPase activity (Fig. 4) despite stimulating decreased NMM-II-B filamentation (Fig. 2). Until recently, traditional thinking was that MgATPase activity depends on homodimerization of the filaments. Accordingly, disassembled filaments, e.g. after treatment with estrogen (Fig. 2A) or CK2 (Fig. 2B), would expect to yield decreased MgATPase activity. Contrary to this thinking, the data in Figs. 2 and 4 showed that estrogen and CK2 increased MgATPase activity despite stimulating decreased filamentation.
To gain a better understanding of the effects of estrogen, the MgATPase activity assays were repeated in the presence of NaCl to block oligomerization of NMM-II-B filaments (19). The method was tested in Fig. 3C, showing that in the presence of increasing concentrations of NaCl, a greater fraction of the NMM-II-B filaments could be recovered in the supernatant, indicating disassembled NMM-II-B filaments. From the curve of Fig. 3C, two concentrations of NaCl were chosen for further assays: 125 and 200 mM, representing, respectively, conditions that would favor maximal and minimal oligomerization.
NaCl modulated rates of MgATPase ATP hydrolysis in response to the different treatments. In NMM-II-B filaments obtained from estrogen-depleted cells, coincubation in 200 mM NaCl abolished ATP hydrolysis (Fig. 5, A and B, filled bars). Coincubation in 125 mM NaCl of NMM-II-B filaments from cells treated with 17β-estradiol or filaments incubated in vitro with CK2 or PKC showed increased MgATPase activity, compared with experiments done in the absence of NaCl (Fig. 5, A and B, filled bars; compare with Fig. 4). Co-incubations in 200 mM NaCl decreased Vmax in filaments obtained from estrogen-treated cells or filaments incubated with CK2, but the Vmax levels were greater than in filaments of control cells (Fig. 5A). NaCl had no significant effect on KATPase rates in filaments obtained from estrogen-treated cells (Fig. 5B). In contrast, NaCl blocked MgATPase activity in filaments treated in vitro with PKC (Fig. 5, A and B).
FIG. 5.
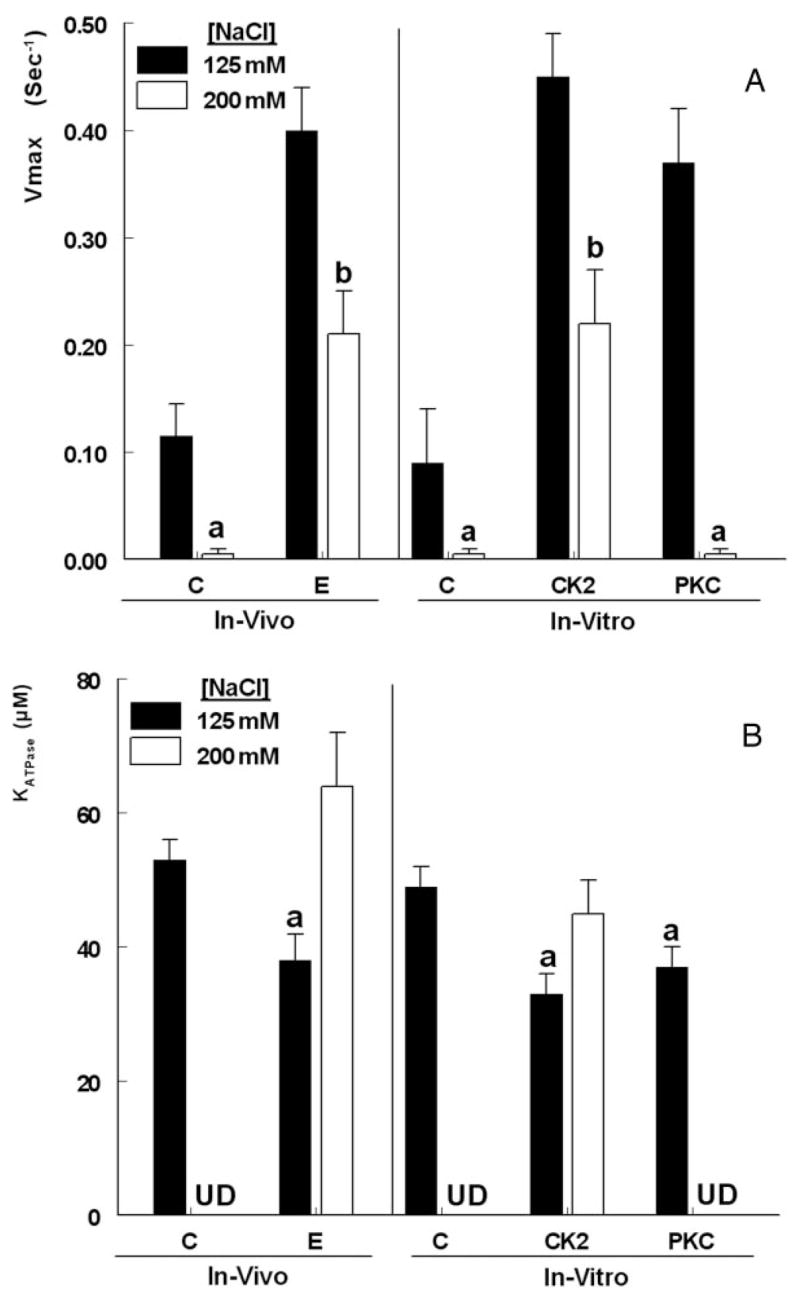
NaCl-dependence of steady-state F-actin-induced ATP hydrolysis by purified NMM-II-B filaments. A, Vmax. B, KATPase. Experiments used NMM-II-B filaments obtained from cells treated in vivo with 17β-estradiol (E) or NMM-II-B filaments obtained from estrogen-depleted cells (C, control) that were coincubated in vitro with CK2 or PKC. Experiments were conducted in the presence of 125 mM NaCl (filled bars) or 200 mM NaCl (empty bars). Shown are means (±SD) of three experiments. a, P < 0.01, compared with C; b, P < 0.05 [125 mM NaCl vs. 200 mM and compared with control (125 mM) conditions]. UD, Undeterminable.
These results suggest that increased solution ionic strength has little effect on NMM-II-B MgATPase activity in filaments obtained from estrogen-treated cells or in filaments treated with CK2 in vitro. In contrast, PKC increase in NMM-II-B MgATPase activity depends on the solution ionic strength and is abrogated under conditions of high NaCl.
Pharmacology and mechanisms of the estrogen effects
Figure 6, A–F, compared the effects of estrogen on NMM-II-B filamentation (filled circles or bars) and MgATPase activity (empty circles or bars). For both end points, the effects of 17β-estradiol were dose related, beginning at about 1 nM and reaching saturation at 20 nM (Fig. 6A); and time dependent, beginning after about 1–6 h of treatment and reaching plateau at about 24 h (Fig. 6B). Treatment with 10 nM diethylstilbestrol mimicked estrogen modulation of NMM-II-B filamentation and MgATPase activity; in contrast, treatments with the weak estrogenic ligand estrone, 17β-estradiol, or the nonpermeable estrogen 17β-E2-BSA had no significant effects (Fig. 6C).
The effects of the ASO for the ERβon ERs levels are shown in Fig. 6, D and E. ASO-ERαdecreased ERαsteady-state mRNA and protein levels without having a significant effect on ERβ, GPR30, and GAPDH steady-state mRNA levels (Fig. 6D) or on ERβ and tubulin protein steady-state levels (Fig. 6E). CLO-ERα had no effects on ERα, ERβ, and GAPDH steady-state mRNA levels and on ERα, ERβ and tubulin protein steady-state levels (Fig. 6, D and E). ASO-ERβ decreased ERβ steady-state mRNA and protein levels without having a significant effect on ERα, GPR30, and GAPDH steady-state mRNA levels and on ERα and tubulin protein steady-state levels (not shown). The ASO-GPR30 decreased GPR30 steady-state mRNA levels without having a significant effect on ERα, ERβ, and GAPDH steady-state mRNA levels and on ERα, ERβ, and tubulin protein steady-state levels (not shown). The CLO of ERβ and GPR30 had no effects on any of the above end points (not shown).
As is discussed in Figs. 1, 2, and 4 and confirmed (7) in Fig. 6F, in estrogen-depleted cells, treatments with the ASO for the ERα or with ICI-182,780 and tamoxifen had no significant effect on NMM-II-B filamentation or NMM-II-B MgATPase activity. Similarly, treatments with the ASO for the ERβ and GPR30 as well as with progesterone (1 μM, 2 d) alone had no significant effects on NMM-II-B MgATPase activity, and cotreatments of these agents/drugs with 17β-estradiol did not significantly affect estrogen action (not shown). In contrast, in estrogen-treated cells, cotreatments with the ASO-ERαICI-182,780 or tamoxifen blocked 17β-estradiol increase in NMM-II-B critical concentration and NMM-II-B MgATPase activity (Fig. 6F).
In estrogen-depleted cells, treatments with the EGFR inhibitor AG1478, ERK/MAPK inhibitor PD98059, protein kinase A H-89 inhibitor, or phosphatidylinositol 3-kinase-Akt inhibitor LY294002 had no significant effects on NMM-II-B filamentation or NMM-II-B MgATPase activity (not shown).
In estrogen-treated cells, cotreatment with H-89 or LY294002 had no significant effect on NMM-II-B filamentation or NMM-II-B MgATPase activity (not shown). In contrast, in estrogen-treated cells, cotreatments with AG1478 or PD98059 inhibited 17β-estradiol increase in NMM-II-B critical concentration and NMM-II-B MgATPase activity (Fig. 6F). Thus, in addition to the ERα and CK2, the data in Figs. 2–6 suggest the involvement also of the EGFR and ERK/MAPK cascades in estrogen decrease in NMM-II-B filamentation and estrogen increase in NMM-II-B MgATPase activity.
To better understand the estrogen signaling pathway, assays determined estrogen activation of CK2 and ERK1–2 using a recently described methodology (7). Treatment with 10 nM 17β-estradiol increased CK2 activity (Fig. 6G) and ERK1–2 activation status (Fig. 6H) in a time-related manner. Effects began about 6 h after treatment and reached plateau after about 24–48 h.
Effects of PKC activation
The CK2 experiments suggested that NMM-II-B MgATPase activity can be regulated independent of NMM-II-B filamentation, but the PKC experiments showed that MgATPase activity depends on NMM-II-B filamentation (Figs. 2 and 5). The estrogen data in Fig. 6 were similar to those of CK2 because treatment of CaSki cells in vivo with 17β-estradiol increased NMM-II-B MgATPase activity despite inhibiting NMM-II-B filamentation (Fig. 6F). To better understand the physiological significance of the PKC data, CaSki cells were treated with sn-1,2-dioctanoyl diglyceride (diC8), a cell permeable analog of diacylglycerol, and effects on NMM-II-B phosphorylation, NMM-II-B critical concentration, and MgATPase activity were determined. The rationale was that diacylglyceride is a signaling intermediate, and it can facilitate various signaling events including activation of PKC (26).
The first experiment tested diC8 activation of the PKC, in terms of translocation of the PKC from the cytoplasm to the cell membrane. Because PKC activation also involves autophosphorylation of the C terminus of the kinase, experiments also tested translocation of the phosphorylated form of the PKC (27). For experiments, estrogen-depleted CaSki cells were treated with 10 μM diC8 in the absence or presence of 15 μM of the Src family kinase inhibitor PP1 (28). In estrogen-depleted cells, treatment with diC8 was associated with increased immuno-reactivities to the anti PKCα and anti phosphopan PKC antibodies in plasma membrane enriched fractions of the cells, and both were suppressed by cotreatment with PP1 (Fig. 7A). Because the main PKC isoform in CaSki cells is PKCα(29), these data suggest activation of PKC by diC8. Treatment with 17β-estradiol had no effect on the translocations of PKC or phosphopan PKC (Fig. 7A). Neither diC8 nor 17β-estradiol produced any significant effect on cellular PKC content (Fig. 7A). These results indicate PKC activation by diC8 but not 17β-estradiol.
FIG. 7.
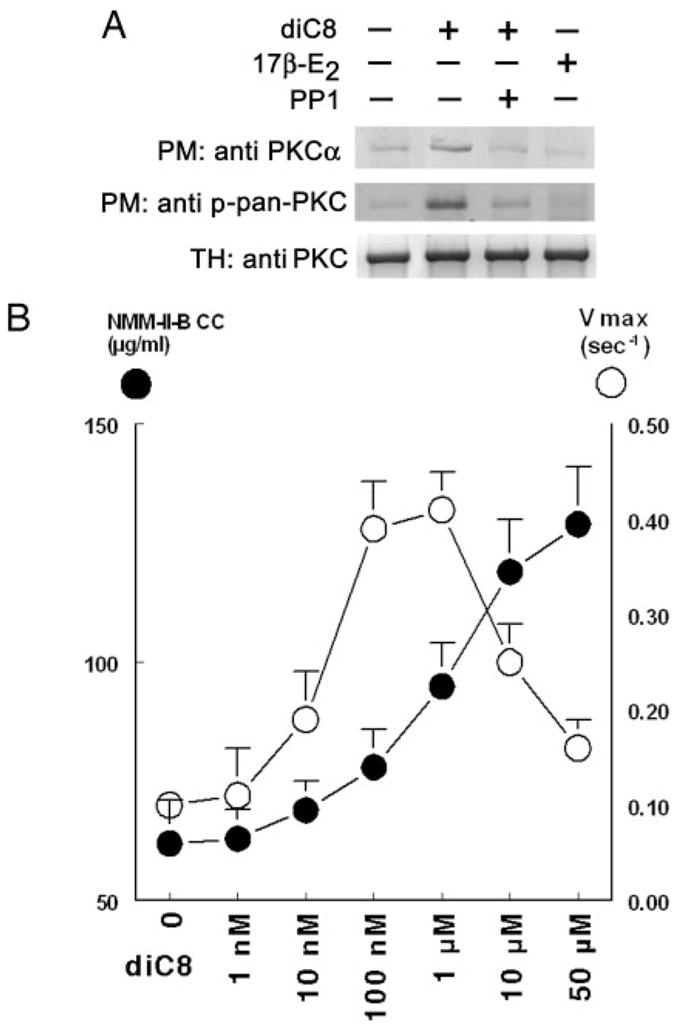
Effects of diC8 on PKC translocation (A) and NMM-II-B filamentation (B). A, Total homogenates (TH) or plasma membrane-enriched fractions (PM) were obtained from estrogen-depleted CaSki cells that were treated with 10 μM diC8 (30 min), 10 nM 17β-estradiol (17β-E2) (48 h), or 15 μM PP1 (12 h), alone or in combination. Western blots were done using the specified primary antibodies against PKCα or the phosphorylated PKC (phospho-pan PKC). Experiments were repeated twice with similar trends. B, Purified NMM-II-B filaments were obtained from estrogen-depleted CaSki cells that were treated 15 min before experiments with one of the indicated concentrations of dic8. NMM-II-B filamentation (filled circles) was determined in terms of NMM-II-B critical concentration (CC), and F-actin-dependent hydrolysis of ATP (filled circles) was determined in terms of Vmax. Shown are means ±SD of three to six repeats.
The second experiment studied the effects of diC8 on NMM-II-B filamentation and MgATPase activity. Treatment with diC8 produced a biphasic increase in NMM-II-B MgATPase activity: an early increase that began already with 1–10 nM diC8, reaching plateau at 0.1–1 μM and decreasing thereafter (Fig. 7B). diC8 also increased NMM-II-B critical concentration; the effect was monophasic and began with higher concentrations of diC8 (10–100 nM); it was saturable (dioC8 EC50 of 1–10 μM) and started to plateau only at about 50 μM (Fig. 7B). These data indicate that NMM-II-B MgATPase is more sensitive to the effects of diC8 than NMM-II-B filamentation and that treatment with diC8 at concentrations of 1 μM or greater will tend to inhibit NMM-II-B filamentation and block NMM-II-B MgATPase-activity.
Discussion
The main objective of the study was to understand the molecular mechanisms by which estrogen modulates NMM-II-B function in epithelial cells. The results in CaSki cells revealed that estrogen increased phosphorylation and inhibited NMM-II-B filamentation. Estrogen also increased NMM-II-B MgATPase activity, and the effect was unrelated to abrogation of NMM-II-B filamentation. These data suggest that NMM-II-B MgATPase activity can be regulated independent of NMM-II-B filamentation and challenge the prevailing hypothesis that MgATPase activity depends on filamentation.
The present results and our previous data (7) suggest that estrogen inhibits filamentation through decreased dimerization and facilitated disassembly of NMM-II-B filaments. Estrogen also increased NMM-II-B MgATPase activity: MgATPase activity of NMMHC-II-B filaments was high, even when assayed in high-salt environment when filaments are supposedly disassembled. Current thinking would dictate that decreased filamentation would abrogate MgATPase activity. However, the present results show the opposite: despite decreased filamentation, treatment with estrogen increased NMMHC-II-B MgATPase activity.
The possibility that the estrogen effect was artifactual is unlikely. Although the methodology of single turnover AT-Pase assay may be affected by the presence of damaged molecules that may lack proper regulatory properties (30), control experiments (not shown) indicated minimal/no yield of damaged NMM-II-B molecules during the purification process. Second, the baseline and the F-actin-induced MgATPase activity rates (Vmax and KATPase) were similar to those reported in other types of cells, suggesting that the purification method yielded intact and undamaged hexamers consisting of a pair of heavy chains plus the two pairs of light chains. Third, the PKC data are in line with the current prevailing hypothesis: in low-salt conditions PKC enhanced phosphorylation and inhibited filamentation of NMMHC-II-B, but it increased MgATPase activity. However, in high-salt conditions, which stimulate maximal degree of defilamentation (Fig. 3C), MgATPase activity was abrogated, and PKC had no additional effect. Moreover, treatments with high concentrations of diC8 (which presumably have induced greater activation of PKC) inhibited NMM-II-B filamentation and blocked NMM-II-B MgATPase activity.
One of the objectives of the study was to better understand estrogen signaling. The data suggest involvement of the ERα as proximal mediator because CaSki cells express ERα mRNA and protein (31), and estrogen modulation of NMM-II-B phosphorylation, filamentation, and MgATPase activity could be blocked with ASOs for the ERα and the ERα antagonists ICI-182,780 and tamoxifen. CaSki cells also express ERβ mRNA and protein (31) and mRNA for the GPR30, but treatment with ASOs for the ERβ and GPR30 did not block estrogen effect.
Less is known about signaling distal to the ERα. The present results suggest that PKC modulates constitutively NMM-II-B phosphorylation, filamentation, and MgATPase activity but ruled out the involvement of PKC as mediator of estrogen action. On the other hand, the data confirmed (7) that CK2 mediates estrogen modulation of NMM-II-B phosphorylation and filamentation. The data also suggest that CK2 is involved in estrogen modulation of NMM-II-B MgAT-Pase activity. This is based on the findings that the CK2 inhibitor DRB blocked estrogen modulation of NMM-II-B MgATPase activity and that CK2 directly modulated NMM-II-B MgATPase activity. CK2 is a ubiquitous, highly pleiotropic, and constitutively active serine/threonine protein kinase (32, 33). Of the more than 300 documented substrates for CK2, most are involved in gene expression, protein synthesis, and signaling but only few in the regulation of the cytoskeleton (32, 33), and only two reports documented the involvement of myosin II in smooth muscle cells (33, 34). The hypothesis that NMMHC-II-B of epithelial cells is a CK2 substrate is also of interest, given the known role of CK2 in cell survival, cell growth, and proliferation (35) and the known mitogenic properties of estrogen.
The present data also confirmed (7) the involvement of the EGFR and ERK/MAPK in estrogen modulation of NMM-II-B filamentation. The EGFR inhibitor AG1478 and the ERK/MAPK inhibitor PD98059 also attenuated estrogen increase of NMM-II-B MgATPase-activity (present results), suggesting involvement of the EGFR and ERK/MAPK mechanisms in estrogen modulation of NMM-II-B MgATPase activity. The involvement of other kinases/phosphatases has not been ruled out, although lack of effects by H-89 and LY294002 does not support the involvement of protein kinase A or the phosphatidylinositol 3-kinase-Akt cascades. Collectively, the present data suggest that estrogen modulation of NMM-II-B MgATPase activity involves the ERα, EGFR, and ERK/MAPK mechanisms as proximal mediators and the CK2 as a terminal mediator.
Estrogen effects on NMM-II-B phosphorylation, filamentation, and NMM-II-B MgATPase activity cannot not be explained by transcription regulation alone and most likely involve a more elaborate network of intermediary steps or ERα-mediated activation of kinase-initiated signaling cascades that participate in the terminal signaling cascade (36). EGFR is transcriptionally regulated by estrogen (37), including in human cervical cells (38), and estrogen up-regulates ERK1–2 activity in human vaginal-cervical cells (7). The interaction between the EGFR and ERK/MAPK pathways is well documented, and CK2 can be a substrate for the EGF/EGFR kinase mechanism (39). Therefore, it is possible that the EGFR is a rate-limiting mediator involved in activation of the CK2, and ERα-up-regulation of cellular EGFR increases activity of this kinase. EGFR, acting possibly via the ERK/MAPK pathway may activate CK2 and induce phosphorylation of NMM-II-B and activation of the NMM-II-B MgATPase. The present data therefore support concomitant actions of estrogen and CK2 on NMM-II-B phosphorylation and MgATPase activity. This hypothesis is supported by findings that CK2 can be a substrate for the EGF/EGFR kinase mechanism (39). The hypothesis may be physiologically relevant because EGF is constitutively secreted by stromal cervical and vaginal cells (40); the EGFR can be activated by changes in cell size (41); and the rate-limiting effect for EGF actions in many tissues is cellular levels of the EGFR (42).
One of the questions that the present data raise is how NMM-II-B MgATPase activity can be regulated independent of NMM-II-B filamentation. A possible explanation, based on studies in other cell types, is of different sets of phosphorylation sites that regulate the two functions. Myosin-II function regulation by phosphorylation of the light chains is relatively well understood (8, 43, 44). However, the present results suggest modulation of NMM-II-B heavy chain phosphorylation. In nonvertebrates, filamentation of myosin-II heavy chains is controlled by myosin heavy chain kinases A–C (45). In vertebrates, the assembly of myosin II monomers into filaments can be regulated by phosphorylation of heavy chains at three threonine residues at the C terminus of the tail (46), but dephosphorylation of the threonines, possibly by Rho-associated kinase-regulated myosin heavy-chain phosphatase (47) is a prerequisite for filament assembly.
A number of kinases are known to modulate myosin-II heavy chains phosphorylation, including Ca2+/calmodu-lin-dependent protein kinase type-II, cyclin-p34cdc2 kinase (48), p21-activated kinase 1 (49), CK2 (50), and PKC (51). The action of the latter can be mediated by the EGF mechanism (52). PKC and CK2 phosphorylation sites are located approximately 30 amino acids apart within the C terminus tail domain (51, 53). Studies by others showed that phosphorylation at or near the C terminus of the heavy chains tends to inhibit filament formation (25), make filaments shorter (54, 55), and inhibit actin-activated ATPase activity by reducing Vmax (54) or decreasing their affinities to actin filaments (55). PKC can also phosphorylate heavy chains at sites within the head region (56), but the sites have not been identified. In dimerized filaments, heads of both myosin molecules have to be phosphorylated before the MgATPase activity of either head can be activated by actin (57).
The present PKC data can be explained based on activation of sets of phosphorylation sites that are located at different domains of the NMMHC-II-B filaments, e.g. tail vs. head. Thus, sites, which regulate NMMHC-II-B MgATPase activity, are more sensitive to PKC than sites that regulate NMM-II-B filamentation. In contrast, the estrogen-CK2 data indicate similar sensitivity to the effects on NMM-II-B filamentation and NMMHC-II-B MgATPase activity. Whether a single CK2 phosphorylation site regulates the two functions or whether disparate sites having similar sensitivities to estrogen-CK2 exert those effects is being studied in the laboratory.
The present results may contribute to our understanding of estrogen regulation of permeability in the female reproductive tract. Estrogen modulation of the RTJ through modulation of occludin (3, 4) and RLIS through actin depolymerization were previously reported (6). The present result and our recent data (7) suggest a different and novel estrogen mechanism of RLIS regulation through myosin regulation of the cortical actomyosin ring. Estrogen-induced decrease of NMM-II-B filamentation and increase in NMMHC-II-B MgATPase activity would tend to favor decreased interaction with actin and enhanced detachment of the myosin from the cortical actin. Estrogen increases metabolic rate and cellular ATP activity (58), which would provide the energetic source for the rapid turnover of the NMMHC-II-B MgATPase.
Because all three estrogen effects lead to decreased para-cellular resistance, the question of specific function vs. redundancy needs discussion. The dose requirements of 17β-estradiol (EC50 of 1–3 nM) and the specificity and agonist profiles of all three effects are similar (Refs. 3, 4, 7 and the present results). However, the time courses are different: modulation of actin is relatively fast (1–6 h); modulation of NMM-II-B phosphorylation and NMMHC-II-B MgATPase-activity are slower (12–24 h), and modulation of occludin is the slowest (24–48 h). Clinically, estrogen modulation of vaginal-cervical permeability also depends on the length of exposure to estrogen. For instance, during the preovulatory phase of the menstrual cycle estrogen surge (which lasts 6–12 h) suffices to induce massive secretion of fluid, but the effect is limited in duration to about 12–24 h. It is possible that this effect, interpreted as increased water transudation secondary to decreased paracellular resistance, is mediated by estrogen modulation of cortical actin. In contrast, treatment of estrogen-deficient postmenopausal women requires prolonged administration of estrogen to induce appropriate lubrication of the lumen. The latter effect may possibly require the concerted actions of modulation of occludin (to decrease the RTJ) and modulation of the cortical actomyosin ring (to decrease RLIS through actin and NMM-II-B effects).
In summary, the data in human vaginal-cervical epithelial cells suggest that estrogen, in an effect mediated by the ERα and CK2, decreases NMM-II-B filamentation and increases NMMHC-II-B MgATPase activity and that the increase in MgATPase activity is unrelated to the modulation of filamentation. Decreased filamentation and increased NMMHC-II-B MgATPase activity could act in concert to destabilize the cytoskeletal cortical actomyosin ring, induce formation of a dynamic-elastic cytoskeleton, and lead to abrogation of the RLIS and increased permeability.
Supplementary Material
Acknowledgments
The technical support of Kimberley Frieden, Brian De-Santis, and Dipika Pal is acknowledged.
This work was supported by National Institutes of Health Grants HD29924 and AG15955.
Abbreviations
- ASO
Antisense oligonucleotide
- CK2
casein kinase-II
- CLO
random control oligonucleotide
- diC8
sn-1,2-dioctanoyl diglyceride
- DRB
5,6-dichloro-1-β-(D)-ribofuranosylbenzimidazole
- EGF
epithelial growth factor
- EGFR
EGF receptor
- ER
estrogen receptor
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- KATP
ATP-sensitive potassium channel
- NMM-II-B
nonmuscle myosin-II-B
- NMMHC-II
NMM-II heavy chain
- PKC
protein kinase C
- PSD
phosphorylation site domain
- RLIS
resistance of the lateral intercellular space
- RTJ
resistance of the tight junctions
- Vmax
maximal velocity
Footnotes
Disclosure Statement: The author has nothing to disclose.
References
- 1.Ussing HH, Zerahn K. Active transport of sodium as the source of electric current in the short-circuited isolated frog skin. Acta Physiol Scand. 1951;23:110–127. doi: 10.1111/j.1748-1716.1951.tb00800.x. [DOI] [PubMed] [Google Scholar]
- 2.Gorodeski GI. The cultured human cervical epithelium: a new model for studying transepithelial paracellular transport. J Soc Gynecol Invest. 1996;3:267–280. doi: 10.1016/s1071-5576(96)00028-7. [DOI] [PubMed] [Google Scholar]
- 3.Zeng R, Li X, Gorodeski GI. Estrogen abrogates transcervical tight junctional resistance by acceleration of occludin modulation. J Clin Endocrinol Metab. 2004;89:5145–5155. doi: 10.1210/jc.2004-0823. [DOI] [PubMed] [Google Scholar]
- 4.Zhu L, Li X, Zeng R, Gorodeski GI. Changes in tight junctional resistance of the cervical epithelium are associated with modulation of content and phosphorylation of occludin 65 kDa and 50 kDa forms. Endocrinology. 2006;147:977–989. doi: 10.1210/en.2005-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gorodeski GI. Estrogen increases the permeability of the cultured human cervical epithelium by modulating cell deformability. Am J Physiol. 1998;275:C888–C899. doi: 10.1152/ajpcell.1998.275.3.C888. [DOI] [PubMed] [Google Scholar]
- 6.Gorodeski GI. cGMP-dependent ADP-depolymerization of actin mediates estrogen increase in human cervical epithelia permeability. Am J Physiol. 2000;279:C2028–C2036. doi: 10.1152/ajpcell.2000.279.6.C2028. [DOI] [PubMed] [Google Scholar]
- 7.Li X, Zhou L, Gorodeski GI. Estrogen regulates epithelial cell deformability by modulation of cortical acto-myosin through phosphorylation of non-muscle myosin-heavy-chain II-B filaments. Endocrinology. 2006;147:5236–5248. doi: 10.1210/en.2006-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sellers JR. Myosins: a diverse superfamily. Biochim Biophys Acta. 2000;1496:3–22. doi: 10.1016/s0167-4889(00)00005-7. [DOI] [PubMed] [Google Scholar]
- 9.Jerdeva GV, Wu K, Yarber FA, Rhodes CJ, Kalman D, Schechter JE, Hamm-Alvarez SF. Actin and non-muscle myosin II facilitate apical exocytosis of tear proteins in rabbit lacrimal acinar epithelial cells. J Cell Sci. 2005;118:4797–4812. doi: 10.1242/jcs.02573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim KY, Kovacs M, Kawamoto S, Sellers JR, Adelstein RS. Disease-associated mutations and alternative splicing alter the enzymatic and motile activity of nonmuscle myosins II-B and II-C. J Biol Chem. 2005;280:22769–22775. doi: 10.1074/jbc.M503488200. [DOI] [PubMed] [Google Scholar]
- 11.Blair SA, Kane SV, Clayburgh DR, Turner JR. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab Invest. 2006;86:191–201. doi: 10.1038/labinvest.3700373. [DOI] [PubMed] [Google Scholar]
- 12.Golomb E, Ma X, Jana SS, Preston YA, Kawamoto S, Shoham NG, Goldin E, Conti MA, Sellers JR, Adelstein RS. Identification and characterization of non-muscle myosin II-C, a new member of the myosin II family. J Biol Chem. 2004;279:2800–2808. doi: 10.1074/jbc.M309981200. [DOI] [PubMed] [Google Scholar]
- 13.Rosenfeld SS, Xing J, Chen LQ, Sweeney HL. Myosin IIb is unconventionally conventional. J Biol Chem. 2003;278:27449–27455. doi: 10.1074/jbc.M302555200. [DOI] [PubMed] [Google Scholar]
- 14.Wang F, Kovacs M, Hu A, Limouze J, Harvey EV, Sellers JR. Kinetic mechanism of non-muscle myosin IIB: functional adaptations for tension generation and maintenance. J Biol Chem. 2003;278:27439–27448. doi: 10.1074/jbc.M302510200. [DOI] [PubMed] [Google Scholar]
- 15.Kovacs M, Wang F, Hu A, Zhang Y, Sellers JR. Functional divergence of human cytoplasmic myosin II: Kinetic characterization of the non-muscle IIA isoform. J Biol Chem. 2003;278:38132–38140. doi: 10.1074/jbc.M305453200. [DOI] [PubMed] [Google Scholar]
- 16.Gorodeski GI, Romero MF, Hopfer U, Rorke E, Utian WH, Eckert RL. Human uterine cervical epithelial cells grown on permeable support—a new model for the study of differentiation and transepithelial transport. Differentiation. 1994;56:107–118. doi: 10.1046/j.1432-0436.1994.56120107.x. [DOI] [PubMed] [Google Scholar]
- 17.Feng YH, Wang L, Wang Q, Li X, Zeng R, Gorodeski GI. ATP ligation stimulates GRK-3-mediated phosphorylation and β-arrestin-2- and dynamin-dependent internalization of the P2X7-receptor. Am J Physiol. 2005;288:C1342–C1356. doi: 10.1152/ajpcell.00315.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murakami N, Singh SS, Chauhan VP, Elzinga M. Phospholipid binding, phosphorylation by protein kinase C, and filament assembly of the COOH terminal heavy chain fragments of nonmuscle myosin II isoforms MIIA and MIIB. Biochemistry. 1995;34:16046–16055. doi: 10.1021/bi00049a019. [DOI] [PubMed] [Google Scholar]
- 19.Gorodeski GI, Eckert RL, Utian WH, Rorke EA. Maintenance of in vivo-like keratin expression, sex steroid responsiveness and estrogen receptor expression in cultured human ectocervical epithelial cells. Endocrinology. 1990;126:399–406. doi: 10.1210/endo-126-1-399. [DOI] [PubMed] [Google Scholar]
- 20.Murakami N, Chauhan VP, Elzinga M. Two nonmuscle myosin II heavy chain isoforms expressed in rabbit brains: filament forming properties, the effects of phosphorylation by protein kinase C and casein kinase II, and location of the phosphorylation sites. Biochemistry. 1998;37:1989–2003. doi: 10.1021/bi971959a. [DOI] [PubMed] [Google Scholar]
- 21.Pollard TD, Korn ED. Acanthamoeba myosin. isolation from acan-thamoeba castellanii of an enzyme similar to muscle myosin. J Biol Chem. 1973;248:4682–4690. [PubMed] [Google Scholar]
- 22.Walter P, Green S, Greene G, Krust A, Bornert JM, Jeltsch JM, Staub A, Jensen E, Scrace G, Waterfield M, Chambon P. Cloning of the human estrogen receptor cDNA. Proc Natl Acad Sci USA. 1985;82:7889–7893. doi: 10.1073/pnas.82.23.7889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ogawa S, Inoue S, Watanabe T, Hiroi H, Orimo A, Hosoi T, Ouchi Y, Muramatsu M. The complete primary structure of human estrogen receptor β(hER β) and its heterodimerization with ER α in vivo and in vitro. Biochem Biophys Res Commun. 1998;243:122–126. doi: 10.1006/bbrc.1997.7893. [DOI] [PubMed] [Google Scholar]
- 24.Carmeci C, Thompson DA, Ring HZ, Francke U, Weigel RJ. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics. 1997;45:607–617. doi: 10.1006/geno.1997.4972. [DOI] [PubMed] [Google Scholar]
- 25.Kuznicki J, Albanesi JP, Cote GP, Korn ED. Supramolecular regulation of the actin-activated ATPase activity of filaments of acanthamoeba myosin II. J Biol Chem. 1983;258:6011–6014. [PubMed] [Google Scholar]
- 26.Exton JH. Phospholipase D: structure, regulation, and function. Rev Physiol Biochem Pharmacol. 2002;144:1–94. doi: 10.1007/BFb0116585. [DOI] [PubMed] [Google Scholar]
- 27.Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ezumi Y, Shindoh K, Tsuji M, Takayama H. Physical and functional association of the Src family kinases Fyn and Lyn with the collagen receptor glycoprotein VI-Fc receptor-γ chain complex on human platelets. J Exp Med. 1998;188:267–276. doi: 10.1084/jem.188.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gorodeski GI, Hopfer U, Wenwu J. Purinergic receptor induced changes in paracellular resistance across cultures of human cervical cells are mediated by two distinct cytosolic calcium related mechanisms. Cell Biochem Biophys. 1998;29:281–306. doi: 10.1007/BF02737899. [DOI] [PubMed] [Google Scholar]
- 30.Ellison PA, Sellers JR, Cremo CR. Kinetics of smooth muscle heavy meromyosin with one thiophosphorylated head. J Biol Chem. 2000;275:15142–15151. doi: 10.1074/jbc.275.20.15142. [DOI] [PubMed] [Google Scholar]
- 31.Gorodeski GI, Pal D. Involvement of estrogen receptors α and β in the regulation of cervical permeability. Am J Physiol. 2000;278:C689–C696. doi: 10.1152/ajpcell.2000.278.4.C689. [DOI] [PubMed] [Google Scholar]
- 32.Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- 33.Pinna LA, Meggio F, Sarno S. Casein kinase-2 and cell signaling. In: Papa S, Tager JM, editors. Biochemistry of cell membranes. Basel: Birkhauser Verlag; 1995. pp. 15–27. [Google Scholar]
- 34.Kelley CA, Adelstein RS. The 204-kDa smooth muscle myosin heavy chain is phosphorylated in intact cells by casein kinase II on a serine near the carboxyl terminus. J Biol Chem. 1990;265:17876–17882. [PubMed] [Google Scholar]
- 35.Unger GM, Davis AT, Slaton JW, Ahmed K. Protein kinase CK2 as regulator of cell survival: implications for cancer therapy. Curr Cancer Drug Targets. 2004;4:77–84. doi: 10.2174/1568009043481687. [DOI] [PubMed] [Google Scholar]
- 36.Edwards DP. Regulation of signal transduction pathways by estrogen and progesterone. Annu Rev Physiol. 2005;67:335–376. doi: 10.1146/annurev.physiol.67.040403.120151. [DOI] [PubMed] [Google Scholar]
- 37.Levin ER. Bidirectional signaling between the estrogen receptor and the epidermal growth factor receptor. Mol Endocrinol. 2003;17:309–317. doi: 10.1210/me.2002-0368. [DOI] [PubMed] [Google Scholar]
- 38.Jacobberger JW, Sizemore N, Gorodeski GI, Rorke EA. Transforming growth factor β regulation of epidermal growth factor receptor in ectocervical epithelial cells. Exp Cell Res. 1995;220:390–396. doi: 10.1006/excr.1995.1330. [DOI] [PubMed] [Google Scholar]
- 39.Ackerman P, Glover CV, Osheroff N. Stimulation of casein kinase II by epidermal growth factor: relationship between the physiological activity of the kinase and the phosphorylation state of its beta subunit. Proc Natl Acad Sci USA. 1990;87:821–825. doi: 10.1073/pnas.87.2.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hom YK, Young P, Wiesen JF, Miettinen PJ, Derynck R, Werb Z, Cunha GR. Uterine and vaginal organ growth requires epidermal growth factor receptor signaling from stroma. Endocrinology. 1998;139:913–921. doi: 10.1210/endo.139.3.5817. [DOI] [PubMed] [Google Scholar]
- 41.Lezama R, Diaz-Tellez A, Ramos-Mandujano G, Oropeza L, Pasantes-Morales H. Epidermal growth factor receptor is a common element in the signaling pathways activated by cell volume changes in isosmotic, hyposmotic or hyperosmotic conditions. Neurochem Res. 2005;30:1589–1597. doi: 10.1007/s11064-005-8837-5. [DOI] [PubMed] [Google Scholar]
- 42.Bazley LA, Gullick WJ. The epidermal growth factor receptor family. Endocr Relat Cancer. 2005;1:S17–S27. doi: 10.1677/erc.1.01032. [DOI] [PubMed] [Google Scholar]
- 43.Trybus KM. Filamentous smooth muscle myosin is regulated by phosphorylation. J Cell Biol. 1989;109:2887–2894. doi: 10.1083/jcb.109.6.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ikebe M, Hartshorne DJ, Elzinga M. Phosphorylation of the 20,000-dalton light chain of smooth muscle myosin by the calcium-activated, phospholipid-dependent protein kinase. Phosphorylation sites and effects of phosphorylation. J Biol Chem. 1987;262:9569–9573. [PubMed] [Google Scholar]
- 45.Yumura S, Yoshida M, Betapudi V, Licate LS, Iwadate Y, Nagasaki A, Uyeda TQP, Egelhoff TT. Multiple myosinii heavy chain kinases: roles in filament assembly control and proper cytokinesis in Dictyostelium. Mol Biol Cell. 2005;16:4256–4266. doi: 10.1091/mbc.E05-03-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luck-Vielmetter D, Schleicher M, Grabatin B, Wippler J, Gerisch G. Replacement of threonine residues by serine and alanine in a phosphorylatable heavy chain fragment of Dictyostelium myosin II. FEBS Lett. 1990;269:239–243. doi: 10.1016/0014-5793(90)81163-i. [DOI] [PubMed] [Google Scholar]
- 47.Ito M, Nakano T, Erdodi F, Hartshorne DJ. Myosin phosphatase: Structure, regulation and function. Mol Cell Biochem. 2004;259:197–209. doi: 10.1023/b:mcbi.0000021373.14288.00. [DOI] [PubMed] [Google Scholar]
- 48.Tanaka E, Fukunaga K, Yamamoto H, Iwasa T, Miyamoto E. Regulation of the actin-activated Mg-ATPase of brain myosin via phosphorylation by the brain Ca2+, calmodulin-dependent protein kinases. J Neurochem. 1986;47:254–262. doi: 10.1111/j.1471-4159.1986.tb02857.x. [DOI] [PubMed] [Google Scholar]
- 49.Even-Faitelson L, Rosenberg M, Ravid S. PAK1 regulates myosin II-B phosphorylation, filament assembly, localization and cell chemotaxis. Cell Signal. 2005;17:1137–1148. doi: 10.1016/j.cellsig.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 50.Murakami N, Matsumura S, Kumon A. Purification and identification of myosin heavy chain kinase from bovine brain. J Biochem. 1984;95:651–660. doi: 10.1093/oxfordjournals.jbchem.a134654. [DOI] [PubMed] [Google Scholar]
- 51.Conti MA, Sellers JR, Adelstein RA, Elzinga M. Identification of the serine residue phosphorylated by protein kinase C in vertebrate nonmuscle myosin heavy chains. Biochemistry. 1991;30:966–970. doi: 10.1021/bi00218a012. [DOI] [PubMed] [Google Scholar]
- 52.Rosenberg M, Ravid S. Protein kinase-Cγ regulates myosin-IIB phosphorylation cellular localization and filament assembly. Mol Biol Cell. 2006;17:1364–1374. doi: 10.1091/mbc.E05-07-0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murakami N, Elzinga M, Singh SS, Chauhann VPS. Direct binding of myosin ii to phospholipid vesicles via tail regions and phosphorylation of the heavy chains by protein kinase C. J Biol Chem. 1994;269:16082–16090. [PubMed] [Google Scholar]
- 54.Collins JH, Kuznicki J, Bowers B, Korn ED. Comparison of the actin binding and filament formation properties of phosphorylated and dephos-phorylated Acanthamoeba myosin II. Biochemistry. 1982;21:6910–6915. doi: 10.1021/bi00269a045. [DOI] [PubMed] [Google Scholar]
- 55.Truong T, Medley QG, Cote GP. Actin-activated Mg-ATPase activity of dictyostelium myosin II. Effects of filament formation and heavy chain phosphorylation. J Biol Chem. 1992;267:9767–9772. [PubMed] [Google Scholar]
- 56.Ikeda N, Yasuda S, Muguruma M, Matsumura S. Protein kinase C phosphorylates both the light chains and the head portion of the heavy chains of brain myosin. Biochem Biophys Res Commun. 1990;169:1191–1197. doi: 10.1016/0006-291x(90)92022-r. [DOI] [PubMed] [Google Scholar]
- 57.Persechini A, Hartshorne DJ. Phosphorylation of smooth muscle myosin: evidence for cooperativity between the myosin heads. Science. 1981;213:1383–1385. doi: 10.1126/science.6455737. [DOI] [PubMed] [Google Scholar]
- 58.Ramamani A, Aruldhas MM, Govindarajulu P. Impact of testosterone and oestradiol on region specificity of skeletal muscle-ATP, creatine phos-phokinase and myokinase in male and female Wistar rats. Acta Physiol Scand. 1991;166:91–97. doi: 10.1046/j.1365-201x.1999.00576.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.