

Abstract
Alkylated perfluorooctanesulfonamides are compounds of environmental concern. To make these compounds available for environmental and toxicological studies, a series of N-alkylated perfluorooctanesulfonamides and structurally related compounds were synthesized by reaction of the corresponding perfluoroalkanesulfonyl fluoride with a suitable primary or secondary amine. Perfluoroalkanesulfonamidoethanols were obtained from the N-alkyl perfluoroalkanesulfonamides either by direct alkylation with bromoethanol or alkylation with acetic acid 2-bromo-ethyl ester followed by hydrolysis of the acetate. N-Alkyl perfluorooctanesulfonamidoacetates were synthesized in an analogous way by alkylation of N-alkyl perfluoroalkanesulfonamides with a bromo acetic acid ester, followed by basic ester hydrolysis. Alternatively, N-alkyl perfluoroalkanesulfonamides can be alkylated with an appropriate alcohol using the Mitsunobu reaction. Perfluorooctanesulfonamide was synthesized from the perfluorooctanesulfonyl fluoride via the azide by reduction with Zn/HCl. All perfluorooctanesulfonamides contained linear as well as branched C8F17 isomers, typically in a 20:1 to 30:1 ratio. The crystal structures of N-ethyl and N,N-diethyl perfluorooctanesulfonamide show that the S-N bond has considerable double bond character. This double bond character results in a significant rotational barrier around the S-N bond (ΔG≠ = 62–71 kJ mol−1) and a preferred solid state and solution conformation in which the N-alkyl groups are oriented opposite to the perfluorooctyl group to minimize steric crowding around the S-N bond.
Keywords: Environmental contaminants, Perfluorooctanesulfonamides, Perfluorobutanesulfonamides, Alkylation, Mitsunobu reaction, X-ray structure
1. Introduction
Perfluorooctanesulfonamides, such as N-alkyl perfluorooctanesulfonamidoethanols, N-alkyl perfluorooctanesulfonamides and perfluorooctanesulfonamide, have been used for over 40 years in a range of applications including pesticides, surfactants, surface treatments for clothes and home furnishings, paper protection and other miscellaneous applications [1, 2]. This group of compounds is ideally suited for these purposes because of unique properties such as excellent spreading characteristics, high surface activity and water and oil repellency. These properties are the result of the high electronegativity of fluorine, its large van der Waals radius compared to hydrogen, and the strong fluorine-carbon bond. Both the strength of the fluorine-carbon bond and the “shielding” effect of several fluorine atoms also result in the stability of perfluoroalkyl chains towards chemical, thermal and biological degradation. Medium-to-long chain perfluorinated surfactants, including perfluorooctanesulfonamides are, therefore, highly persistent in the environment and have been detected worldwide in a large range of environmental matrices and in humans, thus raising human health concerns [2].
Unfortunately, perfluorooctanesulfonamides are not readily available from commercial sources, which limits the ability to study the environmental impact and toxicity of these compounds and creates a need for straightforward and well-documented approaches for their synthesis. Perfluorooctanesulfonamides have been synthesized industrially from perfluorooctanesulfonyl fluoride by reaction with a suitable primary or secondary amine and further modification of the amide, e.g. by alkylation with chloroethanol. Laboratory syntheses of several perfluorooctanesulfonamides, for example perfluorooctanesulfonamidoethanol [3, 4] and N-ethyl perfluorooctanesulfonamide [5], have employed the same synthetic approach; however, a comprehensive description of the synthesis and characterization of these compounds for environmental and toxicological studies has not been reported. We herein report the synthesis and characterization of several environmentally relevant alkylated perfluorooctanesulfonamides from commercially available perfluorooctanesulfonyl fluoride for use in such studies. In addition, we synthesized several analogous perfluorobutanesulfonamides because perfluorobutanesulfonate based materials are emerging as a replacement for perfluorooctanesulfonate based materials [6] and as model compounds for the study of their atmospheric chemistry [7].
2. Results and Discussion
2.1. Synthesis of N-alkyl and N,N-dialkyl perfluoroalkanesulfonamides from perfluoroalkanesulfonyl fluorides
Monoalkylated perfluorooctanesulfonamides are typically synthesized by reaction of perfluorooctanesulfonyl fluoride (1) with an excess of the respective amine, e.g. 3a–c, in diethyl ether or dioxane (for a review see [2]). This reaction initially forms a complex ammonium salt with a proposed formula of C8F17SO2NR− · NRH3+ · NRH3F. The desired alkylated perfluorooctanesulfonamide 5a–c is subsequently isolated after thermal decomposition of this complex salt [8–11] or by treatment of the reaction mixture with hydrochloric or sulfuric acid [11–13]. Following these previous reports, we initially investigated the reaction of perfluorooctanesulfonyl fluoride (1) by using an excess of the respective alkyl amine, e.g. methyl- (3a) or ethylamine (3b), as a base (Scheme 1). Subsequent experiments studied the use of an external base such as triethylamine or pyridine. These efforts and our attempts to further optimize the reaction conditions are summarized in Table 1.
Scheme 1.

Synthesis of mono- and dialkylated perfluorooctanesulfonamides.
Table 1.
Synthesis of mono- and dialkylated perfluorooctanesulfonamides from perfluorooctanesulfonyl fluoride
| Entry | Amine | Base | Solvent | Reaction conditions | Method | Percentage linear: isopropyl branched isomer | Yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | 3a | 3a | None | AT | C | n.d. | 70b,c |
| 2 | 3b | 3b | None | AT | C | n.d. | 70b,c |
| 3 | 3a | 3a | Ether | AT | A | 98:2 | 43 |
| 4 | 3a | 3a | Ether | Reflux | A | 98:2 | 24–28d |
| 5 | 3a | NEt3 | DCM | Reflux | A | 92:8 | 46e |
| 6 | 3a | NEt3 | Ether | Reflux | B | 92:8 | 46 |
| 7 | 3a | NEt3 | Ether | Reflux | B | 98:2 | 43f |
| 8 | 3b | 3b | Ether | AT | A | 96:4 | 43 |
| 9 | 3b | 3b | Ether | Reflux | B | 98:2 | 48–50 |
| 10 | 3b | NEt3 | DCM | AT | B | 95:5 | 57 |
| 11 | 3b | NEt3 | Ether | AT | B | 97:3 | 46g |
| 12 | 3b | NEt3 | Ether | Refluxh | B | 96:4 | 42–58 |
| 13 | 3b | Pyridine | Ether | AT | A | 98:2 | 51i |
| 14 | 3b | NEt3 | Dioxane | Refluxg | B | 99:1 | 56f |
| 15 | 6d | NEt3 | Ether | Reflux | E | 99:1 | 55 |
| 16 | 6e | NEt3 | None | Reflux | E | 94:6 | 69b,c |
| 17 | 6f | NEt3 | Ether | Reflux | B | 94:6 | 53 |
| 18 | 6g | 6g | Ether | Reflux | D | 95:5 | 76 |
The percentage of the linear and isopropyl branched branched isomer was determined based on the integral of the respective CF3 signals;
Slightly yellow, waxy solid with several peaks in the gas chromatogram;
Recrystallized from reagent alcohol/dichloromethane
The low yield is probably due to the ten times smaller scale of these reaction compared to most reactions reported in this Table;
Worked-up with diluted hydrochloric acid to remove traces of pyridine;
Non-aqueous work-up was adopted;
Reaction mixture was hydrolyzed with saturated NaHCO3 solution;
20 hours of reaction time;
Dark brown crude product;
AT = ambient temperature; DCM = dichloromethane; n.d. = not determined.
Initial experiments employed the two gaseous amines 3a and 3b both as reactants and as base at ambient temperature, both with (Method A) and without solvent (Method C). Although apparently higher yields were obtained under the solvent free conditions of Method C (Entries 1 and 2), the products contained significant amounts of impurities (i.e., showed several peaks in the gas chromatogram) and were slightly colored even after Kugelrohr distillation and/or recrystallization. Therefore, we abandoned this approach and focused on reaction conditions employing a solvent such as diethyl ether. Reaction of perfluorooctanesulfonyl fluoride with 3a and 3b at ambient temperature yielded 43% of the desired sulfonamide (Entries 3 and 8). To facilitate the decomposition of the thermally unstable complex ammonium salt we also heated the reaction mixture under reflux after addition of the amine 3b (Entry 9) [9]. In the case of ethylamine (3b) this additional heating step appears to give a slightly higher yield and, therefore, was used in most subsequent reactions.
A major drawback of using excess 3a or 3b as a base is the formation of a white precipitate which made it difficult to monitor the addition of the gaseous amines 3a and 3b and to workup these reactions. Therefore, we investigated the use of an external base, for example triethylamine or pyridine, which improved the yield by 13–16%, reduced the reaction time and gave overall more reproducible yields (Entries 6, 12 and 13). Both pyridine and triethylamine gave similar yields. Although the reaction conditions were identical for the synthesis of 4a and 4b, the yield of N-methyl perfluorooctanesulfonamide 4a was always lower compared to its ethyl analog 4b. This difference in yield is probably due to difficulties in handling the low boiling methyl amine (Entry 4).
We also studied the influence of selected solvents (i.e., ether, dioxane and dichloromethane) on the yield of the reaction (Entries 5, 10 and 11). Pyridine, which has been used previously as solvent for the synthesis of perfluorooctane-1-sulfonic acid (2,2-dimethoxy-ethyl)-amide from perfluorooctanesulfonyl fluoride 1 [3], was not investigated because it is typically difficult to remove residual pyridine traces. We found that the solvent had little-to-no effect on the overall yield. However, dichloromethane as a solvent resulted in a significantly colored reaction mixture and is, therefore, a less suitable solvent for this type of reaction (Entry 10). This is surprising because reactions of shorter chain perfluoroalkylsulfonyl fluorides and chlorides with alkyl amines can be performed using a broad range of solvents (e.g. dichloromethane) and reaction conditions [2].
To facilitate the conversion of the complex ammonium salt to the desired perfluorooctanesulfonamide, we investigated different aqueous workup conditions such as acidic and basic aqueous workup (Entries 5 and 11, respectively). Overall, no improvement in the yield of the reaction was observed suggesting that the complex ammonium salt readily decomposes under the reaction conditions employed. In subsequent experiments we avoided an aqueous workup altogether and directly purified the product by column chromatography as outlined in Method B (Entries 7 and 11). The reaction conditions of Method B, with slight modifications (i.e., acetonitrile as solvent), were also employed to synthesize the corresponding perfluorobutane-1-sulfonamides 5a and 5b from perfluorobutanesulfonyl fluoride 2 in good yields.
The dialkyl perfluoroalkanesulfonamides 7d–g and 8g, compounds of interest as analytical standards, were prepared from fluoride 1 or 2 and the respective dialkyl amines 6d–g using the same strategies employed for the monoalkyl derivatives 4a and 4b (Method D and E, Entries 15–18). The best results with regard to purity were obtained using an excess of the amine as a base (Method D, entries 15 and 17).
2.2. Isomer composition of perfluorooctanesulfonyl amides
Commercially available perfluorooctanesulfonyl fluoride 1 is manufactured by electrochemical fluorination, a process that results in a significant number of organic and inorganic byproducts. Only approximately 70% of perfluorooctanesulfonyl fluoride 1 are the desired linear isomer [2]. Significant amounts of various branched C8F17 isomers with the general structure C8F17SO2F are also present. These include (F3C)2CF(CF2)5SO2F (isopropyl), F3C(CF2)xCF(CF3)(CF2)5–xSO2F (internally branched) and (F3C)3C(CF2)4SO2F (t-butyl). We investigated recrystallization and column chromatography techniques to purify the crude product and, if possible, to remove branched impurities. Among these two purification methods only column chromatography on silica gel was able to remove a significant percentage of the branched perfluorooctanesulfonamides and other fluorinated impurities. Initial fractions were eluted with ethyl acetate-hexane as eluent and, based on 1H and 19F NMR spectroscopy, contained a mixture of branched impurities (major), some linear isomer (minor) and other, uncharacterized impurities. The straight chain isomer and some branched impurities were eluted by further increasing the polarity of the mobile phase (up to 7% of ethyl acetate). In these later fractions the ratios of linear to branched perfluorooctanesulfonamide were determined using 19F NMR spectroscopy. The ratios of linear to isopropyl perfluorooctanesulfonamide (F3C)2CF(CF2)5SO2NRR″ typically ranged from 10:1 to 30:1 in all perfluorooctanesulfonamides synthesized (e.g. Table 1). All samples also contained small quantities of the t-butyl and other branched perfluorooctanesulfonamides. Recrystallization did not remove all branched impurities, but worked well to remove the dark brown color from the crude product.
The perfluorobutanesulfonyl fluoride 2 used in this study was most likely synthesized by electrochemical fluorination of the corresponding butanesulfonyl fluoride [2]. In contrast to its long chain homologue 1, the 19F NMR spectrum of perfluorobutanesulfonyl fluoride 2 showed only the four signals of the perfluorobutyl chain. No impurities could be detected in the 19F NMR spectrum even with neat 2. The high purity of fluoride 2 is not unexpected because the electrochemical fluorination of shorter-chain alkanesulfonyl chlorides and fluorides is known to give the respective fluorides in good purities and excellent yields [1]. As a result, the perfluorobutanesulfonamides 5a–c derived from fluoride 2 are also free of branched impurities.
2.3. Synthesis of N,N-dialkyl perfluoroalkanesulfonyl amides
N-methyl-N-(2-hydroxyethyl)-perfluorooctane-1-sulfonamide 9a and N-ethyl-N-(2-hydroxyethyl)-perfluorooctane-1-sulfonamide 9b were important industrial products and are known environmental contaminants [2]. Both amides have been industrially synthesized by alkylation of the respective amides, for example 4a or 4b, with chloroethanol [14], oxirane [15] or 1,3-dioxolan-2-one (ethylene carbonate) [4]. Following these earlier (patent) reports we initially investigated the synthesis of perfluorooctanesulfonamide 7f by alkylation of N-methyl perfluorooctanesulfonamide 4a with iodoethane. The desired perfluorooctanesulfonamide 7f was obtained in 78% yield when sodium methoxide was employed as a base [16], but the reaction was more straightforward and the yields significantly improved when potassium carbonate was used as a base.
We subsequently investigated the alkylation of the N-alkyl perfluorooctanesulfonyl amides 4a and 4b with bromoethanol using potassium carbonate as base and catalytic amount of sodium iodide in acetone (Scheme 2). Initial experiments gave low yields of the desired sulfonamides 9a and 9b due to impurities present in commercially available bromoethanol; however, good yields ranging from 90 to 99% were obtained by alkylation of 4a and 4b with freshly distilled bromoethanol. In a second approach (Scheme 2), the N-alkyl amide 4a or 4b was alkylated with 2-bromoethyl acetate in the presence of the potassium carbonate base and catalytic amount of sodium iodide. The acetates 10a or 10b were deacetylated with aqueous potassium hydroxide to yield the desired sulfonamides 9a and 9b in 65% and 93% yield, respectively. Because of the overall lower yields, this approach is less suitable for the synthesis of the dialkylated sulfonamides.
Scheme 2.

Synthesis of mono- or N-alkylated perfluorooctanesulfonamidoethanol 9a and 9b.
We also employed a similar alkylation reaction for the synthesis of N-ethyl perfluorooctanesulfonamideacetate 12. As shown in Scheme 3, alkylation of an N-ethyl perfluorooctanesulfonamide with a bromoacetic acid methyl ester yields the desired ester 11 in good yields. We initially investigated the base catalyzed hydrolysis (2–10% aqueous as well as ethanolic KOH solutions) of ester 11 to obtain the desired acid. These severe reaction conditions gave a complex mixture of uncharacterized decomposition products. However, controlled basic hydrolysis [17] yielded the desired acid 12 in moderate yield and acceptable purity.
Scheme 3.

Synthesis of 2-(N-ethyl perflurooctanesulfonamido)acetic acid 12
2.4. Synthesis of N,N-dialkyl perfluoroalkanesulfonamides using the Mitsunobu reaction
The Mitsunobu reaction has been successfully employed for the N-alkylation of derivatives of triflamide (CF3SO2NH2) with various alcohols [18–20]. We investigated this approach as an alternative route to N,N-dialkyl perfluorooctanesulfonamides. As shown in Scheme 4, the Mitsunobu reaction of 4b with methyl glycolate gave ester 11 in good yield. The Mitsunobu reaction of 4b with ethylene glycol did not yield the desired product 9b; however, the Mitsunobu reaction of 4b with TBDMS (t-butyldmethylsilyl) protected ethylene glycol 13 gave the desired product 14 in 98% yield. Hydrolysis of 14 using hydrochloric acid at 25°C yielded 9b in 96% yield. Overall, the Mitsunobu reaction gives the target molecules in good to excellent yields and, thus, is an alternative approach for the alkylation of perfluoroalkanesulfonamides.
Scheme 4.

Synthesis of N,N-dialkylated perfluorooctanesulfonamides 11 and 14 from N-alkylated perfluorooctanesulfonamides using the Mitsunobu reaction.
2.4. Synthesis of perfluorooctanesulfonamide 17 from perfluoroalkanesulfonyl fluoride
Perfluorooctanesulfonamide 17 can be prepared by reaction of ammonia with perfluorooctanesulfonyl fluoride 1 [8–11]. We explored a different approach that avoids the use of ammonia and, as shown in Scheme 5, prepared the amide 17 in a two step synthesis via the azide. In the first step, perfluorooctanesulfonyl fluoride was reacted with an aqueous solution of sodium azide at room temperature to yield the perfluorooctanesulfonyl azide 15. This azide 15 was subsequently converted into the perfluorooctanesulfonamide 16 by treatment with zinc and aqueous hydrochloric acid [21].
Scheme 5.

Synthesis of perfluorooctanesulfonamide 16.
2.5. Structure of N-alkyl and N,N-dialkyl perfluoroalkanesulfonyl amides
To fully understand the biological effects of perfluorooctanesulfonamides it is important to know their three-dimensional structures. Although (partially) fluorinated compounds are often very difficult to crystallize [22], typically forming exceedingly thin, poorly stacked platelets, we were able to determine the crystal structure of N-ethyl-perfluorooctane-1-sulfonamide (4b), N,N-diethyl-perfluorooctane-1-sulfonamide (7e) and 2-(N-ethyl-perfluorooctanesulfonamido) acetic acid (12).
The crystal structure of 4b is shown in Figure 1. Crystals of amide 4b are triclinic (space group P1̄) with a = 6.3411(1), b = 9.5670(3), c = 28.2303(9) Å, and α = 88.579(2)°, β = 85.758(2)°, γ = 87.348(2)°. The asymmetric unit contains two independent molecules that are rotational isomers and are enantiomeric to each other (Figure 1). The crystal structure of 7e is shown in Figure 2. Amide 7e is monoclinic (space group P21/c) with a = 7.3103(7), b = 6.4514(6), c = 39.904(4) Å, and α = 90°, β = 95.089(5)°, γ = 90°. An interesting feature of this crystal structure is that the perfluorooctyl chains of the disorder components are enantiomeric – they spiral around both ways, thus resulting in a disordered crystal. This is not surprising because perfluoroalkane chains typically adopt a helical conformation resulting from the larger van der Waals radius of fluorine relative to hydrogen (see Figure 2B) [23]. The crystal structure of 12 is shown in Figure 3. Amide 12 is also monoclinic (space group Pc; an alternative model in P2/c was less satisfactory) with a = 33.150(3), b = 5.8124(7), c = 10.0365(15) Å, and α = 90°, β = 91.220(6)°, γ = 90°. Since crystallographic study of compounds containing perfluorinated chains is generally very difficult owing to poor quality crystals, the quality of resulting structures is often somewhat lower than for typical small-molecule structure determinations (vide infra). Although this is particularly true of amide 12, it is nevertheless possible to extract chemically meaningful results from the structure models.
Figure 1.

Structure of N-ethyl-perfluorooctane-1-sulfonamide (4b). Displacement ellipsoids are drawn at the 50% probability level.
Figure 2.

View of N,N-diethyl-perfluorooctane-1-sulfonamide (7e) showing the atom-labelling scheme. (A) Structure of 7e showing the atom-labelling scheme. (B) View of 7e along the carbon backbone to illustrate the helical conformation of the perfluorooctyl chain of the major component in the crystal. Displacement ellipsoids are drawn at the 50% probability level.
Figure 3.

View of 2-(N-ethyl-perfluorooctanesulfonamido) acetic acid (12) showing the atom-labelling scheme. Displacement ellipsoids are drawn at the 50% probability level.
As illustrated in Figures 4 to 6, the packing diagrams of all three crystal structures show segregation of the electronically different parts of the perfluorooctanesulfonamide molecules to form bilayers with a perfluoroalkyl core which are separated by the sulfonamide groups. Specifically, both 4b and 7e form bilayers parallel to the a–b plane (Figures 4 and 5), whereas 12 forms bilayers parallel to the b–c plane (Figure 6). The respective perfluorooctanesulfonamides molecules are tilted within the bilayer by approximately 60° (4b), 35° (7e) and 80° (12), respectively. The formation of bilayers by all three perfluorooctanesulfonamides is not a surprising observation because A···B interactions (e.g., interactions between different parts of a molecule) are usually less favorable than the mean of A···A and B···B interactions (i.e., interactions between similar parts of a molecule) [22]. This is particularly true for the hydrophobic and lipophobic perfluoroalkyl chains and, in the case of 12, the -COOH groups which from dimers linked by hydrogen bonds in the crystal structures.
Figure 4.

Packing diagram of N-ethyl-perfluorooctane-1-sulfonamide (4b), viewed down the b axis, illustrating the bilayers of 4b oriented parallel to the a–b plane. H atoms have been omitted for clarity.
Figure 6.
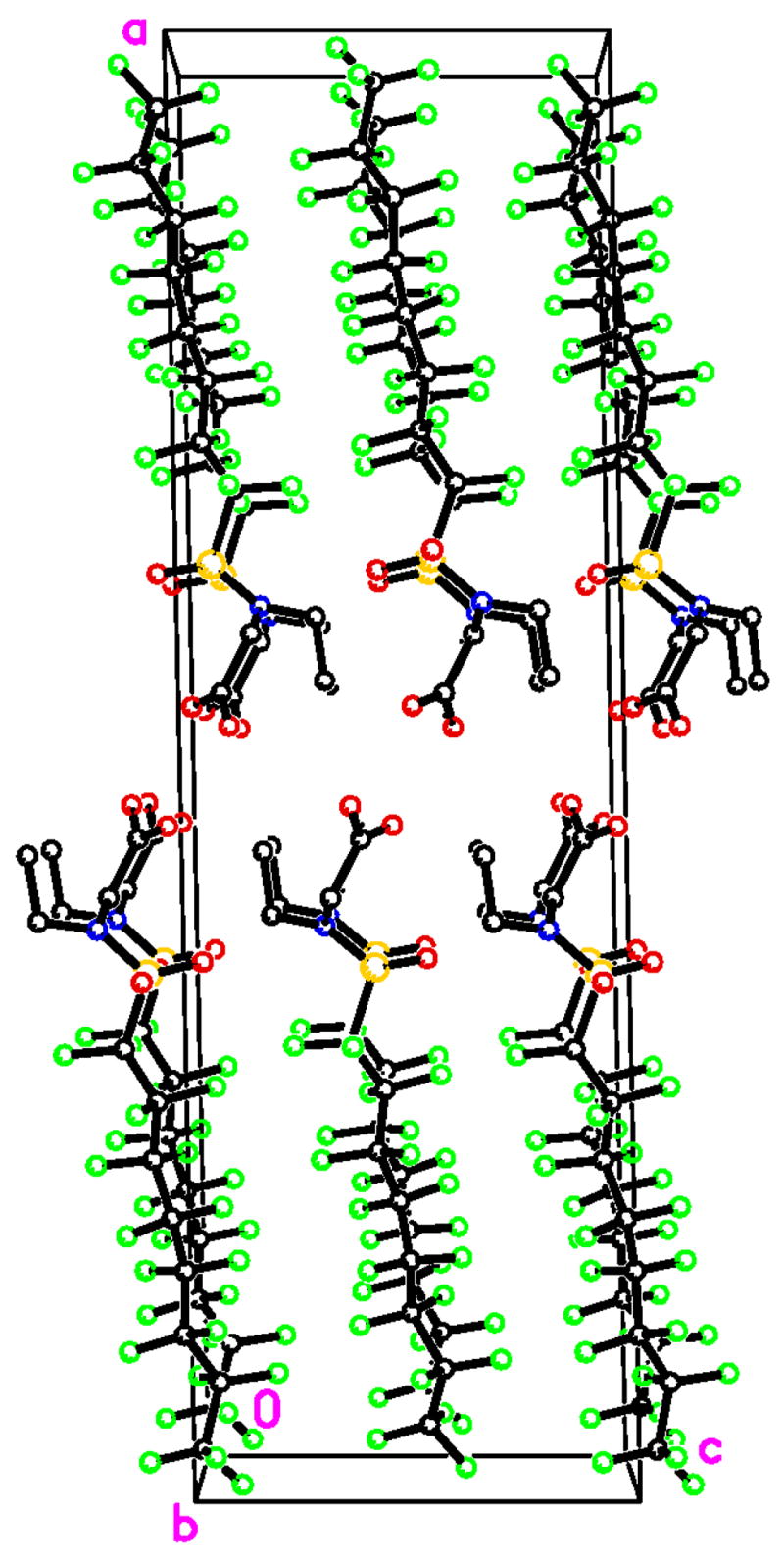
Packing diagram of 2-(N-ethyl-perfluorooctanesulfonamido) acetic acid (12), viewed down the b axis, illustrating the bilayers of 12 oriented paralel to the b–c plane. H atoms have been omitted for clarity.
Figure 5.

Packing diagram of N,N-diethyl-perfluorooctane-1-sulfonamide (7e), viewed down the b axis, illustrating the bilayers of 7e oriented paralel to the a–b plane. H atoms have been omitted for clarity.
The crystal structures of 4b, 7e and 12 have several structural similarities that are the result of both steric and electronic effects. The -CH2N(R)-S(=O)2-CF2- (R = H or CH2) fragment in all three structures adopts a staggered conformation with a trigonal planar nitrogen atom. The ethyl groups in both structures are on the site opposite to the perfluorooctyl group to minimize steric crowding around the S-N bond. A similar conformation has been reported for N,N-dibenzyl-perfluorobutane-1-sulfonamide in both the solid state and in solution [16]. The S-N bonds of 4b (1.574(5) and 1.576(5) Å), 7e (1.581(5) Å) and 12 (1.590(9) Å) are short due to hyperconjugation with the perfluorooctyl chain [16]. For comparison, the unweighted sample mean for C-SO2-N(H)C and C-SO2-N(C)C bonds is 1.633±0.019 and 1.642 ±0.024, respectively [24]. The short S-N bond length suggests an appreciable double-bond character and, thus, a hindered rotation around the S-N bond.
We used 1H NMR spectroscopy to further investigate the hindered rotation around the S-N bond of all dialkylated perfluorooctanesulfonamides. At ambient temperature several of the -N-CH2- systems show magnetically nonequivalent protons and at temperatures below 0°C typical geminal coupling constant of approximately 14.3 to 18.5 Hz were observed (Table 2). The free energy of activation ΔG≠ for several -N-CH2- systems was estimated using variable temperature NMR spectroscopy to be approximately 62–71 kJ mol−1 [16, 25]. These values are in good agreement with literature values [16].
Table 2.
Geminal-coupling-constant values, coalescence temperature, chemical shift difference at the coalescence temperature Δν and barriers of rotation about the S-N bond ΔG≠ for several sulfonamides in d6-acetone
| Sulfonamide | Geminal coupling constanta[Hz] | Coalescence Temperature[K] | Δν [Hz] | Free energy of activation ΔG≠[kJ mol−1] |
|---|---|---|---|---|
| 7e | 14.4 | 309 | 36 | 64 |
| 7f | 14.3 | 312 | 99 | 62 |
| 9ab | n.d. | 319 | 129 | 63 |
| 9bb | 14.6 | 315 | 52 | 64 |
| 10ab,c | 15.1b | 319 | 136 | 63 |
| 12.1c | 307 | 104 | 63 | |
| 10bb | 15.5 | 307 | 47 | 63 |
| 12 | n.d. | 312 | 3 | 71 |
Determined at temperatures below 273 K, i.e. well below the coalescence temperatures of the respective sulfonamide;
NCH2CH2OR;
NCH2CH2OR;
n.d. = not determined.
3. Conclusions
In summary, the synthesis and characterization of several environmentally relevant perfluorooctanesulfonamides and related perfluorobutanesulfonamides from fluoride 1 or 2 is described. These sulfonamides are needed for environmental and toxicological studies but are also useful as analytical standards. The purification of the perfluorooctanesulfonamides represented a challenge because of the impurities present in commercial perfluorooctanesulfonyl fluoride (1). Although fluorinated impurities can largely be removed by column chromatography, some branched C8F17 isomers are still present in the final product. The ratio of these branched isomers to the linear sulfonamides can be easily determined using 19F NMR spectroscopy.
4. Experimental
4.1. General Experimental Procedures
1H, 13C and 19F NMR spectra were recorded on a Bruker Avance-300 spectrometer. All NMR chemical shifts (δ) are reported in parts per million (ppm) and were determined relative to TMS for 1H and 13C NMR spectra and CFCl3 for 19F NMR spectra. Signals of the linear perfluoroalkyl chains were assigned and labeled as described previously [26]. A GC 6890 series (Agilent Technologies, Palo Alto, CA, USA) gas chromatograph equipped with a J&W Scientific HP-1 capillary column (Agilent Technologies, Wilmington, USA) and a flame ionization detector was used to monitor the reactions and to determine the purity of all compounds. The following program was employed: Initial temperature: 50°C, initial time: 1 min, rate: 10°C/min, final temperature: 250°C, final time: 2 min. Combustion analyses were obtained from Atlantic Microlab Inc. (Norcross, GA, USA).
Volatile amides without acidic protons were analyzed in the University of Iowa High Resolution Mass Spectrometry Facility using a ThermoFinnigan Voyager GC-MS system (ThermoFinnigan, San Jose, CA, USA) equipped with a ZB-1 column (Phenomenex, Torrance, CA, USA). Additional structural confirmation for amide with acidic protons was provided by LC/MS/MS. In short, solutions of the purified derivatives were dissolved in methanol at a concentration of 5 μg/mL and subsequently infused into a Micromass Quattro LC/MS/MS system equipped with an electrospray (ESI) interface and operated in the negative ion monitoring mode. Confirmation was obtained by observation of the corresponding deprotonated molecular ions ([M-H]−) coupled with MS/MS product ion scans of the deprotonated molecular ions showing unique and predictable fragmentation of the precursor ions. The observed fragmentation patterns of several perfluorooctanesulfonamides were consistent with spectra obtained from samples that had been provided by the 3M Company [27, 28].
4.2. General Procedures for the synthesis of N-alkyl perfluorooctanesulfonamide derivatives
Method A
Perfluorooctanesulfonyl fluoride 1 (5.0 g, 10 mmol) was placed under a nitrogen atmosphere in a three necked round bottom flask containing dry ether (75 mL) and equipped with a reflux condenser. A two-fold excess of the respective alkyl amine 3a or 3b (30 mmol) was passed slowly through the reaction mixture at 0–5°C over a period of 1 hour, the reaction mixture was allowed to stir for 24 hours at ambient temperature and heated under reflux for 1 hour. The reaction mixture was cooled to room temperature and a white precipitate was filtered off. The precipitate was washed with ether (30 mL). The solvent was removed by rotary evaporation under reduced pressure. The crude product was further purified by column chromatography using silica gel (25–40 μm mesh) with hexane and ethyl acetate as eluent (gradient from 100% to approximately 93% hexane).
Method B
Perfluoroalkanesulfonyl fluoride 1 or 2 (25 g, 50 mmol) and triethylamine (10.1 g, 100 mmol) were placed under a nitrogen atmosphere in a three necked round bottom flask containing dry ether or acetonitrile (100 mL) and equipped with a reflux condenser. The respective alkyl amine 3a or 3b (60 mmol) was passed slowly through the reaction mixture over a period of 3 hours at 0–5°C, the reaction mixture was allowed to stir for 14 hours at ambient temperature and refluxed for 3 hours. The reaction mixture was cooled to room temperature and the solvent was removed from the reaction mixture by rotary evaporation under reduced pressure. The resultant crude product was dissolved in acetone (20 mL), absorbed on silica gel (25–40 μm mesh) and purified by column chromatography as described under Method A.
Method C
An excess of the alkyl amine 3a or 3b was slowly passed through perfluorooctanesulfonyl fluoride 1 (3.65 g, 7.3 mmol). The mixture was stirred at ambient temperature for 5 hours and the resulting dark brown, waxy solid was treated with Zn (2 g) and HCl (5 mL) to decolorize the product. The product was purified by Kugelrohr distillation to yield the product as a slightly brown, waxy solid. Crystals of 4b suitable for crystal structure analysis were obtained by recrystallization from reagent alcohol/dichloromethane at 4°C.
4.2.1. N-methyl-perfluorooctane-1-sulfonamide (4a)
mp 104°C (Lit. 101–103°C [4]); IR (KBr); ν 3335 (-NH-), 1439, 1362, 1241, 1206, 1182, 1152, and 1126 cm−1; 1H NMR (300 MHz, d6-DMSO): δ 1.9 (3H, s, -CH3), 8.4 (1H, br s, -HN-); 13C NMR (75 MHz, d6-DMSO): δ 29.0 (-CH3); 19F NMR (288.8 MHz, d6-DMSO): δ −80.6 (CF3), −112.1 (α-CF2), −119.8 (β-CF2), −121.2 (3 × CF2), −122.2 (ζ-CF2), −125.7 (θ-CF2); MS, m/z (rel. int.): 512 [M-H]+ (100%). Anal. Calcd for C9H4F17NO2S: C, 21.07; H, 0.79; S, 6.25; N, 2.73. Found: C, 21.23; H, 0.72; S, 6.45; N, 2.97.
4.2.2. N-ethyl-perfluorooctane-1-sulfonamide (4b)
mp 87°C (Lit. 87–88.5°C [29] and 120 °C [5]); IR (KBr); ν 3318 (-NH-), 1454, 1363, 1238, 1206, 1152, 1126, and 1072 cm−1; 1H NMR (300 MHz, d6-DMSO): δ 1.2 (3H, t, J = 7.2 Hz, -CH3), 3.2 (2H, “q”, J = 7.2 Hz, -CH2-), 9.3 (1H, br s, -HN-); 13C NMR (75 MHz, d6-DMSO): δ 15.1 (-CH3), 38.7 (-CH2-); 19F NMR (288.8 MHz, d6-DMSO): δ −80.6 (CF3), −112.1 (α-CF2), −119.8 (β-CF2), −121.1 (3 × CF2), −122.2 (ζ-CF2), −125.7 (θ-CF2); MS, m/z (rel. int.): 526 [M-H]+ (100). Anal. Calcd for C10H6F17NO2S: C, 22.78; H, 1.15; S, 6.08; N, 2.66. Found: C, 23.03; H, 1.28; S, 6.15; N, 2.85.
4.2.3. N-methyl-perfluorobutane-1-sulfonamide (5a)
80%; mp 36–37°C; IR (KBr): ν 3338 (-NH-), 1429, 1364, 1235, 1206, 1186, and 1142 cm−1; 1H NMR (300 MHz, CDCl3): δ 3.03 (3H, s, -CH3), 5.13 (1H, br s, -HN-). 13C NMR (75 MHz, CDCl3): δ 30.7 (-CH3); 19F NMR (288.8 MHz, CDCl3): δ −81.28 (CF3), −112.72 (α-CF2), −121.94 (β-CF2), −126.51 (γ-CF2); GC-MS 40eV, m/z (rel. int.): 312(100), 219(22), 112(10). Anal. Calcd for C5H4F9NO2S: C, 19.18; H, 1.29; S, 10.24; N, 4.47. Found: C, 19.11; H, 1.15; S, 10.06; N, 4.38.
4.2.4. N-ethyl-perfluorobutane-1-sulfonamide (5b)
61%; mp 35–36°C (Lit. 40°C [9]); IR (KBr): ν 3321 (-NH-), 1437, 1373, 1238, 1206, 1190, and 1141 cm−1; 1H NMR (300 MHz, CDCl3): δ 1.28 (3H, t, J = 7.2 Hz, -CH3), 3.41 (2H, q, J = 7.2 Hz, -CH2-), 5.06 (1H, br s, -HN-); 13C NMR (75 MHz, CDCl3): δ 16.0 (-CH3), 40.3 (-CH2-); 19F NMR (288.8 MHz, CDCl3): δ −81.30 (CF3), −113.20 (α-CF2), −121.83 (β-CF2), −126.53 (γ-CF2); GC-MS 40eV, m/z (rel. int.): 326(100), 219(25), 126(10). Anal. Calcd for C6H6F9NO2S: C, 22.03; H, 1.85; S, 9.80; N, 4.28. Found: C, 22.31; H, 1.89; S, 9.78; N, 4.31.
4.2.5. N-(2-methoxy-ethyl)-perfluorobutane-1-sulfonamide (5c)
47%; Colorless liquid; IR (Neat): ν 3310–3146 (-NH), 1434, 1376, 1237, 1188, 1140, 1082, and 1036 cm−1; 1H NMR (300 MHz, CDCl3): δ 3.40 (3H, s, -OCH3), 3.48–3.56 (4H, m, -NCH2CH2O-), 6.11 (1H, br s, -NH-); 13C NMR (75 MHz, CDCl3): δ 44.4 (-NCH2-), 59.0 (-OCH3), 71.4 (-CH2O-); 19F NMR (288.8 MHz, CDCl3): δ −81.29 (CF3), −113.25 (α-CF2), −121.80 (β-CF2), −126.00 (γ-CF2); GC-MS 40eV, m/z (rel. int.): 358 [M+H]+ (1), 326 [M-OCH3]+ (5), 312 [M-CH2OCH3]+ (18), 248 (18), 219 (30), 131 (30), 69 (100). Anal. Calcd for C7H8F9NO3S: C, 23.54; H, 2.26; S, 8.98; N, 3.92. Found: C, 23.55; H, 2.26; S, 8.88; N, 3.87.
4.3. General Procedures for the synthesis of N,N-dialkyl perfluorooctanesulfonamides (7d–g and 8g)
Method D
Perfluorooctanesulfonyl fluoride 1 (25 g, 50 mmol) was dissolved in anhydrous ether (100 mL) under a nitrogen atmosphere. The dialkyl amine 6d–g (100 mmol) was added slowly to the reaction mixture over a period of 30 min, allowed to stir for 16 h at room temperature and heated under reflux for 3 h. The reaction mixture was cooled to room temperature and the solvent was removed by rotary evaporation under reduced pressure. The crude product was purified by column chromatography as described above under Method A.
Method E
Perfluorooctanesulfonyl fluoride 1 (3.65 g, 7.3 mmol) and N,N-diethylamine (3.8 mL, 37 mmol) were stirred at ambient temperature for 4 hours. Excess N,N-diethylamine was removed under reduced pressure and the product was recrystallized at 4°C from reagent alcohol/dichloromethane to give 7e as a slightly yellow, waxy solid.
4.3.1. N,N-dimethyl-perfluorooctane-1-sulfonamide (7d)
mp 80–81°C; IR (KBr); ν 2966, 1483, 1369, 1238, 1212, 1149, and 1061 cm−1; 1H NMR (300 MHz, d6-acetone): δ 3.2 (6H, s, -CH3); 13C NMR (75 MHz, d6-acetone): δ 39.6 (-CH3); 19F NMR (288.8 MHz, d6-acetone): δ −81.3 (CF3), −112.0 (α-CF2), −121.0 (β-CF2), 122.1 (3 × CF2), −123.2 (ζ-CF2), −125. 6 (θ-CF2); GC-MS 40 eV, m/z (rel. int.): 526 [M-H]+ (1), 108 (100). Anal. Calcd for C10H6F17NO2S: C, 22.78; H, 1.15; S, 6.08; N, 2.68. Found: C, 22.82; H, 1.04; S, 6.16; N, 2.68.
4.3.2. N,N-diethyl-perfluorooctane-1-sulfonamide (7e)
mp 48–50°C; IR (KBr); ν 2992, 1468, 1385, 1244, 1217, 1155, 1055, and 1024 cm−1; 1H NMR (300 MHz, CDCl3): δ 1.3 (6H, t, J = 7.1 Hz, -CH3), 3.4–3.7 (4H, m, -NCH2-); 13C NMR (75 MHz, CDCl3): δ 14.0 (-CH3), 42.9 (-NCH2-); 19F NMR (288.8 MHz, CDCl3): δ −81.3 (CF3), −113.1 (α-CF2), −120.7 (β-CF2), −122.2 (3 × CF2), −123.2 (ζ-CF2), −126.6 (θ-CF2); GC-MS 40 eV, m/z (rel. int.): 540 [M-CH3]+ (11), 448 [C8F16SO]+ (8). Anal. Calcd for C12H10F17NO2S: C, 25.96; H, 1.82; S, 5.77; N, 2.52. Found: C, 25.95; H, 1.86; S, 5.76; N, 2.64.
4.3.3. N-ethyl-N-methyl-perfluorooctane-1-sulfonamide (7f)
mp 44–46°C; IR (KBr); ν 1375, 1240, and 1150 cm−1; 1H NMR (300 MHz, d6-acetone): δ 1.3 (3H, t, J = 7.2 Hz, -CH3), 3.2 (3H, s, -NCH3), 3.3–3.8 (2H, m, -NCH2-); 13C NMR (300 MHz, d6-acetone): δ 13.9 (-CH3), 35.4 (-NCH3), 47.2 (-NCH2-); 19F NMR (288.8 MHz, d6-acetone): δ −80.5 (CF3), −112.1 (α-CF2), −119.9 (β-CF2), −121.2 (3 × CF2), −122.1 (ζ-CF2), −125.6 (θ-CF2); GC-MS 40 eV, m/z (rel. int.): 526 [M-CH3]+ (2), 462 [C8F16SON]+ (4). Anal. Calcd for C11H8F17NO2S: C, 24.41; H, 1.49; S, 5.92; N, 2.59. Found: C, 24.68; H, 1.41; S, 5.98; N, 2.49.
4.3.4. 4-(Heptadecafluorooctane-1-sulfonyl)-morpholine (7g)
mp 118°C (Lit. 127–129 °C [30]); IR (KBr): ν 1390, 1268, 1184, 1148, 1134, 1113, 1080, and 969 cm−1; 1H NMR (300 MHz, d6-Acetone): δ 3.4–4.1 (8H, m); 13C NMR (75 MHz, d6-Acetone): δ 48.0 (-NCH2-), 67.2 (-CH2O-); 19F NMR (288.8 MHz, d6-Acetone): δ −80.51 (CF3), −112.03 (α-CF2), −119.83 (β-CF2), −121.15 (3 ×-CF2-), −122.11 (ζ-CF2), −125.63 (θ-CF2); GC-MS 40eV, m/z (rel. int.): 569 [M]+ (23), 526 (14), 486 (4), 442 (5), 150 (100), 134 (34), 56 (82); Anal. Calcd for C12H8F17NO3S: C, 25.32; H, 1.42; S, 5.63; N, 2.46. Found: C, 25.45; H, 1.56; S, 5.56; N, 2.65.
4.3.5. 4-(Nonafluorobutane-1-sulfonyl)-morpholine (8g)
61%; mp 81°C; IR (KBr): ν 1390, 1265, 1236, 1187, and 1138 cm−1; 1H NMR (300 MHz, d6-Acetone): δ 3.3–4.1 (8H, m); 13C NMR (75 MHz, d6-Acetone): δ 48.0 (-N-CH2-), 67.2 (-CH2-O-); 19F NMR (288.8 MHz, d6-Acetone): δ −80.62 (CF3), −112.28 (α-CF2), 120.92 (β-CF2), −125.62 (γ-CF2); GC-MS 40eV, m/z (rel. int.): 369 [M]+ (7), 326 (6), 242 (7), 150 (98), 134 (24), 86 (100). Anal. Calcd for C8H8F9NO3S: C, 26.03; H, 2.18; S, 8.68; N, 3.79. Found: C, 26.23; H, 2.22; S, 3.75; N, 8.67.
4.4. General procedure for the N-alkylation of N-alkyl perfluoroalkanesulfonamide derivatives 4 and 5
The respective N-alkyl perfluorooctanesulfonamide 4 or 5 (5.0 mmol), dry potassium carbonate (1.4 g, 10 mmol) and corresponding bromo- or iodo-alkyl derivative (5.5 mmol) were dissolved in acetone (25 mL) and the resultant mixture was heated under reflux until complete conversion. The reaction mixture was allowed to cool to room temperature and filtered. The precipitate was washed with acetone (2 × 10 mL) and the solvent was removed by rotary evaporation under reduced pressure. The crude product was purified by column chromatography using silica gel with hexane and ethyl acetate (96:4) as eluent.
4.4.1. N-(2-hydroxyethyl)-N-methyl-perfluorooctane-1-sulfonamide (9a)
96%; mp 83°C; IR (KBr); ν 3420 (-OH), 1384, 1219, and 1151 cm−1; 1H NMR (300 MHz, d6-acetone): δ 3.2 (3H, s, -CH3), 3.4 (1H, m, -NCH2-), 3.7 (3H, m, -CH2CH2OH), 4.1 (1H, t, J = 5.5 Hz, -CH2OH); 13C NMR (75 MHz, CD3OD): δ 37.2 (-CH3), 54.3 (-NCH2-), 60.6 (-CH2OH); 19F NMR (288.8 MHz, CD3OD): δ-80.5 (CF3), -112.6 (α-CF2), −119.8 (β-CF2), −121.2 (3 × CF2), −122.1 (ζ-CF2), −125.6 (θ-CF2); MS, m/z (rel. int.): 616 (50), 141 (100), 119 (55), 223 (15). Anal. Calcd for C11H8F17NO3S: C, 23.71; H, 1.45; S, 5.75; N, 2.51. Found: C, 23.82; H, 1.33; S, 5.82; N, 2.53.
4.4.2. N-ethyl-N-(2-hydroxyethyl)-perfluorooctane-1-sulfonamide (9b)
99%; mp 70°C (Lit. 65–71°C [4]); IR (KBr); ν 3412 (-OH), 1384, 1212, and 1150 cm−1; 1H NMR (300 MHz, CDCl3): δ 1.3 (3H, t, J = 7.1 Hz, -CH2CH3), 2.2 (1H, br s, -CH2OH), 3.3–3.8 (4H, m, -CH2-), 3.8 (2H, “t”, J = 5.3 Hz, -CH2OH); 13C NMR (75 MHz, CDCl3): δ 13.9 (-CH2CH3), 45.0 (-NCH2CH3), 49.9 (-NCH2CH2OH), 60.8 (-CH2OH); 19F NMR (288.8 MHz, CDCl3): δ −81.3 (CF3), −112.6 (α-CF2), −120.7 (β-CF2), −122.2 (3 ×-CF2-), −123.2 (ζ-CF2), −126.6 (θ-CF2); MS, m/z (rel. int.): 630 (60), 141 (100), 119 (75), 223 (15). Anal. Calcd for C12H10F17NO3S: C, 25.23; H, 1.76; S, 5.61; N, 2.45. Found: C, 25.27; H, 1.83; S, 5.71; N, 2.59.
4.4.5. 2-(N-methyl-perfluorooctylsulfonamido) ethyl acetate (10a)
86%; mp 82–83°C; IR (KBr); ν 1732 (-C=O), 1380, 1373, 1236, 1204, and 1151 cm−1; 1H NMR (300 MHz, d6-Acetone): δ 2.9 (3H, s, -OCOCH3), 3.2 (3H, s, -NCH3), 3.5–4.1 (2H, m, (-NCH2-), 4.2–4.5 (2H, br m, -CH2OCOCH3); 13C NMR (75 MHz, d6-Acetone): δ 21.7 (-OCOCH3), 37.6 (-CH3), 51.8 (-NCH2-), 62.2 (-CH2OCOCH3), 171.9 (-OCOCH3); 19F NMR (288.8 MHz, d6-Acetone): δ −80.5 (CF3), −111.7 (α-CF2), −119.9 (β-CF2), −121.2 (3 ×-CF2-), −122.2 (ζ-CF2), −125.6 (θ-CF2); GC-MS 40eV, m/z (rel. int.): 539 [M-CH3COOH]+ (12), 526 [M-C3H6O2]+ (28), 462 [C8F16SON]+ (40). Anal. Calcd for C13H10F17NO4S: C, 26.06; H, 1.68; S, 5.35; N, 2.34. Found: C, 26.11; H, 1.59; S, 5.57; N, 2.46.
4.4.6. 2-(N-ethyl-perfluorooctylsulfonamido) ethyl acetate (10b)
92%; mp 104°C; IR (KBr); ν 2966, 2934, 1732 (C=O), 1379, 1240, 1214, and 1151 cm−1; 1H NMR (300 MHz, CDCl3): δ 1.3 (3H, t, J = 7.1 Hz, -CH3), 2.1 (3H, s, -OCOCH3), 3.4–3.9 (4H, m, -NCH2-), 4.3 (2H, t, J = 5.6 Hz, -CH2OCOCH3); 13C NMR (75 MHz, CDCl3): δ 13.8 (-CH3), 20.6 (-OCOCH3), 44.4 (-CH2CH3), 46.4 (-NCH2-), 61.2 (-CH2OCOCH3), 170.6 (-OCOCH3); 19F NMR (288.8 MHz, d6-acetone): δ −80.5 (CF3), −112.0 (α-CF2), −119.8 (β-CF2), −121.1 (3 ×-CF2-), −122.1 (ζ-CF2), −125.6 (θ-CF2); GC-MS 40eV, m/z (rel. int.): 553 [M-CH3COOH]+ (5), 540 [M-C3H5O2]+ (42), 448 [C8F16SO]+ (27). Anal. Calcd for C14H12F17NO4S: C, 27.42; H, 1.97; S, 5.23; N, 2.28. Found: C, 27.37; H, 1.79; S, 5.26; N, 2.35.
4.4.7. Methyl 2-(N-ethyl-perfluorooctanesulfonamido) acetate (11)
90%; mp 55–56°C; IR (KBr); ν 1756, 1379, 1202, 1180, 1167, and 1124 cm−1; 1H NMR (300 MHz, CD3OD): δ 1.24 (3H, t, J = 7.1 Hz, -CH3), 3.5–3.7 (2H, m, -CH2CH3), 3.78 (2H, s, -CH2CO2-), 4.27 (2H, s, -CO2CH3); 13C NMR (75 MHz, CD3OD): δ 13.9 (-CH3), 46.9 (-CH2CH3), 49.2 (-CH2CO2-), 53.1 (-CO2CH3), 170.1 (-CO2CH3); 19F NMR (288.8 MHz, CD3OD): δ −80.48 (CF3), −112.41 (α-CF2), −119.69 (β-CF2), −121.15 (3 × CF2), −122.13 (ζ-CF2), −125.59 (θ-CF2); GC-MS 40 eV, m/z (rel. int.): 600 [M+H] (68), 540 (50), 448 (40), 56 (100); Anal. Calcd for C13H10F17NO4S: C, 26.06; H, 1.68; S, 5.35; N, 2.34. Found: C, 26.22; H, 1.73; S, 5.32; N, 2.41.
4.5. Synthesis of 2-(N-ethyl-perfluorooctanesulfonamido)acetic acid 12
Methyl 2-(N-ethyl-perfluorooctanesulfonamido) acetate 11 was dissolved in 1N NaOH (1.5 eq., 1.4 mL) and 1,4-dioxane (3 mL), stirred for 2 h at 65°C, diluted with water (20 mL) and filtered. The filtrate was acidified with 1 N HCl (10 mL) and extracted with ethyl acetate (~20 mL). The organic layer was washed with water (2 × 20 mL) and dried over sodium sulfate. The solution was treated with charcoal, filtered and concentrated under reduced pressure to give 12 as a white solid in 40% yield.
mp 156–157°C (Lit. 162°C [31]); IR (KBr); ν 1728, 1379, 1201, 1168, 1149, and 1124 cm−1; 1H NMR (400 MHz, CD3OD): δ 1.25 (3H, t, J = 7.1 Hz, -CH3), 3.63 (2H, q, J = 7.1 Hz, -CH2CH3), 4.24–4.56 (2H, m, - CH2CO2-); 13C NMR (75 MHz, CD3OD): δ 13.9 (-CH3), 46.8 (-CH2CH3), 49.1 (-CH2CO2-), 170.1 (-CO2H); 19F NMR (288.8 MHz, CD3OD): δ −80.64 (CF3), −112.38 (α-CF2), −119.0 (β-CF2), −121.13 (3 × CF2), −122.04 (ζ-CF2), −125.59 (θ-CF2); MS, m/z (rel. int.): 584(95), 141(35), 369(20), 499(20), 223(10), 217(10). Anal. Calcd for C12H8F17NO4S: C, 24.63; H, 1.38; S, 5.48; N, 2.39. Found: C, 24.37; H, 1.59; S, 5.71; N, 2.29.
4.6. Synthesis of N,N-dialkyl perfluorooctanesulfonamides using the Mitsunobu reaction
4.6.1. General procedure for the Mitsunobu reaction [18]
A solution of DIAD (diisopropyl azodicarboxylate, 0.3 g, 15 mmol) in ether (2 mL) was added slowly to a sonicated mixture of alcohol 13 [32] or methyl glycolate (10 mmol), N-ethyl-perfluorooctanesulfonamide 4b (10 mmol) and triphenylphosphine (0.4 g, 15 mmol) in anhydrous ether (3 mL) at 0°C. After completion of the addition, the reaction mixture was sonicated at 25°C until the starting material had disappeared. The solvent was removed under reduced pressure and the product was purified by column chromatography using silica gel using ethyl acetate (0–5%) and hexane (100-98%) as eluent.
4.6.2. 1,1,2,2,3,3,4,4,5,5,6,6,7,7,8,8,8-Heptadecafluoro-octane-1-sulfonic acid [2-(tert-butyl-dimethyl-silanyloxy)-ethyl]-ethyl-amide (14)
98%; mp 29°C; IR (KBr); ν 2932, 1391, 1243, 1215, and 1150 cm−1; 1H NMR (300 MHz, CDCl3): δ 0.08 (6H, s, 2 ×-SiCH3), 0.90 (9H, s, -C(CH3)3), 1.28 (3H, t, J = 7.2 Hz, -CH2CH3), 3.35–3.75 (4H, m, -N(CH2CH3)CH2CH2-), 3.81 (2H, t, J = 5.5 Hz, -CH2O-); 13C NMR (75 MHz, CDCl3): δ −5.6 (2 ×-SiCH3), 13.9, 18.1, 25.7 (-C(CH3)3), 45.1, 49.7, 62.2; 19F NMR (288.8 MHz, CDCl3): δ −81.26 (CF3), −112.95 (α-CF2), −120.78 (β-CF2), −122.18 −122.22 (3 × CF2), 123.20 (ζ-CF2), −126.63 (θ-CF2); Anal. Calcd for C18H24F17NO3SSi: C, 31.54; H, 3.53; S, 4.68; N, 2.04. Found: C, 31.64; H, 3.48; S, 4.57; N, 2.07.
4.7. Procedure for the hydrolysis of TBDMS protected ether 14
The TBDMS protected ether 14 (0.15 mmol) was dissolved in 1N HCl and methanol (5 mL, 1:1, v/v) and stirred for 2 days at ambient temperature. The solvent was removed under reduced pressure. The product was extracted with ethyl acetate (5 mL), the combined organic extracts were washed with water (2 × 5 mL) and dried over sodium sulfate. The solvent was removed under reduced pressure to give 9b in 96% yield.
4.8. Synthesis of perfluorooctane-1-sulfonamide (16)
Perfluorooctanesulfonyl fluoride 1 (2.0 g, 3.9 mmol) was dissolved in ether (5 mL) and an aqueous solution of sodium azide (1.2 g, 19 mmol, in 1 mL of water) was added. The reaction mixture was allowed to stir at room temperature for 12 hours. Extraction with ether (15 mL) and evaporation of the solvent gave crude perfluorooctane-1-sulfonyl azide 15 as a colorless liquid. The crude azide 15 was directly converted into the amide 16 without further purification. The azide 16 was added to a suspension of Zn dust (1.3 g, 20 mmol) in ether (5 mL). Hydrochloric acid solution (5N, 3 mL) was added slowly to this suspension until the Zn dust was completely dissolved. The mixture was stirred for 24 hours at room temperature and extracted with ether (3 × 10 mL). Evaporation of the solvent under reduced pressure gave a waxy, brown solid. This solid was dissolved in reagent alcohol and the sulfonamide 16 was precipitated as a white waxy solid with dichloromethane.
IR (KBr); ν 3344, 3176, 3058, 1378, 1229, 1203, and 1147 cm−1; 1H NMR (300 MHz, CDCl3): δ 8.2 (2H, br s, NH2); 19F NMR (288.8 MHz, CDCl3): δ −80.7 (CF3), −112.8 (α-CF2), −119.8 (β-CF2), −121.1 ζ-CF2), −122.2 (3 ×-CF2-), −125.7 (θ-CF2); GC-MS m/z, (rel. int.): 499.9 [M]+ (30). Anal. Calcd for C8H2F17NO2S: C, 19.25; H, 0.40; S, 6.42; N, 2.81. Found: C, 19.49; H, 0.41; S, 6.35; N, 2.90.
4.9. X-Ray Crystallography [33]
Flaky platelet crystals of 4b (synthesized using Method C), 7e (synthesized using Method E) and 12 were obtained by recrystallization from reagent alcohol/dichloromethane at 4°C. These crystals proved far too small for analysis on conventional small-molecule diffraction equipment but gave recordable, albeit weak, diffraction using CuKα x-rays on a specially configured hybrid small/macromolecule diffraction system based on the Bruker-Nonius X8 Proteum (Nonius FR-591 rotating anode x-ray generator, Bruker Helios graded multilayer optics, Nonius Kappa goniometer, Bruker SMART 6000 CCD detector, CryoCool LN2 low temperature device from CryoIndustries of America).
For each crystal, initial unit cell parameters were obtained using APEX2 software [34] from ω -scans at six different φ and χ angles. Final cell parameters were obtained (program SaintPlus in APEX2 [34]) using spot positions from all data collection frames. Crystal decay (negligible) was checked in each case by re-measurement of a portion of the first data collection scan. A total of 17923, 19116 and 3455 reflections were collected for 4b, 7e and 12, respectively. Merging of symmetry equivalents resulted in 6071 (4288 with I > 2σ (I)), 3491 (2988 with I > 2σ (I)) and 1951 (1594 with I > 2σ (I)) reflections for 4b, 7e and 12, respectively. Correction of Lorentz and polarization effects, data reduction, merging and an empirical absorption correction for each dataset were performed within the APEX2 package (programs SaintPlus and Sadabs [34]). The structures were solved by direct methods using SHELXS97 [35] and refined by full-matrix least-squares against F2 using SHELXL97 [35]. All non-hydrogen atoms in both structures were refined with anisotropic displacement parameters (ADPs).
The structure of 7e was extensively disordered and refinement required strong restraints. Similar bond lengths and angles within and between each disordered pair (major:minor component ratio 62:38) were restrained to similar values (commands ‘SADI’ and ‘SAME’ in SHELXL) and anisotopic displacement parameters (ADPs) were subject to rigid-body (‘DELU’ in SHELXL97) and approximate isotropic (‘ISOR’ in SHELXL97) restraints. Further, the ADPs of disordered pairs of atoms in close proximity (C3, C3′; C6, C6′; C8, C8′) were constrained to be the same.
All hydrogens atoms in both 4b, 7e and 12 were found in difference Fourier maps and were subsequently placed at calculated positions using appropriate riding models with distances of 0.98Å (C-H3), 0.99Å (C-H2) and 0.88Å (N-H in 4b). Isotropic displacement parameters were fixed at either 1.2 times (C-H2, N-H), or 1.5 times (C-H3) the Ueq of the carrier atom.
Acknowledgments
The authors would like to thank Air Products and Chemicals, Inc. (Allentown, PA, USA) for a donation of free lecture bottles of methyl and ethyl amine. This work was supported by grants from the National Institute of Environmental Health Sciences (ES12475 (HJL)) and the National Science Foundation (NIRT 0210517 (HJL) and NSF MRI grant #0319176 (SP)). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the funding agencies.
References
- 1.Kissa E. Fluorinated surfactants and repellents (Surfactant Science Series) Vol. 97. Marcel Dekker; New York: 2001. [Google Scholar]
- 2.Lehmler HJ. Chemosphere. 2005;58:1471–1496. doi: 10.1016/j.chemosphere.2004.11.078. [DOI] [PubMed] [Google Scholar]
- 3.Xu L, Krenitsky DM, Seacat AM, Butenhoff JL, Anders MW. Chem Res Toxicol. 2004;17:767–775. doi: 10.1021/tx034222x. [DOI] [PubMed] [Google Scholar]
- 4.Niederpruem H, Voss P, Wechsberg M. Liebigs Ann Chem. 1973:11–19. [Google Scholar]
- 5.Benefice-Malouet S, Blancou H, Teissedre R, Commeyras A. J Fluorine Chem. 1986;31:319–332. [Google Scholar]
- 6.Organization for Economic Co-operation and Development (OECD), Environment Directorate, Results of survey on production and use of PFOS, PFAS and PFOA, related substances and products/mixtures containing these substances, Report ENV/JM/MONO(2005)1, 2005.
- 7.Martin JW, Ellis DA, Mabury SA, Hurley MD, Wallington TJ. Environ Sci Technol. 2006;40:864–872. doi: 10.1021/es051362f. [DOI] [PubMed] [Google Scholar]
- 8.Bussas R, Kresze G. Liebigs Ann Chem. 1982:545–563. [Google Scholar]
- 9.Podol’skii AV, Kachalkova MI, Ilatovskii RE, Kodess MI, Kolenko IP. Russ J Org Chem. 1990:1242–1244. [Google Scholar]
- 10.Roesky HW, Holtschneider G, Giere HH. Z Naturforsch. 1970;25b:252–254. [Google Scholar]
- 11.Meussdoerffer JN, Niederpruem H. Chemiker-Zeitung. 1972;96:582–583. [Google Scholar]
- 12.Zhu S-Z, Zhang J, Xu B, Li A. Phosphorus, Sulfur Silicon Relat Elem. 1994;89:77–82. [Google Scholar]
- 13.DeChristopher PJ, Adamek JP, Lyon GD, Klein SA, Baumgarten RJ. J Org Chem. 1974;39:3525–3532. [Google Scholar]
- 14.A.H. Ahlbrecht, H.A. Brown, US 2803656 (1957).
- 15.K.H. Mitschke, K. Geisler, H. Niederpruem, DE 2832346 (1980).
- 16.Lyapkalo IM, Reissig HU, Schäfer A, Wagner A. Helv Chim Acta. 2002;85:4206–4215. [Google Scholar]
- 17.Kawase M, Motohashi N, Niwa M, Nozaki M. Heterocycles. 1997;45:1121–1129. [Google Scholar]
- 18.Balint AM, Bodor A, Gömöry A, Vekey K, Szabo D, Rabai J. J Fluorine Chem. 2005;126:1524–1530. [Google Scholar]
- 19.Bell KE, Knight DW, Gravestock MB. Tetrahedron Lett. 1995;36:8681–8684. [Google Scholar]
- 20.Edwards ML, Stemerick DM, McCarthy JR. Tetrahedron. 1994;50:5579–5590. [Google Scholar]
- 21.Zhu SZ, Xu Y, Wang YL, Peng WM. Chin J Chem. 2001;19:1259–1262. [Google Scholar]
- 22.Lehmler HJ, Parkin S, Brock CP. Acta Cryst B. 2004;60:325–332. doi: 10.1107/S0108768104005609. [DOI] [PubMed] [Google Scholar]
- 23.Bunn CW, Howells ER. Nature. 1954;174:549–551. [Google Scholar]
- 24.E. Prince, International Tables for Crystallography, Volume C: Mathematical, Physical and Chemical Tables; Third Edition 2004.
- 25.Ahmed A, Bragg RA, Clayden J, Lai LW, McCarthy C, Pink JH, Westlund N, Yasin SA. Tetrahedron. 1998;54:13277–13294. [Google Scholar]
- 26.Bossev DP, Matsumoto M, Sato T, Watanabe H, Nakahara M. J Phys Chem B. 1999;103:8259–8266. [Google Scholar]
- 27.Boulanger B, Vargo J, Schnoor JL, Hornbuckle KC. Environ Sci Technol. 2004;38:4064–4070. doi: 10.1021/es0496975. [DOI] [PubMed] [Google Scholar]
- 28.Boulanger B, Vargo JD, Schnoor JL, Hornbuckle KC. Environ Sci Technol. 2005;39:5524–5530. doi: 10.1021/es050213u. [DOI] [PubMed] [Google Scholar]
- 29.E.K. Kleiner, German patent 2015332 (1970).
- 30.T.J. Brice, P.W. Trott, US patent 2732398 (1956).
- 31.H.A. Brown, US patent 2809990 (1957).
- 32.Azumaya I, Uchida D, Kato T, Yokoyama A, Tanatani A, Takayanagi H, Yokozawa T. Angew Chem Int Ed. 2004;43:1360–1363. doi: 10.1002/anie.200352788. [DOI] [PubMed] [Google Scholar]
- 33.Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC...... Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, (fax: +44 1223 336033 or e-mail: deposit@ccdc.cam.ac.uk).
- 34.Bruker-Nonius, APEX2: software suite for data collection and processing of single crystal x-ray diffraction data, 2004.
- 35.G.M. Sheldrick, SHELX-97: Programs for the solution (SHELXS97) and refinement (SHELXL97) of crystal structures from diffraction data, 1997.