

Several mutations at Glu81 located on the crystal contact of human acidic fibroblast growth factor were studied in an effort to improve crystal growth. Mutation to Ser and Thr resulted in crystallization of a rather bulky form of the wild type, whereas mutation to Val prohibited crystallization. These results suggest that crystal growth may be controlled by designing a new interface by protein engineering.
Keywords: acidic fibroblast growth factor, crystal contacts, mutagenesis
Abstract
An attempt has been made to improve a crystal contact of human acidic fibroblast growth factor (haFGF; 140 amino acids) to control the crystal growth, because haFGF crystallizes only as a thin-plate form, yielding crystals suitable for X-ray but not neutron diffraction. X-ray crystal analysis of haFGF showed that the Glu81 side chain, located at a crystal contact between haFGF molecules, is in close proximity with an identical residue related by crystallographic symmetry, suggesting that charge repulsion may disrupt suitable crystal-packing interactions. To investigate whether the Glu residue affects the crystal-packing interactions, haFGF mutants in which Glu81 was replaced by Ala, Val, Leu, Ser and Thr were constructed. Although crystals of the Ala and Leu mutants were grown as a thin-plate form by the same precipitant (formate) as the wild type, crystals of the Ser and Thr mutants were grown with increased thickness, yielding a larger overall crystal volume. X-ray structural analysis of the Ser mutant determined at 1.35 Å resolution revealed that the hydroxy groups of Ser are linked by hydrogen bonds mediated by the formate used as a precipitant. This approach to engineering crystal contacts may contribute to the development of large protein crystals for neutron crystallography.
1. Introduction
Protein crystallization constitutes a key limiting step in structural characterization by diffraction methods. Proteins of interest are screened against a multitude of prepared solutions because rational prediction of protein crystallization conditions has been impossible, despite knowledge of the physics and thermodynamics of protein crystallization. Development of high-throughput crystallization devices, able to set up over 100000 samples per day, accelerates screening of protein crystallization conditions (Stevens, 2000 ▶). It is, however, reported that the success rates of crystallization of proteins that are expressed as a soluble form in Escherichia coli are less than 30% (Dale et al., 2003 ▶).
Proteins in a crystal lattice align with each other through weak interactions on the surface of the protein. Controlling this weak interaction by protein engineering is very important to produce successful crystal packing or control the crystal growth. Recent studies show that targeted mutagenesis of surface patches containing residues with large flexible side chains (i.e. Lys and Glu) and their replacement with smaller amino acids (typically Ala) (Derewenda & Vekilov, 2006 ▶), derivatization by methylation (Walter et al., 2006 ▶) or complexation with antibodies (Ostermeier et al., 1995 ▶; Kuroki et al., 2002 ▶; Feese et al., 2004 ▶) can lead to effective preparation of X-ray quality crystals of proteins. Moreover, creation of symmetric crystal contacts by mutagenesis has also been attempted by introduction of a disulfide bond (Heinz & Matthews, 1994 ▶; Banatao et al., 2006 ▶) and leucine zipper (Yamada et al., 2007 ▶). Therefore, it is considered that engineering of the crystal contact is an important approach to increase the volume of protein crystals, particularly for neutron diffraction studies.
Human acidic fibroblast growth factor (haFGF) is a member of a family of heparin-binding mitogens and hormones (Johnson et al., 1991 ▶; Jaye et al., 1992 ▶), and the X-ray structure has already been determined to 1.10 Å resolution (Bernett et al., 2004 ▶). However, haFGF crystallizes as a thin-plate form with corresponding limitations for neutron diffraction studies. By X-ray crystal structure analysis, it is hypothesized that the proximity of the side chains of Glu81, located at a crystal contact between haFGF molecules related by crystallographic symmetry, may disrupt suitable crystal-packing interactions (along the b axis) by charge repulsion and/or entropy effects. To investigate whether the Glu residue affects crystal-packing interactions, we constructed haFGF mutants in which Glu81 was replaced by Ala, Val, Leu, Ser and Thr.
2. Materials and methods
2.1. Expression and purification
A synthetic polynucleotide coding the 140-amino-acid form of human FGF-1 (Gimenez-Gallego et al., 1986 ▶; Linemeyer et al., 1990 ▶; Ortega et al., 1991 ▶; Blaber et al., 1996 ▶) with the addition of an amino-terminal six-residue His tag (Brych et al., 2001 ▶) was used in this study. The QuikChange site-directed mutagenesis protocol (Stratagene) was used to introduce the mutations (Glu81 to Ala, Val, Leu, Ser and Thr) using mutagenic oligonucleotides of 25 to 31 bases in length (Biomolecular Analysis Synthesis and Sequencing Laboratory, Florida State University). All haFGF mutants were expressed using the pET21a(+) plasmid/BL21(DE3) E. coli host expression system (Invitrogen). Expression and purification of haFGF were performed following previously described procedures (Blaber et al., 1999 ▶; Culajay et al., 2000 ▶; Brych et al., 2001 ▶).
2.2. Crystallization
The purified wild-type and mutant haFGFs were dialyzed against 50 mM sodium phosphate buffer (pH 7.5) containing 100 mM NaCl, 10 mM ammonium sulfate, 2 mM DTT and 0.5 mM EDTA, and then concentrated to 38 to 40 mg ml−1. Crystallization was performed by hanging-drop vapor diffusion using formic acid as a precipitant. 2 µl drops consisting of 1 µl of protein solution and 1 µl of mother liquor were equilibrated against 1 ml of reservoir solution at 293 K for one week. To find suitable crystallization conditions, the crystallization phase diagrams for the wild-type and all mutant haFGFs were drawn from the result of crystallization using various concentrations of haFGF (6, 10, 14, 18, 22, 26, 30 and 34 mg ml−1) and formic acid (1.4, 1.8, 2.2, 2.6, 3.0, 3.4, 3.8, 4.2, 4.6, 5.0, 5.4 and 5.8 M).
2.3. Data collection and refinement
Diffraction data of the haFGF mutants were collected at BL41XU (SPring-8), Hyogo, Japan. The crystals were mounted using a nylon cryo loop (Hampton Research), frozen in a stream of liquid nitrogen and cooled to 100 K during data collection. The intensity data were processed using DENZO and merged with SCALEPACK (Otwinowski & Minor, 1997 ▶). The crystals of Ser mutant belong to the same space group (C2221) as the wild-type haFGF, with unit-cell parameters a = 73.5, b = 97.3, c = 108.5 Å (Table 1 ▶). The structures of the haFGF mutants were solved using the coordinates (PDB code: 1rg8) of haFGF determined at 1.10 Å resolution (Bernett et al., 2004 ▶) as an initial model. Refinement was carried out using the program REFMAC5 in the CCP4 program suite (Collaborative Computational Project, Number 4, 1994 ▶). An atomic model was built using the graphics program QUANTA (Accelrys Inc., San Diego, CA, USA).
Table 1. Data collection and refinement statistics for E81S mutant haFGF.
Values in parentheses correspond to the highest-resolution shell (1.40–1.35).
| Data collection | |
| Space group | C2221 |
| Unit-cell dimensions (Å) | a = 73.5, b = 97.3, c = 108.5 |
| No. of molecules per asymmetric unit | 2 |
| Solvent content (%) | 58 |
| Resolutions (Å) | 1.35 |
| No. of observed reflections | 530712 |
| No. of unique reflections | 81087 |
| Redundancy | 6.5 (5.7) |
| Completeness (%) | 95.0 (82.3) |
| 〈I/σ(I)〉 | 49.0 (4.1) |
| Rmerge† | 0.043 (0.349) |
| Wilson plot B factor (Å2) | 15.7 |
| Refinement statistics | |
| Resolutions (Å) | 20.0–1.35 |
| No. of reflections | 76 990 |
| R factor/Rfree‡ | 0.182/0.211 |
| R.m.s.d. bonds (Å) | 0.015 |
| R.m.s.d. angles (°) | 1.545 |
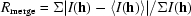 , where 〈I(h)〉 is the average intensity of reflection h and symmetry-related reflections.
, where 〈I(h)〉 is the average intensity of reflection h and symmetry-related reflections.
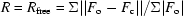 , calculated for the reflections of the working and test (5%) sets.
, calculated for the reflections of the working and test (5%) sets.
3. Results and discussion
X-ray crystal analysis of the wild-type haFGF (Bernett et al., 2004 ▶) showed that symmetry-related side chains of Glu81, located at a crystal contact between haFGF molecules, were in close proximity. This contact suggests that charge repulsion may disrupt suitable crystal-packing interactions. To investigate whether the Glu residue affects crystal formation, we constructed haFGF mutants in which Glu81 was replaced by Ala, Val and Leu (potentially promoting crystallization via hydrophobic effects) and Ser and Thr (potentially promoting crystallization via reduction of side-chain entropy). All mutant haFGF proteins were purified to apparent homogeneity for crystallization trials.
From crystallization trials utilizing 96 different conditions, crystals were grown in drops containing 4.2–4.6 M formate and 18–34 mg ml−1 of haFGF; however, E81V did not crystallize under any conditions. If the slow crystal growth along the b axis is caused by the charge repulsion at Glu81 or the surface conformational entropy, all mutations (E81A, E81L, E81S and E81T) to remove these effects should result in crystals with an improved thickness along the thin cell edge. The crystals of the E81A and E81L mutants were, however, grown as a thin plate similar to that of the wild type (Fig. 1 ▶ a). The E81S (26 mg ml−1) and E81T (30 mg ml−1) mutants were crystallized to thicker-shaped crystals with dimensions of 0.4 × 0.4 × 0.3 mm for 4.2 M formate and 0.2 × 0.3 × 0.8 mm for 4.6 M formate, respectively (Figs. 1 ▶ b and 1 ▶ c).
Figure 1.

The largest crystals of the wild-type and mutant haFGFs obtained during screening: (a) haFGF, (b) E81S mutant and (c) E81T mutant.
The crystal structure of the E81S mutant haFGF was determined to 1.35 Å resolution in the same space group as the wild type by X-ray crystallography. The refined structure was compared with that of the wild-type haFGF previously determined (Bernett et al., 2004 ▶). The overall structure of E81S mutant haFGF was quite similar to that of the wild type, including the noncrystallographic interaction between monomers within an asymmetric unit. It was also found that one formate molecule mediates a twofold crystal contact through four hydrogen-bonding interactions involving side-chain hydroxy groups of Ser81 (at a distance of 2.6 Å) and ∊ amino groups of Lys101 (at a distance of 3.5 Å) between crystallographically related molecules (Fig. 2 ▶ b). In this case, the formate molecule lies directly on the twofold axis of symmetry (the C atom being centrosymmetric) and is thought to be necessary to maintain the local electrostatic charge neutrality, notably with the neighboring Lys101 side chains. Therefore, the results suggest that the improvement of crystal growth along the b axis may be caused not only by the removal of negative charge repulsion of Glu81 but also by the formation of a hydrogen-bond network in this crystal contact of haFGF.
Figure 2.

The crystal contact region of the wild-type and E81S mutant haFGFs: (a) wild-type haFGF and (b) E81S mutant.
In conclusion, the introduction of suitable molecular interactions by protein engineering may allow improvement of the crystal growth. Since the incorporation of hydrophobic residues to the interface of haFGF rather prohibits the crystal growth, short hydrophilic residues such as serine or threonine may be suitable for rebuilding the crystal contact.
Acknowledgments
We thank M. Kawamoto for help in collection of data with synchrotron radiation at SPring-8 (proposal No. 2006A2700). This work was supported by grant Nos. MCB 0314740 (MB) from the National Science Foundation and 065513B (MB) from the American Heart Association.
References
- Banatao, D. R., Cascio, D., Crowley, C. S., Fleissner, M. R., Tienson, H. L. & Yeates, T. O. (2006). Proc. Natl Acad. Sci. USA, 103, 16230–16235. [DOI] [PMC free article] [PubMed]
- Bernett, M. J., Somasundaram, T. & Blaver, M. (2004). Proteins, 57, 626–634. [DOI] [PubMed]
- Blaber, M., DiSalvo, J. & Thomas, K. A. (1996). Biochemistry, 35, 2086–2094. [DOI] [PubMed]
- Blaber, S. I., Culajay, J. F., Khurana, A. & Blaber, M. (1999). Biophys. J.77, 470–477. [DOI] [PMC free article] [PubMed]
- Brych, S. R., Blaber, S. I., Logan, T. M. & Blaber, M. (2001). Protein Sci.10, 2587–2599. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Culajay, J. F., Blaber, S. I., Khurana, A. & Blaber, M. (2000). Biochemistry, 10, 7153–7158. [DOI] [PubMed]
- Dale, G. E., Oefner, C. & D’Arcy, A. (2003). J. Struct. Biol.142, 88–97. [DOI] [PubMed]
- Derewenda, Z. S. & Vekilov, P. G. (2006). Acta Cryst. D62, 116–124. [DOI] [PubMed]
- Feese, M. D., Tamada, T., Kato, Y., Maeda, Y., Hirose, M., Matsukura, Y., Shigematsu, H., Muto, T., Matsumoto, A., Watarai, H., Ogami, K., Tahara, T., Kato, T., Miyazaki, H. & Kuroki, R. (2004). Proc. Natl Acad. Sci. USA, 101, 1816–1821. [DOI] [PMC free article] [PubMed]
- Gimenez-Gallego, G., Conn, G., Hatcher, V. B. & Thomas, K. A. (1986). Biochem. Biophys. Res. Commun.128, 611–617. [DOI] [PubMed]
- Heinz, D. W. & Matthews, B. W. (1994). Protein Eng.7, 301–307. [DOI] [PubMed]
- Jaye, M., Schlessinger, J. & Dionne, C. A. (1992). Biochim. Biophys. Acta, 1135, 185–199. [DOI] [PubMed]
- Johnson, D. E., Lu, J., Chen, H., Werner, S. & Williams, L. T. (1991). Mol. Cell. Biol.11, 4627–4634. [DOI] [PMC free article] [PubMed]
- Kuroki, R., Hirose, M., Kato, Y., Feese, M. D., Tamada, T., Shigematsu, H., Watarai, H., Maeda, Y., Tahara, T., Kato, T. & Miyazaki, H. (2002). Acta Cryst. D58, 856–858. [DOI] [PubMed]
- Linemeyer, D. L., Menke, J. G., Kelly, L. J., Disalvo, J., Soderman, D., Schaeffer, M.-T., Ortega, S., Gimenez-Gallego, G. & Thomas, K. A. (1990). Growth Factors, 3, 287–298. [DOI] [PubMed]
- Ortega, S., Schaeffer, M.-T., Soderman, D., DiSalvo, J., Linemeyer, D. L., Gimenez-Gallego, G. & Thomas, K. A. (1991). J. Biol. Chem.266, 5842–5846. [PubMed]
- Ostermeier, C., Iwata, S., Ludwig, B. & Michel, H. (1995). Nat. Struct. Biol.10, 842–846. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Stevens, R. C. (2000). Curr. Opin. Struct. Biol.10, 558–563. [DOI] [PubMed]
- Walter, T. S., Meier, C., Assenberg, R., Au, K. F., Ren, J., Verma, A., Nettleship, J. E., Owens, R. J., Stuart, D. J. & Grimes, J. M. (2006). Structure, 14, 1617–1622. [DOI] [PMC free article] [PubMed]
- Yamada, H. et al. (2007). Protein Sci.16, 1389–1397. [DOI] [PMC free article] [PubMed]