

Abstract
Management of chronic pain is a real challenge, and current treatments focusing on blocking neurotransmission in the pain pathway have only resulted in limited success. Activation of glia cells has been widely implicated in neuroinflammation in the central nervous system, leading to neruodegeneration in many disease conditions such as Alzheimer’s and multiple sclerosis. The inflammatory mediators released by activated glial cells, such as tumor necrosis factor-α and interleukin-1β can not only cause neurodegeneration in these disease conditions, but also cause abnormal pain by acting on spinal cord dorsal horn neurons in injury conditions. Pain can also be potentiated by growth factors such as BDNF and bFGF that are produced by glia to protect neurons. Thus, glia cells can powerfully control pain when they are activated to produce various pain mediators. We will review accumulating evidence supporting an important role of microglia cells in the spinal cord for pain control under injury conditions (e.g. nerve injury). We will also discuss possible signaling mechanisms in particular MAP kinase pathways that are critical for glia control of pain. Investigating signaling mechanisms in microglia may lead to more effective management of devastating chronic pain.
Keywords: microglia, MAP kinase, chemokines, cytokines, intracellular signaling, chronic pain, nerve injury
Introduction
In the last several years we have seen exponentially increasing number of research articles studying the role of glial cells in pain control especially in conditions involving inflammation. Inflammation in any part of our body is associated with pain, due to the release of inflammatory mediators such as prostaglandin E2 (PGE2), the proinflammatory cytokines tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), and nerve growth factor (NGF). These mediators, produced by non-neural cells or immune cells, can stimulate nociceptor terminals in the peripheral tissue to increase pain sensitivity (Julius & Basbaum, 2001; Scholz & Woolf, 2002). Inflammation also occurs in the central nervous system (CNS) after brain trauma, brain infection, and in neurodegenerative diseases, such as Alzheimer’s, Parkinson’s, or multiple sclerosis. This so-called neuroinflammation is characterized by activation of glial cells (e.g., microglia and astrocytes) in the CNS and contributes importantly to the development of neurodegeneration by releasing inflammatory mediators from glial cells (Block et al., 2007; Lobsiger & Cleveland, 2007). This glia-mediated neuroinflammation also plays an important role in pain control under pathological conditions.
Pain is described as an unpleasant sensory and emotional experience associated with actual or potential tissue damage. It is an essential sense to avoid a dangerous environment. Individuals lacking pain sensation sustain injuries, bite their tongue and lips, or get infections (Cox et al., 2006). When tissue damage (e.g., surgical incision) occurs, a reversible increase in pain sensitivity can be observed in the inflamed and surrounding tissue, which helps wound healing by avoiding any contact. However, after peripheral nerve lesions, spinal cord injury or rheumatoid arthritis, the increase in pain sensitivity can be long lasting and sometimes irreversible, becoming pathological pain or abnormal pain. Two major neuronal mechanisms are responsible for the pain hypersensitivity observed in these injury conditions. First, peripheral sensitization, increased sensitivity in the peripheral nervous system, results from the local action of inflammatory mediators on the peripheral terminals of high-threshold sensory neurons. Hyperactivity of transduction molecules [e.g., transient receptor potential subtype V1 and A1 (TRPV1 and TRPA1)] and conduction molecules (e.g., TTX-resistant sodium channels Nav1.8/1.9) leads to increased sensitivity and excitability of nociceptors (Julius & Basbaum, 2001; Levine & essandri-Haber, 2007). Second, central sensitization is caused by an increase in synaptic efficacy at the spinal cord level. It is caused by chemical, structural, and functional plasticity in spinal cord dorsal horn neurons, which modulates and amplifies signals from the periphery and is responsible for secondary hyperalgesia (spread of sensitivity well beyond the peripheral site of injury). Central sensitization embraces different forms including long-term potentiation (LTP) that is implicated in long-term memory when LTP occurs in the hippocampus and persistent pain when LTP occurs in the spinal cord (Ji et al., 2003; Scholz & Woolf, 2002).
However, the role of glial cells cannot be ignored even in this neuronal view of pain control, because neural plasticity is triggered by many inflammatory mediators, which in the CNS are mainly produced by glial cells. Indeed in the past several years, more and more attention has been paid to neuron-glia interaction as a driving force for the development and maintenance of abnormal pain (reviewed in Ji & Strichartz, 2004; Marchand et al., 2005; Scholz & Woolf, 2007; Tsuda et al., 2005; Watkins et al., 2001). Glia activation in the spinal cord after nerve injury was first associated with pain behavior in 1991 (Garrison et al., 1991). Later, it was found that spinal injection of an glial inhibitor fluorocitrate can reduce hyperalgesia (Meller et al., 1994). However, the specific role of microglia in nerve injury-induced neuropathic pain was demonstrated a decade later (Tsuda et al., 2003). Glial activation has been studied in different animal models such as neuropathic pain after injuries to peripheral nerves (Colburn et al., 1997) or the spinal cord (Hains & Waxman, 2006), inflammatory pain after injection of inflammatory substances (e.g. formalin) into a hindlimb (Fu et al., 1999), cancer pain after inoculation of tumor cells (Honore et al., 2000), or orofacial pain after lesion of joint muscle (Sessle, 2007). Both microglia and astrocytes are activated in these pain models and interact with neurons in the complex pain pathophysiology. We have recently discussed the role of astrocytes in pain control in this journal (Ji et al., 2006). In this review, we will focus on how activation of microglia controls abnormal pain.
Microglia activation and proliferation in the spinal cord in chronic pain conditions
Microglia comprise around 5–12% of the cells in the CNS and are heterogeneously distributed (Lawson et al., 1990). They originate from bone marrow-derived monocytes migrating during perinatal time (Nakajima & Kohsaka, 2001). Quiescent microglia are not passive cells but rather actively sense their environment with their ramified processes (Nimmerjahn et al., 2005; Raivich, 2005; Hanisch and Kettenmann, 2007). Microglia become activated by various stimuli. Microglia activation is also described in various ways (Tsuda et al., 2005), such as changes in morphology from ramified to amoeboid (Eriksson et al., 1993), increase in the expression of microglial markers [e.g., MHC II, CD 11b (Coyle, 1998; Eriksson et al., 1993; Liu et al., 1995)], and increase in the number of microglia (proliferation). These slow changes often take hours to days to manifest. However, some rapid changes in microglia, such as changes in second messengers and changes in signal transduction are often ignored.
Proliferation in the spinal cord is an important feature of glia activation, as in resting state very few glial cells divide. Cell proliferation in the spinal cord was mainly investigated using bromodeoxyuridine (BrdU), a nucleotide analog that can be incorporated into the DNA of duplicating cells. Fortunately, antibodies are available to recognize BrdU by immunohistochemistry. Microglia are rarely dividing in the intact spinal cord (Horner et al., 2000). However, microglia proliferation occurs after nerve injury (Graeber et al., 1988) and was found in diverse models of neuropathic pain. We used the spared nerve injury (SNI) model (Decosterd & Woolf, 2000) in which two of the three terminal branches of the sciatic nerve are ligated leaving the third brand sural nerve intact. Neuropathic pain symptoms develop in the hindpaw territory of the spared sural nerve. Using BrdU staining, we found a massive proliferation in the dorsal and ventral horn of the spinal cord on the ipsilateral side after SNI. However, no staining was seen in control animals or on the contralateral dorsal horn of animals with SNI (Fig. 1). The proliferation was already very prominent 2 days after nerve injury. Double staining showed that most BrdU positive cells were also labeled by Iba1, a microglial marker (Fig. 1). Therefore, the proliferating cells in the spinal cord are predominantly microglia, in agreement with previous studies with sciatic nerve transection (Liu et al., 2000), sciatic nerve constriction (Echeverry et al., 2007) or partial sciatic nerve ligation (Narita et al., 2006). The proliferating response of glia to nerve injury might be different depending on the lesion site. After rhizotomy (transection of the central axons) the proliferating populations differ: 30% proliferating cells are astrocytes, in addition to 60% of microglia (Liu et al., 2000). After spinal cord injury, a condition known to induce central neuropathic pain, proliferation involves mostly progenitor cells (developing into oligodendrocytes) as well as microglia (Horky et al., 2006; Lytle & Wrathall, 2007). Consistently in all the studies, neurons never show proliferation. However, some report also suggests that after peripheral nerve injury the most abundant proliferating cells are stem cells (Xu et al., 2007b). Unfortunately, the specific role of glia proliferation in pain control has not been clearly demonstrated. Nevertheless, increasing number of glial cells such as microglia may result in increasing production of inflammatory mediators, leading to abnormal pain.
Figure 1. (a–d). Spared nerve injury (SNI) induces proliferation of microglial cells in the spinal cord.
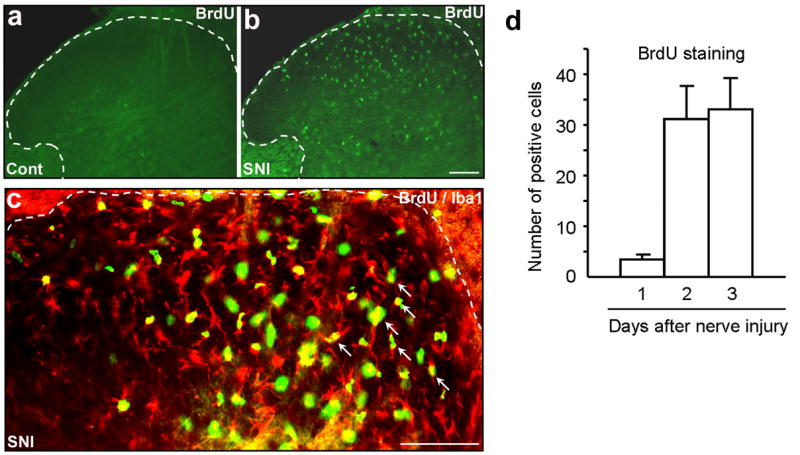
(a) Sham control (Cont) animals show almost no BrdU immunostaining. (b) Three days following SNI, there is a profound proliferation in the ipsilateral dorsal horn. Scale, 100 μm. (c) Double immunostaining of BrdU with the microglial marker Iba1 in the dorsal horn. Note that most BrdU positive cells also express Iba1 (indicated with arrows). White lines show the borders of the dorsal horn. Scale, 100 μm. (d) Number of BrdU-positive cells (per 30 μm-thick section) in the spinal cord dorsal horn. Note that proliferation starts rapidly after SNI.
Probably the most studied feature for glia activation is increased expression of specific glia markers, such as complement receptor 3 (CR3 or CD11b, recognized by OX-42 antibody) or Iba1. Upregulation of OX-42 or Iba1 is indicative of microglia activation. In inflammatory pain models that do not involve nerve injury, such as carrageenan model (Hua et al., 2005) and zymosan model (Clark et al., 2007a; Sweitzer et al., 1999), there is only a very moderate increase of OX42-immunoreactivity (IR) in the spinal cord. Whereas other models such as complete Freund’s adjuvant model (Clark et al., 2007a; Zhang et al., 2003) and mustard oil model (Molander et al., 1997) fail to demonstrate any increase of OX-42-IR. The inflammatory pain model that shows significant OX-42 increase is the formalin model (Aumeerally et al., 2004; Fu et al., 1999; Fu et al., 2000; Sweitzer et al., 1999). But formalin injection into a hindpaw will also produce nerve injury in the peripheral axons. Undoubtedly, nerve injury, such as lesions of the sciatic nerve or the spinal nerve produces robust upregulation of OX-42 (Fig. 2, Clark et al., 2007b; Lin et al., 2007; Wen et al., 2007; Zhang et al., 2003). Nerve injury-induced upregulation of OX-42 and Iba1 appears to be age-dependent: young rats show weak responses, which may explain why neuropathic pain is less robust in these animals (Moss et al., 2007; Vega-Avelaira et al., 2006). Although OX-42 is the most used marker for microglia activation in different pain conditions, pain states can be dissociated from OX-42 changes in several cases (Colburn et al., 1997; Obata et al., 2006; Winkelstein & DeLeo, 2002). Indeed, OX-42 increase is more associated with nerve injury than correlated with the pain symptoms.
Figure 2. (a–d). SNI induces CD11b (OX-42) upregulation in the spinal cord dorsal horn area that is terminated by injured nerve branches.
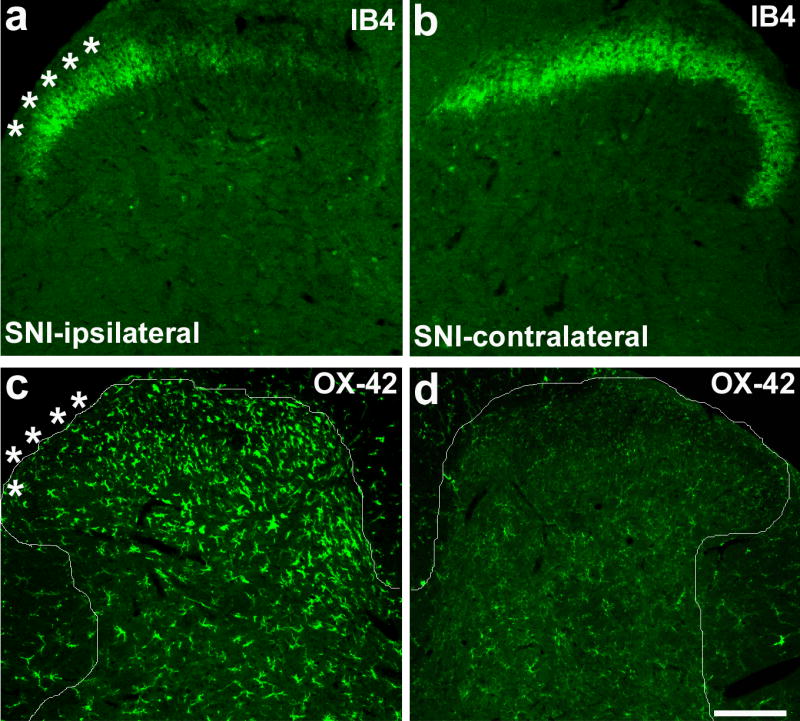
(a, b) Isolectin B4 (IB4) staining for primary afferents shows a reduction of the staining in the medial ipsilateral dorsal horn (L4 level), an area terminated by the injured branches (tibial and peroneal) of the sciatic nerve, at 3 days after SNI. (c, d) Immunohistochemistry shows increased expression of OX-42 in the medial dorsal horn on the ipsilateral side 3 days after SNI. White lines indicate the borders of the dorsal horn. White stars indicate the spinal area terminated by the intact sural nerve. Scale, 100 μm. Modified from Wen et al., 2007.
To better correlate microglia activation to pain-related behavior, we need to explore functional markers for microglia activation. Fortunately, in last several years, we have seen several potential markers, such as the ATP receptor P2X4 (Tsuda et al., 2003), the chemokine receptors CCR2 (Abbadie et al., 2003) and CX3CR1 (Verge et al., 2004; Zhuang et al., 2007), as well as Toll-like recepotor-4 (TLR4) (Tanga et al., 2005).
The role of microglia in pain control
In clinics, patients have a variety of complaints depending on the pathophysiological mechanisms underlying their pain. Pain can be spontaneous or evoked by diverse stimuli (cold, heat, touch). In animal models, we try to mimic these clinical characteristics. For example, we observe flinching or licking behavior of an affected paw and regard it as a spontaneous pain. We also use cold, heat, or mechanical stimuli to measure evoked pain. After injury, animals develop hyperalgesia, i.e. increased responses to a noxious stimulus. The most robust behavior phenomenon after injury is allodynia, painful behavior induced by a normally innocuous stimulus, such as light touch. For example, patients feel pain even with the simple contact of the clothes when they put it on.
Behavioral studies on glia regulation of pain began with the glia inhibitor fluorocitrate, an anti-metabolic drug which selectively disrupts the Krebs energy cycle of glia by inhibiting the glia-specific enzyme aconitase. Fluorocitrate has been shown to alleviate pain behavior in animal models of inflammatory pain, neuropathic pain, and postoperative pain (Clark et al., 2007a; Meller et al., 1994; Milligan et al., 2003; Obata et al., 2006; Watkins et al., 1997). However, it fails to inhibit muscle pain, a pain condition that does not show any glial activation (Ledeboer et al., 2006).
Minocycline, a tetracycline antibiotic, has been used as a microglia inhibitor and shows efficacy in several neurodegenerative conditions (Tikka & Koistinaho, 2001; Yong et al., 2004). Spinal injection of minocycline via intrathecal route was shown to attenuate neuropathic pain at early times but not at late times, suggesting a unique role of microglia in the development of nerve injury-induced neuropathic pain (Ledeboer et al., 2005b; Raghavendra et al., 2003). However, in a central neuropathic pain condition induced by spinal cord injury, minocycline is also capable of reversing neuropathic pain several weeks after injury (Hains & Waxman, 2006). Thus, the time course of microglia activation may be different after peripheral nerve injury and spinal cord injury. Minocycline has also been shown to inhibit OX-42 upregulation in the spinal cord under different pain conditions (Hains & Waxman, 2006; Lin et al., 2007; Raghavendra et al., 2003). The mechanisms underlying minocycline’s action are still not clear and may involve different targets, such as p38 MAP kinase (Nikodemova et al., 2006). Minocycline also inhibits matrix metalloproteinase (MMP), an important player in neuroinflammation. For example, our data have shown that minocycline can suppress MMP-9 expression in dissociated primary sensory neurons following TNF-α stimulation (unpublished results).
To determine the role of spinal microglia in neuropathic pain after spinal cord injury, Zhao et al. have tried to selectively target dorsal horn microglia with the Mac-1-SAP immunotoxin, a chemical conjugate of mouse monoclonal antibody to CD11b and the ribosome-inactivating protein saporin. They found reduced microglia staining and reversed pain-related behaviors after the treatment of the immunotoxin (Zhao et al., 2007a).
Since 2003, molecular mechanisms underlying microglia control of pain have begun to be revealed (Table 1). Tsuda et al. found that the purinergic receptor P2X4 is upregulated in spinal microglia after nerve injury, which is required for the development of neuropathic pain. Interestingly, intrathecal injection of ATP-activated microglia in the spinal cord is sufficient to induce pain (Tsuda et al., 2003). Further studies indicate that ATP-activated microglia can enhance pain by producing the growth factor BDNF (Coull et al., 2005). BDNF can act on lamina I neurons in the spinal cord to remove GABA inhibition by altering chloride reverse potential (Coull et al., 2005). ATP may also activate P2X7 receptors in spinal microglia to enhance pain (Donnelly-Roberts et al., 2007; Inoue, 2006).
Table 1.
| Chemokine receptors | |||
| CX3CR1 (Fractalkine) | Up-regulation | Enhance pain | Verge et al., 2004; Zhuang et al., 2007 |
| CCR2 (MCP-1) | Up-regulation | Enhance pain | Abaadie et al., 2003; White et al., 2005
Zhang et al., 2007 |
| CCR7 (CCL21) | Unknown | Enhance pain | Zhao et al., 2007b |
| ATP receptors | |||
| P2X4 | Up-regulation | Enhance pain | Tsuda et al., 2003 |
| P2X7 | Unknown | Enhance pain |
Donnelly-Roberts et al., 2007
Inoue, 2006 |
| Toll like receptors | |||
| TLR-4 | Unknown | Enhance pain | Tanga et al., 2005 |
| TLR-2 | Unknown | Enhance pain | Kim et al., 2007 |
| Cannabinoid receptor | |||
| CB2 | Up-regulation | Reduce pain | Zhang et al., 2003 |
| Romero-Sandoval & Eisenach, 2007 | |||
| Complement cascades | |||
| C1q | Up-regulation | Unknown | Griffin et al., 2007 |
| C3 | Up-regulation | Unknown | |
| C4 | Up-regulation | Unknown | |
| C5 | Up-regulation | Enhance pain | |
| C5aR | Up-regulation | Enhance pain | |
| Protein kinases | |||
| p-p38 | Up-regulation | Enhance pain | Jin et al., 2003; Tsuda et al., 2004 |
| pERK1/2 | Up-regulation | Enhance pain | Zhuang et al., 2005 |
| pERK5 | Up-regulation | Enhance pain | Obata et al., 2007 |
| pSrc family | Up-regulation | Enhance pain | Katsura et al., 2006 |
| Proteinases | |||
| Cathepsin S | Up-regulation | Enhance pain | Clark et al., 2007b |
| Transcriptional factors | |||
| NF-κB | Up-regulation | Enhance pain | Fig. 5, Ledeboer et al., 2005 |
| Pain mediators | |||
| BDNF | Unknown | Enhance pain | Coull et al., 2005 |
| TNF-α | Up-regulation | Enhance pain | Kim et al., 2007 |
| COX-1 | Up-regulation | Enhance pain | Zhu and Eisenach, 2003 |
| COX-2 | Up-regulation | Enhance pain | Zhao et al., 2007a |
Recent studies have shown that chemokines play important roles in microglia activation and pain sensitization (White et al., 2007). Chemokines are important for mediating neural-glia interaction, since they can be produced by neurons and act on microglia. For example, fractalkine (CXCL1) is produced in dorsal root ganglion (DRG) neurons and able to produce pain hypersensitivity (Verge et al., 2004). Nerve injury induces a rapid cleavage of fractalkine from the membrane (Zhuang et al., 2007), which involves a cysteine protease cathepsin S that is expressed in microglia (Clark et al., 2007b). Notably, the fractalkine receptor CX3CR1 is only expressed in spinal microglia and upregulated after nerve injury (Verge et al., 2004; Zhuang et al., 2007). Intrathecal injection of a CX3CR1 neutralizing antibody has been shown to inhibit neuropathic pain (Milligan et al., 2004) and inflammatory pain (Sun et al., 2007). Monocyte chemoattractant protein-1 (MCP-1, also called CCL2) is a well known chemoattractant for microglia. Nerve injury increases MCP-1 expression in the DRG (Tanaka et al., 2004; White et al., 2005; Zhang & De Koninck, 2006). Mice lacking CCR2 show an impaired neuropathic pain (Abbadie et al., 2003). Intrathecal injection of MCP-1 activates microglia in wild-type but not in CCR2-deficient mice (Zhang et al., 2007). In addition, the chemokine CCL21 was shown to activate microglia in the thalamus and induce pain-related behaviors (Zhao et al., 2007b).
It is generally believed that Toll like receptors (TLRs) normally respond to pathogens to regulate innate immunity. However, recent studies show that TLRs in the spinal cord are also involved in microglia activation and pain sensitization. For example, TLR2 and TLR4 are required for nerve injury-induced microglia activation in the spinal cord. Mice lacking TLR2 and TLR4 show attenuated neuropathic pain (Kim et al., 2007; Tanga et al., 2005). However, the cellular localization and injury-induced regulation of TLRs in the spinal cord remain to be investigated.
Activation of complement cascade in spinal microglia also plays a role in neuropathic pain (Griffin et al., 2007). A recent microarray study shows that the most regulated transcripts among different nerve injury models are related to complement components (e.g., C1q, C3, and C4). In addition, the terminal complement component C5 and its receptor C5aR are upregulated in spinal microglia after peripheral nerve injury and are required for neuropathic pain sensitivity (Griffin et al., 2007).
Although these TLRs and receptors of chemokines, complement components, and ATP are important for microglia activation and development of chronic pain, the signaling mechanisms linking these receptors to the production of inflammatory mediators are still missing.
MAP kinase activation in microglia and pain control
Mounting evidence suggests that mitogen-activated protein kinases (MAPKs), including extracellular signal-regulated kinase (ERK), p38, and c-Jun N-terminal kinase (JNK), play important roles in pain sensitization (Ji et al., 2007). Specific antibodies against activated (phosphorylated) forms of MAPKs and specific inhibitors available for all three MAPK pathways have allowed us to study both the activation of MAPKs in the spinal cord and behavioral consequences of the activation. Inhibition of all three MAPK pathways at the spinal level have been shown to alleviate inflammatory pain and neuropathic pain (reviewed in Ji et al., 2007; also see Daulhac et al., 2006; Obata et al., 2004; Schafers et al., 2003; Svensson et al., 2003; Zhao et al., 2007a). Interestingly, some glial inhibitors such as minocycline can inhibit the activation of MAPKs (Nikodemova et al., 2006). Among three major MAPKs, p38 and ERK are activated in spinal microlgia under injury conditions, and the activation of these two MAPKs are important for microglial signaling.
Many studies from different groups have shown that activation of p38 MAPK in spinal microglia is essential for the pathogenesis of chronic pain (Ji & Suter, 2007). p38 is required for central sensitization in the spinal cord dorsal horn and in the trigeminal subnucleus caudalis (medullary dorsal horn) (Svensson et al., 2003; Xie et al., 2007). p38 is normally activated by stress signals, such as heat shock, ultraviolet light, and ischemia, as well as proinflammatory cytokines (Widmann et al., 1999). After spinal nerve ligation, we have shown that p38 is only activated in OX-42-positive microglia in the spinal cord. This activation is evident as early as 12 hours and peaks after 3 days (Jin et al., 2003). Another groups using the same model also shows p38 activation in spinal microglia with slightly delayed time course (Tsuda et al., 2004). Other groups also demonstrate p38 activation in spinal microglia in different pain conditions (Hains & Waxman, 2006; Hua et al., 2005; Kim et al., 2002; Svensson et al., 2003; Svensson et al., 2005b; Xu et al., 2007a; Xu et al., 2007b). In the SNI model of neuropathic pain, the microglia activation of p38 is mostly seen in the medial part of the dorsal horn, where the injured tibial and peroneal nerves terminate (Wen et al., 2007).
Several molecules, such as proinflammatory cytokines and chemokines have been implicated in injury-induced p38 activation in the spinal cord. Intrathecal administration of TNF-α inhibitor suppresses p38 activation after spinal nerve ligation (Svensson et al., 2005b), ventral root transection (Xu et al., 2007a), and joint arthritis (Boyle et al., 2006). We have recently demonstrated that spinal fractalkine injection causes p38 activation. Conversely, antagonizing CX3CR1 inhibits nerve injury-induced p38 activation and neuropathic pain (Zhuang et al., 2007). Consistently, cysteine protease cathepsin S activates p38 in spinal microglia by cleavage of fractalkine (Clark et al., 2007b). ATP may also causes p38 activation in spinal microglia (Inoue, 2006).
Nerve injury induces spontaneous electrical activity in the axons and cell bodies of DRG neurons, which is important for the genesis of neuropathic pain. We have shown that spontaneous activity is also required for the activation of p38 in spinal cord microglia. Blocking peripheral nerve activity by bupivacaine, a local anesthetic, can prevent nerve injury-induced p38 activation in the spinal cord (Wen et al., 2007). Injury-evoked hyperactivity in DRG neurons is likely to release several microglia activators, such as ATP, cytokines, and chemokines for activating microglia (Ji & Suter, 2007).
Injury-induced spontaneous activity can also release the matrix metalloproteinase-9 (MMP-9) from DRG neurons. We have shown that MMP-9 upregulation following nerve injury is required for the early phase development of neuropathic pain (Kawasaki et al., 2008). Intrathecal injection of MMP-9 induces both microglia activation (p38 phosphorylation and OX-42 upregulation) in the spinal cord and neuropathic pain symptoms such as mechanical allodynia. Importantly, MMP-9-induced allodynia is blocked by spinal inhibition of p38. Furthermore, MMP-9 activates microglia via cleavage of pro-IL-1β to produce active IL-1β (Kawasaki et al., 2008), although MMP-9 may also have other substrates such as fractalkine. Thus MMP-9 is another important player for mediating neural-glia interaction.
How can p38 activation in spinal microglia result in pain sensitization? Increasing evidence supports a role of IL-1β in inducing central sensitization. Nerve injury increases the synthesis of IL-1β in the spinal cord and blocking IL-1β signaling inhibits neuropathic pain sensitization (Sweitzer et al., 2001). IL-1β in the spinal cord can directly enhance excitatory synaptic transmission and suppress inhibitory synaptic transmission (Kawasaki and Ji, unpublished results). IL-1β expression in the spinal cord is mediated by p38, because intrathecal p38 inhibitor can suppress nerve injury-induced IL-1β upregulation, presumably via NF-κB mechanisms. The transcription factor NF-κB has been strongly implicated in regulating the transcription of inflammatory mediators such as IL-1β (Ji & Suter, 2007) and pain hypersensitivity (Ledeboer et al., 2005a). After nerve injury, NF-κB is induced in spinal microglia (Fig. 5). p38 activation should also regulate the transcription of other inflammatory mediators such as TNFα and cyclooxygenase-2 (Bhat et al., 1998; Jana et al., 2003). In addition, activation of spinal p38 also causes rapid IL-1β release in spinal cord slices following LPS stimulation (Clark et al., 2006) and PGE2 release in the spinal cord following intrathecal injection of A-dynorphin (Svensson et al., 2005a).
Figure 5. (a–c). Spinal nerve ligation (SNL) increases NF-κB expression in spinal microglia.

(a, b) SNL increases the immunoreactivity of NF-κB (65kD unit) on day 3 in the medial superficial spinal cord, compared to non-injured control (Cont) spinal cord. Scale, 50 μm. (c) Double immunofluorescence shows colocalization of NF-κB and OX-42. Scale, 20 μm.
Compared with p38 activation, ERK activation in the spinal cord is more dynamic (Ji, 2007). Following intense noxious stimulation, acute or chronic inflammation, ERK is activated in dorsal horn neurons and contributes importantly to central sensitization (Ji, 2007). However, after spinal nerve ligation, ERK is sequentially activated in neurons (first several hours), microglia (first several days), and astrocytes (after 3 weeks) (Zhuang et al., 2005). Consistently, ERK is predominantly activated in spinal microglia three days after SNI, as seen by colocalization of pERK with Iba-1 (Figs 3, 4). In this model, spinal inhibition of ERK activation with U0126, an ERK kinase (MEK) inhibitor, prevents the development of mechanical allodynia (Fig. 3e). Intrathecal MEK inhibitors were also shown to alleviate neuroapthic pain after spinal nerve ligation at time points when ERK is activated in spinal microglia (Obata et al., 2004; Zhuang et al., 2005). Thus, both ERK and p38 activation in spinal microglia are important for the pathogenesis of neuropathic pain. Most studies on ERK pathway focus on ERK1/2 that are activated by MEK1/2. However, ERK subfamily also contains ERK5 that is activated by MEK5. Notably, the MEK inhibitor U0126 inhibits the activation of both ERK1/2 and ERK5. ERK5 has recently been shown activated in dorsal horn microglia after nerve injury. This activation is also necessary for neuropathic pain development, since inhibition of ERK5 with antisense knockdown shows reversal of allodynia (Obata et al., 2007).
Figure 3. (a–e). Activation of p38 and ERK in the spinal cord after SNI is required for neuropathic pain development.
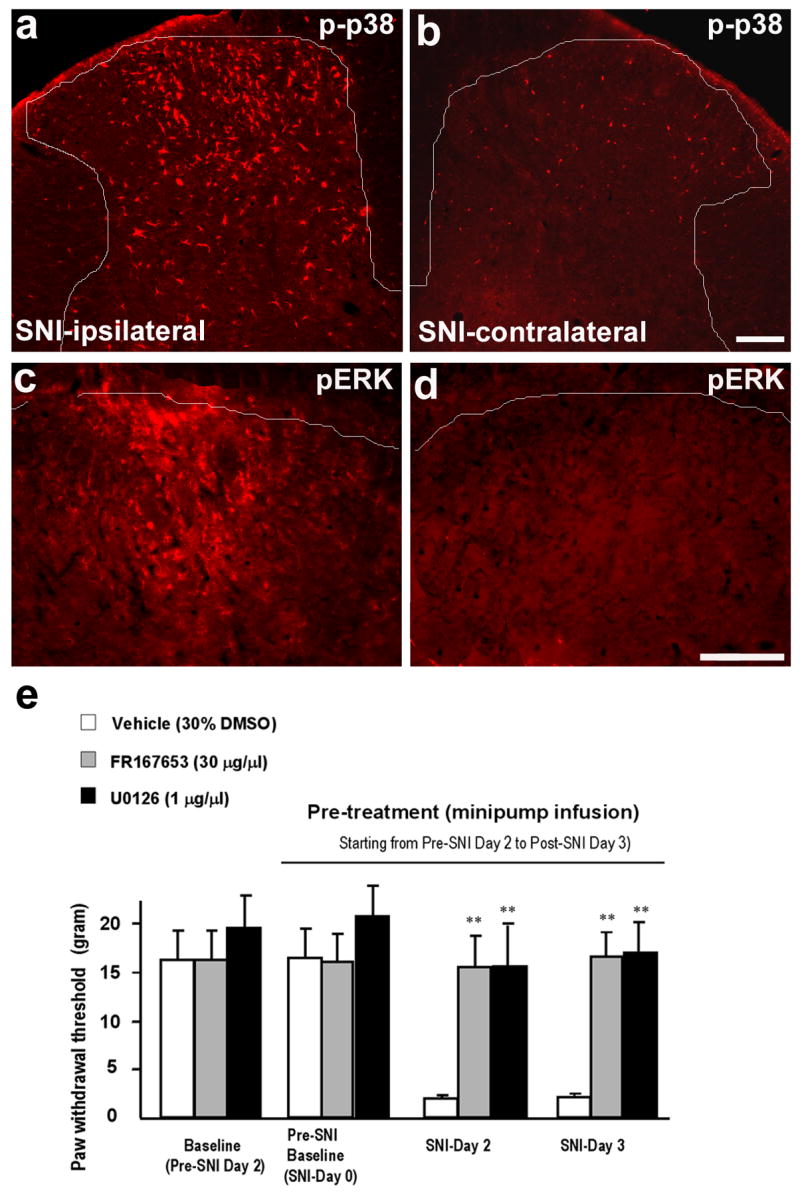
(a–d) Immunohistochemistry shows an increase in phosphorylation of p38 (p-p38, a, b) and ERK (pERK, c, d) in the medial dorsal horn on the ipsilateral side (L4) 3 days after SNI. White lines indicate the borders of the dorsal horn. Scales, 100 μm. (e) SNI-induced mechanical allodynia is prevented by the p38 inhibitor FR167653 or the MEK (ERK kinase) inhibitor U0126. FR167653 (30 μg/μl) or U0126 (1 μg/μl) was infused into intrathecal space via an osmotic pump (0.5 μl/h for 5 days) starting 2 days before SNI. Note that the basal mechanical sensitivity does not change after FR167653 or U0126 infusion. **, P < 0.01, t test, compared to corresponding vehicle controls (30% DMSO), n = 4. Modified from Wen et al., 2007.
Figure 4. (a–g). p38 and ERK are activated in different populations of microglia in the spinal cord following SNI.

(a–f) Double immunofluorescence shows colocalization of p-p38 and OX-42 (a–c) and colocalization of pERK and OX-42 (d–f) in the medial superficial dorsal horn. (g) Double immunofluorescence shows that p-p38 and pERK are not activated in the same cells. c is an overlay of a and b, f is an overlay of d and e. Scales, 50 μm.
The tyrosine kinase Src appears to be upstream of ERK. The Src-family kinases are hyperphosphorylated in spinal microglia after nerve injury. Inhibiting Src can attenuate neuroapthic pain by suppressing ERK activation in the spinal cord. However, p38 activation in the spinal cord is not affected by Src inhibitor (Katsura et al., 2006). ERK is also downstream of Src activation in cultured microglia. However, ERK only mediates Src-induced chemokine release, but not Src-induced microglia phagocytosis. Instead, phagocytois is mediated by the phosphatidylinositol-3 kinase (PI-3K) pathway. Downstream, ERK activation in microglia results in the activation of TNF-α converting enzyme (TACE or ADAM17), leading to the cleavage of pro-TNF-α and subsequent release of mature TNF-α (Jana et al., 2003). Our electrophysiological studies in spinal cord slices show that TNF-α enhances glutamate synaptic transmission and potentiates NMDA currents in superficial dorsal horn neurons (Kawasaki and Ji, unpublished observation). Activation of ERK in spinal microglia can also induce transcription of many genes, further maintaining enhanced pain states (Ji, 2007). For example, ERK activation leads to COX-2 upregulation in spinal microglia leading to increased production of PGE2 (Zhao et al., 2007a), another important pain mediator for central sensitization (Scholz & Woolf, 2002). In addition to COX-2, COX-1 was also shown to be activated in spinal glia presumably microglia after nerve injury and essential for neuropathic pain sensitization (Zhu & Eisenach, 2003).
There is little evidence demonstrating the existence of different populations of microglia in the brain (Hanisch and Kettenmann, 2007). However, microglia are not homogenous. Different microglia can display diverse phenotypes, expressing different markers and probably exhibit different functions. Even though both p38 and ERK are activated in microglia at early times after nerve injury, they appear to be activated in different populations of microglia. As shown in Fig. 4, phosphorylated ERK and p38 (pERK and p-p38) are not expressed in the same cells 3 days post SNI, in agreement with another study showing a small portion (33%) of overlapping after spinal nerve ligation (Katsura et al., 2006). Although blocking either ERK or p38 pathway can attenuate neuropathic pain, nerve injury-induced Iba1 increase in the spinal cord is not suppressed by inhibition of ERK or p38. However, a combination of MEK and p38 inhibitors can reduce Iba1 levels (Suter and Ji, unpublished observation). Thus, it is likely that blocking both MAPK pathways is a more effective treatment for neuropathic pain. The heterogeneity of microglia is also supported by a recent study showing that Iba1-positive microglia population in the spinal cord is composed of a mixture of resident microglia and invading monocytes (Zhang et al., 2007).
Conclusions and clinical implications
Chronic pain is an increasing burden for the society, affecting 20% of the population. Unfortunately, current treatments that focus mostly on targeting neuronal excitability and transmission are not satisfying. The emergence of glial cells as important players in pain control and also as new targets for pain medicine has brought great excitement to the pain research field. We have discussed the role of astrocytes in the previous review (Ji et al., 2006) and the role of microglia in the current review. In the normal conditions, there seems to be limited role of glia, as none of the treatments cited above really changes the baseline pain thresholds in animals. It is important that a drug can alleviate pathological pain without inhibiting normal pain perception, as this protective pain is important for our survival as a warning for danger.
According to a current view, microglia are not dormant in the normal conditions by having an active surveying function. After injury, microglia just shift their activation status (being reactive) rather than “become activated” (Hanisch and Kettenmann, 2007). In this review, we refer “microglia activation” to a dramatic change of activation status in the spinal cord that is associated with different injury conditions (e.g., nerve injury) that can lead to chronic pain. Microglia have dual role in diverse neurologic pathologies and can be both destructive and protective, depending on whether microglia release neurotoxic factors (e.g., TNF-α, IL-1β, reactive oxygen species) or neuroprotective factors (e.g., growth factors BDNF, NGF, bFGF). It is important to emphasize that, in the spinal cord, all these mediators promote pain (Fig. 6). Thus, microglia activation is a long-standing problem for chronic pain, and targeting microglia should be an effective strategy for clinical management of chronic pain
Figure 6. (a–c). Schematic representation of microglial control of pain.
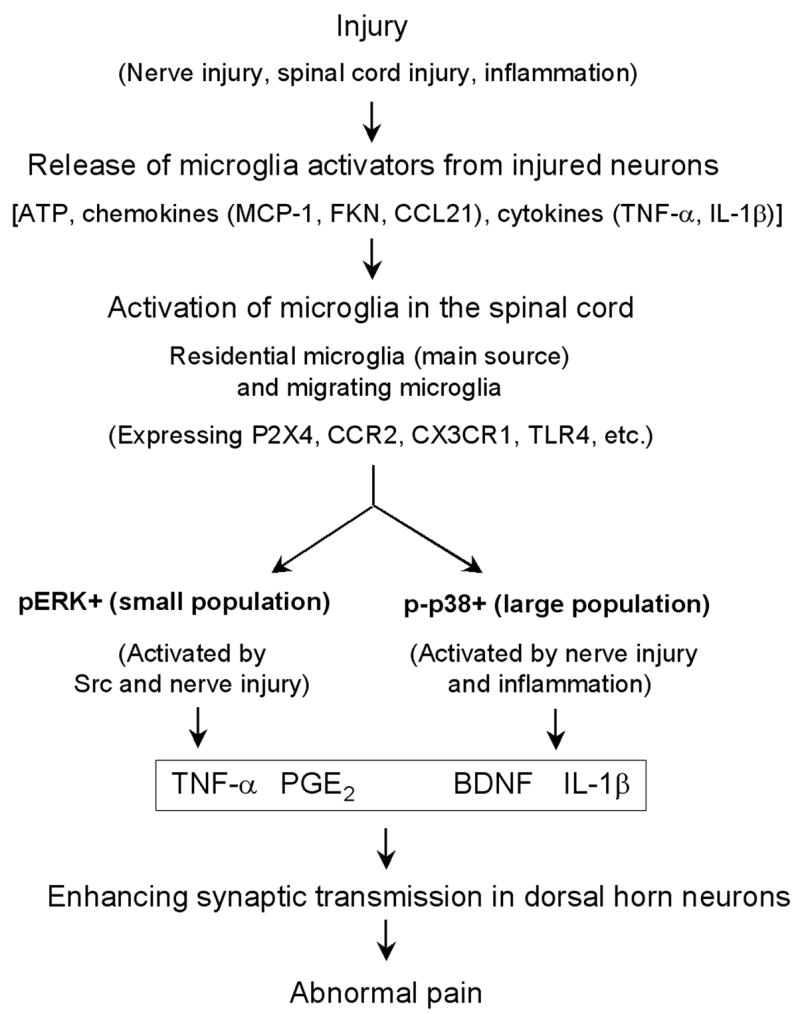
After various injury conditions (such as nerve injury, spinal cord injury, and inflammation), injured or affected neurons (e.g., DRG or spinal cord neurons) can release factors that are capable of activating microglia in the spinal cord. These factors include ATP, chemokines such as MCP-1, fractalkine (FKN), and CCL21, and proinflammatory cytokines such as TNF-α and IL-1β. These microglia activators can bind their receptors on microglia, leading to the activation of microglia. These activated spinal microglia are mainly residential microglia, but may also be migrating microglia from circulation. Activated microglia contain three subpopulations in the spinal cord: pERK + population, p-p38 + population, and unknown population. While ERK is activated by Src and nerve injury, p38 is activated by both nerve injury and inflammation. ERK activation releases TNF-α via activation of TNF-α converting enzyme, whereas p38 activation enhances IL-1β release. Activation of p38 and ERK can also regulate the synthesis of the inflammatory mediators via transcription factor (e.g., NF-κB). Microglia also produce the growth factor BDNF. Finally, these pain mediators collaborate to induce and maintain abnormal pain in injury conditions by enhancing synaptic strength and central sensitization in dorsal horn neurons.
There are several types of “microglia-targeting” drugs that can be developed and tested for pain control. First, many receptors, such as chemokine receptors (CCR2, CX3CR1), ATP receptors (P2X4, P2X7), TLRs (TLR2, TLR4), and complement receptor (C5aR), are expressed in spinal microglia and regulated after injury (Table 1). Development of antagonists for these receptors may lead to new therapy. Also, cannabinoid receptor type 2 (CB2) is expressed in spinal microglia, and CB2 agonist can reduce incision-induced microglial activation in spinal cord and reverse incisional pain without inducing behavioral and psychological side effects as CB1 agonists do (Romero-Sandoval and Eisenach, 2007). Second, drugs that target downstream common pathways such as MAPK pathways or TNF-α and IL-1β signaling (Fig. 6) could be very effective, if the side effects can be tolerated (Watkins and Maier, 2003; Ji et al., 2007). p38 MAPK inhibitors show high efficacy in different animal models, but they may produce side effects when high doses or long-term treatment are employed. The beta isoform of p38 MAPK (p38β) could be a valid target due to its relatively restricted expression compared to p38α isoform (Svensson 2005; Xu et al., unpublished observation). Third, those drugs, such as minocycline, propentofylline, and methotrexate, which have already been tested clinically for other indications, are certainly easier to get approved for pain treatment than new compounds. Unfortunately, minocycline fails in a recent trial for treating multiple sclerosis (Gordon et al., 2007), in part due to delayed treatment. Minocycline may show efficacy when given during the early phase of disease development. Propentofylline, a glial modulating agent, shows efficacy in different pain models including neuropathic pain model (Watkins and Maier., 2003; Tawfik et al, 2007). Methotrexate has recently been shown to reduce neuropathic pain and microglia activation in animals (Scholz et al., 2008). However, it is difficult for clinical studies to choose appropriate patients, times, doses, and route of administration. Finally, co-administration of a glial modulating agent (e.g., minocycline or p38 inhibitor) with an analgesic (e.g., morphine) would improve the efficacy of that analgesic. Since opioids are still the most used analgesics for chronic pain management, enhancing morphine analgesia with a glial activation inhibitor will be therapeutically practical. Long-term morphine treatment produces glial activation and morphine tolerance (Song and Zhao, 2001; Cui et al., 2006; Watkins et al., 2005). Interestingly, p38 inhibitor has been shown to attenuate morphine tolerance (Cui et al., 2006) and enhance morphine analgesia after nerve injury (Cheng and Ji, unpublished results). It would be a major advance in pain treatment even by reducing tolerance and side effects of opioids.
Acknowledgments
This study was funded by NIH grants DE17794, NS 54932, TW7180, and a collaboration grant from International Association for the Study of Pain. MRS was supported by Swiss National Science Foundation PBLAB118504 and UPSA Pain Institute Switzerland Postdoctoral Fellowship 2007.
References
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci USA. 2003;100(13):7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aumeerally N, Allen G, Sawynok J. Glutamate-evoked release of adenosine and regulation of peripheral nociception. Neuroscience. 2004;127(1):1–11. doi: 10.1016/j.neuroscience.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-alpha gene expression in endotoxin-stimulated primary glial cultures. J Neurosci. 1998;18(5):1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Boyle DL, Jones TL, Hammaker D, Svensson CI, Rosengren S, Albani S, Sorkin L, Firestein GS. Regulation of peripheral inflammation by spinal p38 MAP kinase in rats. PLoS Med. 2006;3(9):e338. doi: 10.1371/journal.pmed.0030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AK, D’Aquisto F, Gentry C, Marchand F, McMahon SB, Malcangio M. Rapid co-release of interleukin 1beta and caspase 1 in spinal cord inflammation. J Neurochem. 2006;99(3):868–880. doi: 10.1111/j.1471-4159.2006.04126.x. [DOI] [PubMed] [Google Scholar]
- Clark AK, Gentry C, Bradbury EJ, McMahon SB, Malcangio M. Role of spinal microglia in rat models of peripheral nerve injury and inflammation. Eur J Pain. 2007a;11(2):223–230. doi: 10.1016/j.ejpain.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, Bevan S, Malcangio M. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci USA. 2007b doi: 10.1073/pnas.0610811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colburn RW, DeLeo JA, Rickman AJ, Yeager MP, Kwon P, Hickey WF. Dissociation of microglial activation and neuropathic pain behaviors following peripheral nerve injury in the rat. J Neuroimmunol. 1997;79(2):163–175. doi: 10.1016/s0165-5728(97)00119-7. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438(7070):1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444(7121):894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle DE. Partial peripheral nerve injury leads to activation of astroglia and microglia which parallels the development of allodynic behavior. Glia. 1998;23(1):75–83. [PubMed] [Google Scholar]
- Daulhac L, Mallet C, Courteix C, Etienne M, Duroux E, Privat AM, Eschalier A, Fialip J. Diabetes-induced mechanical hyperalgesia involves spinal mitogen-activated protein kinase activation in neurons and microglia via N-methyl-D-aspartate-dependent mechanisms. Mol Pharmacol. 2006;70(4):1246–1254. doi: 10.1124/mol.106.025478. [DOI] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87(2):149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- Donnelly-Roberts D, McGaraughty S, Shieh CC, Honore P, Jarvis MF. Painful Purinergic Receptors. J Pharmacol Exp Ther. 2007 doi: 10.1124/jpet.106.105890. [DOI] [PubMed] [Google Scholar]
- Echeverry S, Shi XQ, Zhang J. Characterization of cell proliferation in rat spinal cord following peripheral nerve injury and the relationship with neuropathic pain. Pain. 2007 doi: 10.1016/j.pain.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Eriksson NP, Persson JK, Svensson M, Arvidsson J, Molander C, Aldskogius H. A quantitative analysis of the microglial cell reaction in central primary sensory projection territories following peripheral nerve injury in the adult rat. Exp Brain Res. 1993;96(1):19–27. doi: 10.1007/BF00230435. [DOI] [PubMed] [Google Scholar]
- Fu KY, Light AR, Maixner W. Relationship between nociceptor activity, peripheral edema, spinal microglial activation and long-term hyperalgesia induced by formalin. Neuroscience. 2000;101(4):1127–1135. doi: 10.1016/s0306-4522(00)00376-6. [DOI] [PubMed] [Google Scholar]
- Fu KY, Light AR, Matsushima GK, Maixner W. Microglial reactions after subcutaneous formalin injection into the rat hind paw. Brain Res. 1999;825(1–2):59–67. doi: 10.1016/s0006-8993(99)01186-5. [DOI] [PubMed] [Google Scholar]
- Garrison CJ, Dougherty PM, Kajander KC, Carlton SM. Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Res. 1991;565(1):1–7. doi: 10.1016/0006-8993(91)91729-k. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Tetzlaff W, Streit WJ, Kreutzberg GW. Microglial cells but not astrocytes undergo mitosis following rat facial nerve axotomy. Neurosci Lett. 1988;85(3):317–321. doi: 10.1016/0304-3940(88)90585-x. [DOI] [PubMed] [Google Scholar]
- Griffin RS, Costigan M, Brenner GJ, Ma CH, Scholz J, Moss A, Allchorne AJ, Stahl GL, Woolf CJ. Complement induction in spinal cord microglia results in anaphylatoxin C5a-mediated pain hypersensitivity. J Neurosci. 2007;27(32):8699–8708. doi: 10.1523/JNEUROSCI.2018-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26(16):4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore P, Rogers SD, Schwei MJ, Salak-Johnson JL, Luger NM, Sabino MC, Clohisy DR, Mantyh PW. Murine models of inflammatory, neuropathic and cancer pain each generates a unique set of neurochemical changes in the spinal cord and sensory neurons. Neuroscience. 2000;98(3):585–598. doi: 10.1016/s0306-4522(00)00110-x. [DOI] [PubMed] [Google Scholar]
- Horky LL, Galimi F, Gage FH, Horner PJ. Fate of endogenous stem/progenitor cells following spinal cord injury. J Comp Neurol. 2006;498(4):525–538. doi: 10.1002/cne.21065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner PJ, Power AE, Kempermann G, Kuhn HG, Palmer TD, Winkler J, Thal LJ, Gage FH. Proliferation and differentiation of progenitor cells throughout the intact adult rat spinal cord. J Neurosci. 2000;20(6):2218–2228. doi: 10.1523/JNEUROSCI.20-06-02218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua XY, Svensson CI, Matsui T, Fitzsimmons B, Yaksh TL, Webb M. Intrathecal minocycline attenuates peripheral inflammation-induced hyperalgesia by inhibiting p38 MAPK in spinal microglia. Eur J Neurosci. 2005;22(10):2431–2440. doi: 10.1111/j.1460-9568.2005.04451.x. [DOI] [PubMed] [Google Scholar]
- Inoue K. The function of microglia through purinergic receptors: neuropathic pain and cytokine release. Pharmacol Ther. 2006;109(1–2):210–226. doi: 10.1016/j.pharmthera.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Jana M, Dasgupta S, Saha RN, Liu X, Pahan K. Induction of tumor necrosis factor-alpha (TNF-alpha) by interleukin-12 p40 monomer and homodimer in microglia and macrophages. J Neurochem. 2003;86(2):519–528. doi: 10.1046/j.1471-4159.2003.01864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR. The Role of ERK/MAPK in Spinal Glia for Neuropathic Pain: Signal Transduction in Spinal Microglia and Astrocytes after Nerve Injury. In: DeLeo JA, Sorkin LS, Watkins LR, editors. Immune and Glial Regulation of Pain. IASP Press; Seattle: 2007. pp. 269–282. [Google Scholar]
- Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Decosterd I. Possible role of spinal astrocytes in maintaining chronic pain sensitization: review of current evidence with focus on bFGF/JNK pathway. Neuron Glia Biol. 2006;2(4):259–269. doi: 10.1017/S1740925X07000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Zhang YQ. Protein kinases as potential targets for the treatment of pathological pain. Handb Exp Pharmacol. 2007;(177):359–389. doi: 10.1007/978-3-540-33823-9_13. [DOI] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26(12):696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Sci STKE. 2004;2004(252):reE14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- Ji RR, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain. 2007;3(1):33. doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci. 2003;23(10):4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413(6852):203–210. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- Katsura H, Obata K, Mizushima T, Sakurai J, Kobayashi K, Yamanaka H, Dai Y, Fukuoka T, Sakagami M, Noguchi K. Activation of Src-family kinases in spinal microglia contributes to mechanical hypersensitivity after nerve injury. J Neurosci. 2006;26(34):8680–8690. doi: 10.1523/JNEUROSCI.1771-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Xu Z, Wang X, Park JY, Zhuang ZY, Tan PH, Gao YJ, Roy K, Corfas G, Lo EH, Ji RR. Distinct roles of matrix metalloproteinases in early and late phase development of neuropathic pain. Nat Med. doi: 10.1038/nm1723. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Kim MA, Cho IH, Kim MS, Lee S, Jo EK, Choi SY, Park K, Kim JS, Akira S, Na HS, Oh SB, Lee SJ. A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J Biol Chem. 2007;282(20):14975–14983. doi: 10.1074/jbc.M607277200. [DOI] [PubMed] [Google Scholar]
- Kim SY, Bae JC, Kim JY, Lee HL, Lee KM, Kim DS, Cho HJ. Activation of p38 MAP kinase in the rat dorsal root ganglia and spinal cord following peripheral inflammation and nerve injury. Neuroreport. 2002;13(18):2483–2486. doi: 10.1097/00001756-200212200-00021. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39(1):151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Gamanos M, Lai W, Martin D, Maier SF, Watkins LR, Quan N. Involvement of spinal cord nuclear factor kappaB activation in rat models of proinflammatory cytokine-mediated pain facilitation. Eur J Neurosci. 2005a;22(8):1977–1986. doi: 10.1111/j.1460-9568.2005.04379.x. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Mahoney JH, Milligan ED, Martin D, Maier SF, Watkins LR. Spinal cord glia and interleukin-1 do not appear to mediate persistent allodynia induced by intramuscular acidic saline in rats. J Pain. 2006;7(10):757–767. doi: 10.1016/j.jpain.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005b;115(1–2):71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Levine JD, essandri-Haber N. TRP channels: targets for the relief of pain. Biochim Biophys Acta. 2007;1772(8):989–1003. doi: 10.1016/j.bbadis.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Lin CS, Tsaur ML, Chen CC, Wang TY, Lin CF, Lai YL, Hsu TC, Pan YY, Yang CH, Cheng JK. Chronic intrathecal infusion of minocycline prevents the development of spinal-nerve ligation-induced pain in rats. Reg Anesth Pain Med. 2007;32(3):209–216. doi: 10.1016/j.rapm.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Liu L, Rudin M, Kozlova EN. Glial cell proliferation in the spinal cord after dorsal rhizotomy or sciatic nerve transection in the adult rat. Exp Brain Res. 2000;131(1):64–73. doi: 10.1007/s002219900273. [DOI] [PubMed] [Google Scholar]
- Liu L, Tornqvist E, Mattsson P, Eriksson NP, Persson JK, Morgan BP, Aldskogius H, Svensson M. Complement and clusterin in the spinal cord dorsal horn and gracile nucleus following sciatic nerve injury in the adult rat. Neuroscience. 1995;68(1):167–179. doi: 10.1016/0306-4522(95)00103-p. [DOI] [PubMed] [Google Scholar]
- Lobsiger CS, Cleveland DW. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat Neurosci. 2007;10(11):1355–1360. doi: 10.1038/nn1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytle JM, Wrathall JR. Glial cell loss, proliferation and replacement in the contused murine spinal cord. Eur J Neurosci. 2007;25(6):1711–1724. doi: 10.1111/j.1460-9568.2007.05390.x. [DOI] [PubMed] [Google Scholar]
- Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci. 2005;6(7):521–532. doi: 10.1038/nrn1700. [DOI] [PubMed] [Google Scholar]
- Meller ST, Dykstra C, Grzybycki D, Murphy S, Gebhart GF. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33(11):1471–1478. doi: 10.1016/0028-3908(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Twining C, Chacur M, Biedenkapp J, O’Connor K, Poole S, Tracey K, Martin D, Maier SF, Watkins LR. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci. 2003;23(3):1026–1040. doi: 10.1523/JNEUROSCI.23-03-01026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Zapata V, Chacur M, Schoeniger D, Biedenkapp J, O’Connor KA, Verge GM, Chapman G, Green P, Foster AC, Naeve GS, Maier SF, Watkins LR. Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur J Neurosci. 2004;20(9):2294–2302. doi: 10.1111/j.1460-9568.2004.03709.x. [DOI] [PubMed] [Google Scholar]
- Molander C, Hongpaisan J, Svensson M, Aldskogius H. Glial cell reactions in the spinal cord after sensory nerve stimulation are associated with axonal injury. Brain Res. 1997;747(1):122–129. doi: 10.1016/s0006-8993(96)01230-9. [DOI] [PubMed] [Google Scholar]
- Moss A, Beggs S, Vega-Avelaira D, Costigan M, Hathway GJ, Salter MW, Fitzgerald M. Spinal microglia and neuropathic pain in young rats. Pain. 2007;128(3):215–224. doi: 10.1016/j.pain.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Kohsaka S. Microglia: activation and their significance in the central nervous system. J Biochem(Tokyo) 2001;130(2):169–175. doi: 10.1093/oxfordjournals.jbchem.a002969. [DOI] [PubMed] [Google Scholar]
- Narita M, Yoshida T, Nakajima M, Narita M, Miyatake M, Takagi T, Yajima Y, Suzuki T. Direct evidence for spinal cord microglia in the development of a neuropathic pain-like state in mice. J Neurochem. 2006;97(5):1337–1348. doi: 10.1111/j.1471-4159.2006.03808.x. [DOI] [PubMed] [Google Scholar]
- Nikodemova M, Duncan ID, Watters JJ. Minocycline exerts inhibitory effects on multiple mitogen-activated protein kinases and IkappaBalpha degradation in a stimulus-specific manner in microglia. J Neurochem. 2006;96(2):314–323. doi: 10.1111/j.1471-4159.2005.03520.x. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Obata H, Eisenach JC, Hussain H, Bynum T, Vincler M. Spinal glial activation contributes to postoperative mechanical hypersensitivity in the rat. J Pain. 2006;7(11):816–822. doi: 10.1016/j.jpain.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Obata K, Katsura H, Mizushima T, Sakurai J, Kobayashi K, Yamanaka H, Dai Y, Fukuoka T, Noguchi K. Roles of extracellular signal-regulated protein kinases 5 in spinal microglia and primary sensory neurons for neuropathic pain. J Neurochem. 2007;102(5):1569–1584. doi: 10.1111/j.1471-4159.2007.04656.x. [DOI] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Kobayashi K, Dai Y, Mizushima T, Katsura H, Fukuoka T, Tokunaga A, Noguchi K. Role of mitogen-activated protein kinase activation in injured and intact primary afferent neurons for mechanical and heat hypersensitivity after spinal nerve ligation. J Neurosci. 2004;24(45):10211–10222. doi: 10.1523/JNEUROSCI.3388-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306(2):624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Raivich G. Like cops on the beat: the active role of resting microglia. Trends Neurosci. 2005;28(11):571–573. doi: 10.1016/j.tins.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Schafers M, Svensson CI, Sommer C, Sorkin LS. Tumor necrosis factor-alpha induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci. 2003;23(7):2517–2521. doi: 10.1523/JNEUROSCI.23-07-02517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. Can we conquer pain? Nat Neurosci. 2002;5(Suppl 1):1062–1067. doi: 10.1038/nn942. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10(11):1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- Sessle BJ. Glia: non-neural players in orofacial pain. J Orofac Pain. 2007;21(3):169–170. [PubMed] [Google Scholar]
- Sun S, Cao H, Han M, Li TT, Pan HL, Zhao ZQ, Zhang YQ. New evidence for the involvement of spinal fractalkine receptor in pain facilitation and spinal glial activation in rat model of monoarthritis. Pain. 2007;129(1–2):64–75. doi: 10.1016/j.pain.2006.09.035. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Hua XY, Powell HC, Lai J, Porreca F, Yaksh TL. Prostaglandin E2 release evoked by intrathecal dynorphin is dependent on spinal p38 mitogen activated protein kinase. Neuropeptides. 2005a;39(5):485–494. doi: 10.1016/j.npep.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Marsala M, Westerlund A, Calcutt NA, Campana WM, Freshwater JD, Catalano R, Feng Y, Protter AA, Scott B, Yaksh TL. Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem. 2003;86(6):1534–1544. doi: 10.1046/j.1471-4159.2003.01969.x. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Schafers M, Jones TL, Powell H, Sorkin LS. Spinal blockade of TNF blocks spinal nerve ligation-induced increases in spinal P-p38. Neurosci Lett. 2005b;379(3):209–213. doi: 10.1016/j.neulet.2004.12.064. [DOI] [PubMed] [Google Scholar]
- Sweitzer S, Martin D, DeLeo JA. Intrathecal interleukin-1 receptor antagonist in combination with soluble tumor necrosis factor receptor exhibits an anti-allodynic action in a rat model of neuropathic pain. Neuroscience. 2001;103(2):529–539. doi: 10.1016/s0306-4522(00)00574-1. [DOI] [PubMed] [Google Scholar]
- Sweitzer SM, Colburn RW, Rutkowski M, DeLeo JA. Acute peripheral inflammation induces moderate glial activation and spinal IL-1beta expression that correlates with pain behavior in the rat. Brain Res. 1999;829(1–2):209–221. doi: 10.1016/s0006-8993(99)01326-8. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Minami M, Nakagawa T, Satoh M. Enhanced production of monocyte chemoattractant protein-1 in the dorsal root ganglia in a rat model of neuropathic pain: possible involvement in the development of neuropathic pain. Neurosci Res. 2004;48(4):463–469. doi: 10.1016/j.neures.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci USA. 2005;102(16):5856–5861. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol. 2001;166(12):7527–7533. doi: 10.4049/jimmunol.166.12.7527. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci. 2005;28(2):101–107. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia. 2004;45(1):89–95. doi: 10.1002/glia.10308. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424(6950):778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Vega-Avelaira D, Moss A, Fitzgerald M. Age-related changes in the spinal cord microglial and astrocytic response profile to nerve injury. Brain Behav Immun. 2006 doi: 10.1016/j.bbi.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Verge GM, Milligan ED, Maier SF, Watkins LR, Naeve GS, Foster AC. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur J Neurosci. 2004;20(5):1150–1160. doi: 10.1111/j.1460-9568.2004.03593.x. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Milligan ED, Maier SF. “Listening” and “talking” to neurons: Implications of immune activation for pain control and increasing the efficacy of opioids. Brain Res Rev. 2007;56(1):148–169. doi: 10.1016/j.brainresrev.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Martin D, Ulrich P, Tracey KJ, Maier SF. Evidence for the involvement of spinal cord glia in subcutaneous formalin induced hyperalgesia in the rat. Pain. 1997;71(3):225–235. doi: 10.1016/s0304-3959(97)03369-1. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001;24(8):450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- Wen YR, Suter MR, Kawasaki Y, Huang J, Pertin M, Kohno T, Berde CB, Decosterd I, Ji RR. Nerve Conduction Blockade in the Sciatic Nerve Prevents but Does Not Reverse the Activation of p38 Mitogen-activated Protein Kinase in Spinal Microglia in the Rat Spared Nerve Injury Model. Anesthesiology. 2007;107(2):312–321. doi: 10.1097/01.anes.0000270759.11086.e7. [DOI] [PubMed] [Google Scholar]
- White FA, Jung H, Miller RJ. Chemokines and the pathophysiology of neuropathic pain. Proc Natl Acad Sci USA. 2007 doi: 10.1073/pnas.0709250104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Sun J, Waters SM, Ma C, Ren D, Ripsch M, Steflik J, Cortright DN, LaMotte RH, Miller RJ. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc Natl Acad Sci USA. 2005;102(39):14092–14097. doi: 10.1073/pnas.0503496102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79(1):143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- Winkelstein BA, DeLeo JA. Nerve root injury severity differentially modulates spinal glial activation in a rat lumbar radiculopathy model: considerations for persistent pain. Brain Res. 2002;956(2):294–301. doi: 10.1016/s0006-8993(02)03560-6. [DOI] [PubMed] [Google Scholar]
- Xie YF, Zhang S, Chiang CY, Hu JW, Dostrovsky JO, Sessle BJ. Involvement of glia in central sensitization in trigeminal subnucleus caudalis (medullary dorsal horn) Brain Behav Immun. 2007;21(5):634–641. doi: 10.1016/j.bbi.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Xu JT, Xin WJ, Wei XH, Wu CY, Ge YX, Liu YL, Zang Y, Zhang T, Li YY, Liu XG. p38 activation in uninjured primary afferent neurons and in spinal microglia contributes to the development of neuropathic pain induced by selective motor fiber injury. Exp Neurol. 2007a;204(1):355–365. doi: 10.1016/j.expneurol.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Xu M, Bruchas MR, Ippolito DL, Gendron L, Chavkin C. Sciatic nerve ligation-induced proliferation of spinal cord astrocytes is mediated by kappa opioid activation of p38 mitogen-activated protein kinase. J Neurosci. 2007b;27(10):2570–2581. doi: 10.1523/JNEUROSCI.3728-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong VW, Wells J, Giuliani F, Casha S, Power C, Metz LM. The promise of minocycline in neurology. Lancet Neurol. 2004;3(12):744–751. doi: 10.1016/S1474-4422(04)00937-8. [DOI] [PubMed] [Google Scholar]
- Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem. 2006;97(3):772–783. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hoffert C, Vu HK, Groblewski T, Ahmad S, O’Donnell D. Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. Eur J Neurosci. 2003;17(12):2750–2754. doi: 10.1046/j.1460-9568.2003.02704.x. [DOI] [PubMed] [Google Scholar]
- Zhang J, Shi XQ, Echeverry S, Mogil JS, De KY, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27(45):12396–12406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Waxman SG, Hains BC. Extracellular signal-regulated kinase-regulated microglia-neuron signaling by prostaglandin E2 contributes to pain after spinal cord injury. J Neurosci. 2007a;27(9):2357–2368. doi: 10.1523/JNEUROSCI.0138-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Waxman SG, Hains BC. Modulation of thalamic nociceptive processing after spinal cord injury through remote activation of thalamic microglia by cysteine cysteine chemokine ligand 21. J Neurosci. 2007b;27(33):8893–8902. doi: 10.1523/JNEUROSCI.2209-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Eisenach JC. Cyclooxygenase-1 in the spinal cord is altered after peripheral nerve injury. Anesthesiology. 2003;99(5):1175–1179. doi: 10.1097/00000542-200311000-00026. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114(1–2):149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav Immun. 2007;21(5):642–651. doi: 10.1016/j.bbi.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]