

Summary
The dynamics of CD4+ effector T (Teff)cells and CD4+Foxp3+ regulatory T (Treg)cells during diabetes progression in non-obese diabetic mice was investigated to determine if an imbalance of Treg cells and Teff cells contributes to the development of type 1 diabetes. Our results demonstrated a progressive decrease in the Treg:Teff ratio in inflamed islets, but not in pancreatic lymph nodes. Intra-islet Treg cells expressed reduced amounts of CD25 and Bcl-2 suggesting that their decline was due to increased apoptosis. Additionally, administration of low dose interleukin-2 (IL-2) promoted Treg cell survival and protected mice from developing diabetes. Together, these results suggest intra-islet Treg cell dysfunction secondary to defective IL-2 production is a root cause of the progressive breakdown of self-tolerance and development of diabetes in the non-obese diabetic mice.
Introduction
Type 1 diabetes (T1D), also known as autoimmune diabetes, results from autoimmune destruction of insulin-producing β cells in the pancreatic islets of Langerhans. The disease is characterized by a loss of blood glucose homeostasis accompanied by inflammatory infiltrates in the islets. Both genetic and environmental factors contribute to the development of the disease. Hallmarks of human T1D are observed in the non-obese diabetic (NOD) mouse, a strain identified 27 years ago in an inbred colony in Japan (Makino et al., 1980). NOD mice provide a valuable tool for dissecting the pathogenesis of T1D. Over 20 genetic loci, termed insulin-dependent diabetes (Idd) loci, have been linked to disease susceptibility in both human patients and NOD mice (Todd and Wicker, 2001). Among these genes, those that encode MHC class II, insulin, CTLA-4, and IL-2 have been identified as contributors to the breakdown of central and/or peripheral tolerance, although the precise mechanisms associated with pathogenesis remain to be defined (Chentoufi and Polychronakos, 2002; Nakayama et al., 2005; Prochazka et al., 1987; Todd et al., 1987; Ueda et al., 2003; Yamanouchi et al., 2007).
Circumstantial evidence suggests that regulatory T (Treg) cells control the progression of diabetes. Disruption of Treg cell development and homeostasis by blocking the CD28-B7 pathway or IL-2 activity in NOD mice leads to acceleration of diabetes (Salomon et al., 2000; Setoguchi et al., 2005). In addition, Tcrα-/-, Rag1-/- or Foxp3-deficient scurfy BDC2.5 T cell receptor transgenic mice, which are completely devoid of Treg cells, display no delay between onset of insulitis and overt diabetes (Chen et al., 2005). These findings suggest that diabetes onset may be associated with a reduction in Treg cell numbers and/or functions. An alternative, but not mutually exclusive explanation is that the onset of diabetes is a result of the emergence of regulation-resistant effector T (Teff) cells. Studies on Treg cell dynamics in human T1D patients have produced disparate results, ranging from reduced Treg cell frequency (Kukreja et al., 2002) or function (Brusko et al., 2005; Lindley et al., 2005), to no changes when comparing diabetics to healthy controls (Putnam et al., 2005). Three independent investigations in NOD mice uniformly found an age-dependent decline in Treg cell function (Gregori et al., 2003; Pop et al., 2005; You et al., 2005). Two of these reports also demonstrated a reciprocal age-dependent increase in Teff cell resistance to regulation. However, data on the frequency of Treg cells in NOD mice is inconclusive at present. One study reported an age-dependent decline in the frequency of Foxp3-mRNA expressing CD4+ cells in the pancreatic lymph nodes (PLN) and islets (Pop et al., 2005), whereas another found an increase in Foxp3 mRNA at the time of diabetes onset compared to 6-week old pre-diabetic controls (You et al., 2005).
Here we report on our study examining the population dynamics of CD4+ Teff cells and Treg cells during the progression of T1D in NOD mice. Our investigation revealed a paradoxical increase of Treg cells in the PLN at the time of diabetes onset and demonstrated that disease progression was associated with a loss of Treg:Teff balance in the inflamed islets and concomitant reduction of CD25 and Bcl-2 expression on intra-islet Treg cells. Additionally, administration of IL-2 promoted Treg cell survival and protected NOD mice from diabetes. This study demonstrates that cellular dynamics in local tissues play the key role in determining the balance of immune homeostasis and disease progression. Our finding that IL-2 deficiency contributes to intra-islet Treg cell dysfunction and progressive breakdown of peripheral self-tolerance in the NOD mouse may have important implications for the use of IL-2 modulating therapies for the treatment of diabetes and other autoimmune diseases.
Results
Preservation of Treg:Teff balance in the PLN at diabetes onset
To determine whether the onset of diabetes in NOD mice reflects a general decline in Treg cells and an excess of autoreactive Teff cells, we examined the Treg:Teff balance in the PLN of the NOD mouse at different stages of disease progression in vivo. Unexpectedly, the Treg cell fraction of the total CD4+ T cell population in the PLN did not decrease, but rather increased by 50 to 70% at the time of diabetes onset (Figure 1A and Supplemental Material Table S1). No significant change in the percentage of Treg cells was found in distal inguinal LN (ILN, Supplemental Material Table S1). The absolute number of total Treg cells in PLN at the time of disease onset varied from mouse to mouse, mostly due to the variation in the cellularity of the PLN. However, the increase in the percentage of Treg cells at the time of disease onset was consistently reproduced in a large number of mice followed during a three-year study period, using in situ detections by immunohistochemistry (Figure 1B) and immunofluorescence (see below) or more quantitative analyses by flow cytometry using two different clones of anti-Foxp3 mAb conjugated with various fluorochromes.
Figure 1. Preserved Treg:Teff balance in PLN during T1D progression.
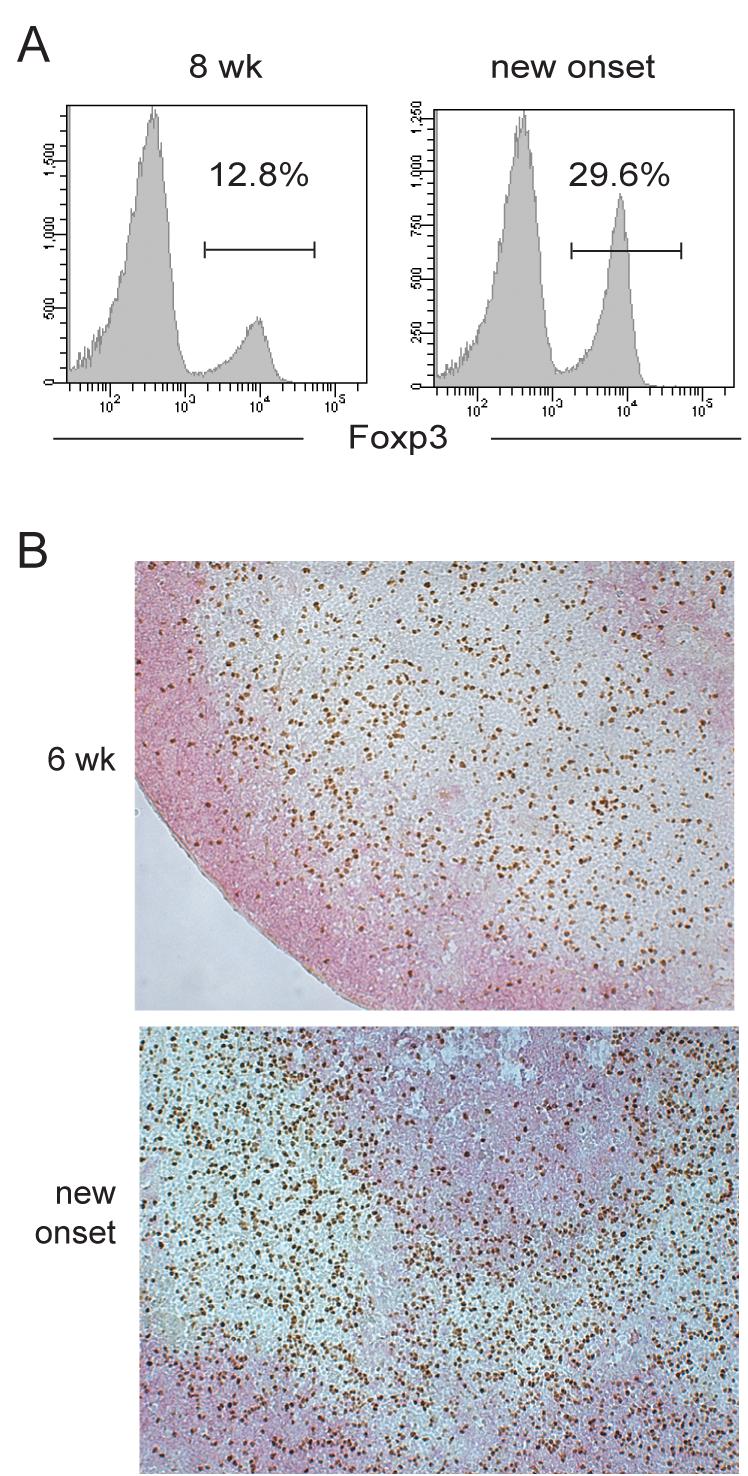
(A.) Flow-cytometric analysis of the percentage of Foxp3+ cells in the total CD4+ T cell population of the PLN of NOD mice. Representative histograms from an 8 week old prediabetic and a new-onset diabetic NOD are shown. (B.) Immunohistochemistry of Foxp3 (brown) and B200 (pink) expression in the PLN of 6-week old and new onset diabetic NOD mice. Data represents five independent experiments with total of 10-12 mice from each disease group.
Sustained activation of PLN Treg cells
To determine whether Treg cells present in the PLN at the time of diabetes onset were functionally suppressive, we examined the activation status of Treg cells. Higher fraction of PLN Treg cells showed reduced CD62L expression in newly diabetic mice when compared to Treg cells in 6-week old pre-diabetic animals (Figure 2A), suggesting that Treg cells were more activated in the PLN at time of disease onset (Huehn et al., 2004; Tang et al., 2004). Our previous work showed that Treg cells effectively prevent interaction of Teff cells with dendritic cells (DCs) (Tang et al., 2006). Thus, we transferred CD4+CD25-CD62Lhi naïve T cells from BDC2.5 TCR transgenic mice and examined their interaction with endogenous DCs in newly diabetic and 6-week old prediabetic mice by two-photon microscopy. Clustering and swarming of the islet-reactive BDC2.5 T cells, indicative of their interaction with endogenous DCs, was absent in newly diabetic mice, in sharp contrast with that found in the PLN of 6-week old prediabetic mice (Figure 2B and Supplementary videos online). Consistently, proliferation of the adoptively transferred BDC2.5 T cells was markedly reduced at the time of diabetes onset (Figure 2C). Sections of PLN were stained with mitotic marker Ki67 in addition to CD4 and Foxp3 to compare levels of priming of endogenous Teff cells in the PLN of young prediabetic and newly diabetic NOD mice. A marked reduction in the number of proliferating endogenous Teff cells (CD4+Foxp3-) in the PLN at the time of disease onset was evident correlating with a concomitant increased number of CD4+Foxp3+ Treg cells (Figure 2D). These results suggest that the imbalance in Treg cells in the PLN of newly diabetic NOD mice prevented naïve effector cells from becoming activated when recruited to the PLN at this late time point in disease progression, although the reduced proliferation of Teff cells could also be contributed by reduced availability of islet antigens due to the reduction in β cell mass in overtly diabetic mice. It is important to point out that the increase in Treg cells was specific to the PLN of the new onset diabetic NOD mice (Supplemental Material Table S1) and was not observed in chronically diabetic animals (data not shown) suggesting that the increase in Treg cells was dependent on the presence of residual islet autoantigens. Taken together, our results demonstrated an increased number and function of Treg cell in PLN as a result of sustained Treg cell activation leading to reduced Teff cell priming over the course of disease progression.
Figure 2. Preservation of Treg cell functions and reduced Teff cell priming in the PLN at the time of disease onset.

(A.) Flow cytometric analysis of CD62L expression on CD4+ Foxp3+ cells in the PLN of 6-week old and new-onset diabetic NOD mice. The graph on the right is a summary of four independent experiments comparing CD62L expression on CD4+CD25+ cells in pancreatic LN of young prediabetic (n=9) and newly diabetic (n=11) female NOD mice. (B.) CD4+CD62L+CD25- cells from BDC2.5 T cell receptor transgenic mice were purified by FACS, labeled with CFSE and transferred to 6-week old NOD mice and to mice within three days of diabetes onset. The movement dynamics of transferred Treg cells in explanted PLN were monitored using two-photon laser scanning microscopy. The normalized average CFSE fluorescent intensity over a 30-minute imaging period is illustrated in the “heat maps” shown. The large aggregates of strong fluorescent intensity represented by the yellow-red-white colors indicates restricted movement of cell clusters due to antigen recognition and the lower intensity represented by the blue-purple-black colors indicates random movement in the absence of antigen recognition. (C.) In Vivo Proliferation of transferred CD4+CD62L+CD25- cells from a BDC2.5 T cell receptor transgenic mice in PLN of 6-week old or new-onset diabetic mice were determined by CFSE dilution assay. Two representative histograms are shown. (D.) Proliferation of endogenous T cells in the PLN of 6-week-old and new-onset diabetic NOD mice was determined by co-staining PLN sections for the mitotic marker Ki67 (green), anti-CD4 (blue), and anti-Foxp3 (red). Representative micrographs are shown (left) and the average numbers of Foxp3+ cells and Foxp3- Ki67+ cells in three randomly selected objective fields in the T cell zones of the PLN are summarized (right, mean±sd, n=3 mice). Results in each panel represent two to four independent experiments.
Loss of Treg:Teff balance in the islets
Expansion of Treg cells during chronic inflammation has been reported to be associated with disease resolution (Knoechel et al., 2005; Korn et al., 2007). However, our results showed a relative increase of Treg cells in the PLN with disease progression. This paradoxical observation prompted us to analyze Treg cell dynamics in the islets of NOD mice. Because leukocyte infiltration and tissue destruction progress asynchronously in different islets (Anderson and Bluestone, 2005), confocal microscopic analyses of immunofluorescent antibody-stained pancreatic sections were performed to characterize the Foxp3+CD4+ Treg cell population within individual islets (Supplementary Material Figure S1). Such analyses revealed marked heterogeneity in the percentage of Treg cells, from 5 to 30%, in individual islets. Although the total numbers of Treg cells in each islet increased as the number of intra-islet Teff CD4+ cells expanded (Supplemental Figure 2S), the percentage of Treg cells dropped precipitously, revealing a striking inverse relationship between the magnitude of infiltration and the percentage of Treg cells in the individual islets (Figure 3A). The reduction in Treg cell percentages was most pronounced in islets from prediabetic mice that exhibited mild (grade 1) and moderate (grade 2) insulitis (less than 500 CD4+ cells/islet section), suggesting the relative loss of Treg cells in the islets preceded the escalation of inflammation and islet destruction. These results demonstrated a uncoupling of the PLN and the pancreas in terms of Treg:Teff balance over the course of the disease and suggested that the progressive dysregulation of immune homeostasis in islets led to β cell destruction and development of diabetes.
Figure 3. Dynamics of Teff and Treg cells in the islets of NOD mice during diabetes progression.

(A.) Numbers of CD4+ and Foxp3+ cells in individual islets were quantified by manual counting of frozen pancreatic sections strained with immunofluorescent labled antibodies. The percentages of Foxp3+ Treg cells among CD4+ T cells in individual islet sections are plotted against the total numbers of CD4+ cells in the corresponding islet sections. Correlation analysis revealed a significant inverse correlation (Spearman r = - 0.707) between the numbers of total CD4+ T cells per islet section and percentage of Treg cells in the corresponding islet (p<0.0001). (B.) Numbers of Ki67+CD4+Foxp3-, Ki67-CD4+Foxp3-, Ki67+CD4+Foxp3+, and Ki67-CD4+Foxp3+ cells in individual islets were quantified as described in A. Percentages of proliferating (Ki67+) Treg cells vs. proliferating Teff cells in islets were calculated and plotted. For both panels, blue symbols represent islets from 6-8 week old NOD mice, green symbols are from 10-12 week old mice and red symbols are from mice with recent diabetes onset.
Differential proliferation, trafficking, and/or survival of Teff cells and Treg cells may explain the loss of Treg:Teff balance in the islets. To compare the proliferation rate of intra-islet Teff and Treg cells, we stained pancreatic sections for Ki67 and determined the percentages of proliferating Teff cells (CD4+Foxp3-Ki67+) and Treg cells (CD4+Foxp3+Ki67+) in individual islets. The total numbers of proliferating Teff and Treg cells increased as the intra-islet infiltrate expanded (supplemental Figure 3S and 4S). A direct comparison of Foxp3+ and Foxp3- cell proliferation rate in the same islets demonstrated that a higher percentage of Treg cells entered mitosis in most of the islets examined independent of the age of the mice (Figure 3B). Flow cytometry analysis of Ki67 expression by intra-islet Treg and Teff cells also demonstrated that a higher fraction of Treg cells were in cycle as compared to Teff cells (data not shown). Thus, the relative decline of Treg cells seen in islets with increased infiltration was not due to a reduction in Treg cell proliferation compared to Teff cells.
Defective survival of intra-islet Treg cells
We tested whether a defect in intra-islet Treg cell survival could explain the relative loss of Treg cells. Because CD25 expression is essential for Treg cell survival in response to IL-2 in the periphery (D’Cruz and Klein, 2005; Fontenot et al., 2005a; Tang et al., 2003), we measured CD25 expression on Foxp3+ cells in ILN, PLN, and islets. Greater than 85% of Treg cells in ILN of prediabetic and newly diabetic NOD mice expressed CD25, similar to that reported for C57BL/6 mice (Fontenot et al., 2005b). Treg cells in the PLN exhibited a moderate reduction of CD25 expression when compared to the Treg cells in ILN (Figure 4A). In contrast, only ∼50% of intra-islet Treg cells expressed CD25 (Figure 4A). More strikingly, the mean fluorescent intensity of CD25 staining on the intra-islet Foxp3+ cells was 20% of their ILN counterparts (Figure 4B). The reduced CD25 expression on intra-islet Treg cells was evident in young prediabetic mice coinciding with the shift in Treg:Teff balance in the islets at the early phase of the pathogenic response (Figure 3A). Foxp3+ cells from other inflammatory site, such as lachrymal and salivary glands, also exhibited lower CD25 expression (Supplementary Figure S5), demonstrating that the loss of CD25 expression in Treg cells at inflammatory site was not an islet-specific phenomenon, but rather a general deficiency in the NOD mice.
Figure 4. Loss of CD25 and Bcl-2 expression on intra-islet Treg cells.

(A and B.) Flow cytometric analyses of CD25 expression on CD4+Foxp3+ cells. Representative CD25 versus Foxp3 dot plots of CD4+ cells in ILN, PLN and islets and are shown (A) along with a graph summarizing the the mean fluorescent intensities of CD25 on CD4+Foxp3+ cells (B). The open symbols represent 8 week old mice and filled symbols represent mice with recent diabetes onset. Results are representative of four independent experiments. (C and D.) Flow cytometric analysis of Bcl-2 expression in CD4+Foxp3+ cells. Representative Bcl-2 (top) and isotype control staining (bottom) histograms of CD4+Foxp3+ cells in ILN, PLN, and islets are shown (C) along with a bar graph summarizing the mean fluorescent intensities of Bcl-2 expression in CD4+Foxp3+ cells (D, mean±sd, n=4). Results are representative of five independent experiments. (E.) real-time RT-PCR analysis of IL-2 mRNA in various lymphoid organs and islets. Tissue samples from prediabetic NOD female mice were assayed individually (mean±sd, n=4). Student t test was performed to determine the statistical significance of the difference and p values for the significantly different sample pairs are shown on the graph.
These data, combined with the increased proliferation of these cells, are consistent with the notion that intra-islet Treg cells are more prone to apoptosis. However, current methods for measuring T cell apoptosis, including TUNEL, annexin V or caspase 3 staining, were not sensitive enough for in vivo analysis because of the efficient removal of apoptotic cells by the scavenging system (Krysko et al., 2006). Therefore, we examined the expression of an intracellular biochemical marker of IL-2-driven cell survival, Bcl-2. Bcl-2 is an anti-apoptotic effector molecule (Miyazaki et al., 1995), and its loss occurs at an early state of programmed cell death before the loss of cytoplasm membrane integrity and the subsequent sensing by the scavenging system. Bcl-2 was expressed at similar high amounts in both Treg cells and Teff cells in inguinal and pancreatic LNs (Figure 4C). In contrast, Bcl-2 expression was significantly lower in islet infiltrating T cells, suggesting that IL-2 was limiting in the inflamed tissue. Most importantly, the reduction in Bcl-2 expression was more severe in intra-islet Treg cells (66% reduced as compared to LN-resident Treg cells) than in intra-islet Teff cells (36% reduced as compared to LN-resident Teff cells, Figure 4D). We have examined Bcl-2 expression in mice of wide age range, from 8 to 25 week old, and found the loss of Bcl-2 on intra-islet Treg cells was evident at earliest time examined and persisted through diabetes onset (data not shown).
To directly determine if there is a local deficiency of IL-2 production in the inflamed islets relative to LNs and spleens, we purified CD4+ T cells and measured IL-2 mRNA expression by real-time RT-PCR. Without in vitro stimulation, IL-2 mRNA was present at a low amount in all tissues just above the detection threshold (Figure 4E). IL-2 mRNA increased and become readily detectable after three hour stimulation with anti-CD3 and anti-CD28. CD4+ cells from islets produced significantly lower amount of IL-2 mRNA comparing to CD4+ cells from spleen or inguinal LN (Figure 4E). Together, these findings suggest that a defect in the survival of Treg cells secondary to a deficiency in IL-2 production, accounts for the selective loss of the Treg:Teff balance in the islets.
Low dose IL-2 promotes Treg cell survival and protects NOD mice against diabetes
The hypothesis that the selective loss of the Treg:Teff balance in the inflamed islets is due to enhanced apoptosis of intra-islet Treg cells as a consequence of local IL-2 deficiency is supported by previous studies demonstrating that IL-2 binding to CD25 and subsequent STAT5 signaling are essential for the survival of Foxp3+ Treg cells in the periphery (D’Cruz and Klein, 2005; Fontenot et al., 2005a; Furtado et al., 2002). In addition, IL-2 is one of the most potent inducers of CD25 and Bcl-2 expression in T cells (Shi et al., 2001), thus loss of CD25 and Bcl-2 strongly suggests an IL-2 deficiency. Further, several publications have reported a relative IL-2 deficiency in NOD mice, a defect mapped to the Idd3 locus on chromosome 3 (Denny et al., 1997; Yamanouchi et al., 2007). To test our hypothesis that enhanced intra-islet Treg cell apoptosis secondary to an IL-2 deficiency led to islet destruction, we initiated studies to determine if correction of the IL-2 deficiency would improve Treg cell survival and confer disease protection. One week treatment of prediabetic NOD mice with a cocktail of 5 μg recombinant IL-2 and 50 μg IL-2 mAbs, a regimen previously shown to selectively expand Treg cells without increasing CD8+ memory T cells in C57BL/6 mice (Boyman et al., 2006), led to a marked increase in CD25 expression on Treg cells (Figure 5A) and a systemic increase in the percentage of Treg cells (Figure 5B). At this dose, the treatment increased CD25 expression on CD4+Foxp3- and CD8+ T cells in prediabetic NOD mice (Figure 5C and D). In addition, a substantial expansion of NK cells were observed in LN, spleen and among islet infiltrates (Figure 5E). Notably, one week treatment of 10 week old female prediabetic mice at this dose rapidly precipitated diabetes (Figure 5F). Thus, high dose IL-2 treatment enhanced functions of pathogenic Teff cells and the number of Treg cells with a net result of accelerating autoimmune tissue destruction.
Figure 5. Effect of high dose IL-2 treatment on prediabetic NOD mice.

A cohort of 10-week old female NOD mice were treated with daily intra-peritoneal injections of 5 μg IL-2 and 50 μg of anti-IL-2 complex or irrelevant rat IgG as control for five consecutive days (n=3/condition). One week after the initiation of the treatment, the composition of the spleen, PLN and intra-islet infiltrates were analyzed by flow cytometry. (A.) Dot plots displaying Foxp3 and CD25 expression on CD4+ cells in control- (Top) and IL-2 complex-(bottom) treated mice. (B) Bar graphs summarizing the frequency of Treg cells and (C) CD25hiFoxp3- Teff cells among CD4+ cells, (D) CD25hi cells among CD8+ cells and (E) percentage of NK cells among all CD45+ leukocytes are shown (mean±sd, n=3). The differences between control and IL-2 treated samples in panels B through E in all organs analyzed are significant (p<0.05) by student t test analysis. Results are representative of three independent experiments. (F.) A separate cohort of 10-week old female NOD mice were treated with control rat IgG or IL-2 complex as in A (n=10/group). The outcome one week after the treatment is summarized. Result represents three separate experiments.
Because Treg cells constitutively express CD25, lower dose of IL-2 treatment may selectively act on Treg cells with minimal effect on CD4+ or CD8+ Teff cells. With careful titration, we found that one tenth of the dose used in the experiments shown in Figure 5 led to a moderate increase in Treg cell percentage (Figure 6A and D) correlating with an increase in their CD25 and Bcl-2 expression (Figure 6A, B and C). At this dose, mild increase in CD4+ (Foxp3-CD25hi) or CD8+ (CD25hi) Teff cells was observed (Figure 6E, and F) and there was no marked effect on NK cells (Figure 6G). To determine the long-term effect of the low dose IL-2 therapy on diabetes development, a cohort of female NOD mice was treated between 10 and 20 weeks of age. Control mice were treated with an anti-human HLA-Bw6 rat mAb or saline for the same period. Low dose IL-2 treatment prevented diabetes development in majority of the mice (Figure 7A). Similar results were observed when mice were treated with recombinant human IL-2 from 5 to 20 weeks of age (Figure 7B). These data were confirmed by histological examination of the mice that remained free of diabetes at the end of the experiment. There was a marked reduction in the severity of insulitis in the mice that received IL-2 treatment (Figure 7C). Taken together, our study suggest that the lower expression of CD25 on intra-islet Treg cells and their reduced survival were not due to defects intrinsic to NOD Treg cells. Rather, the reduced availability of IL-2 from the Teff cells is most likely the root cause of the progressive loss of Treg:Teff balance in the islet leading to β cell destruction.
Figure 6. Low dose IL-2 therapy restores Treg:Teff balance.

A cohort of ten week-old female NOD mice were treated with daily injections of 0.5 μg IL-2 and 5 μg anti-IL-2 complex or irrelevant rat IgG as control for five consecutive days. Expression of (A) CD25 and (B and C) Bcl-2 on Treg cells in PLN and islets were determined by flow cytometry. Percentages of (D) Treg cells, (E) Foxp-CD25hi CD4+ Teff cells, (F) CD25hi CD8+ Teff cells, and (G) NK cells are summarized (mean±sd, n=5 for panel C, D, and G; n=3 for panels E and F). P-values for the samples that showed significant differences between control and IL-2 treated mice are indicated in the graphs, and all others were not significantly different by student t test (p>0.05). Results are representative of at least three independent experiments.
Figure 7. Low dose IL-2 therapy prevents diabetes.

Diabetes progression in NOD mice treated with (A) 0.5 μg IL-2 and 5 μg anti-IL-2 complex or (B) recombinant human IL-2. Control mice received saline. Shaded areas inside the graphs indicate the duration of the treatments, between 10-20 weeks for A and between 5 and 20 for B. P-values between the control and the IL-2-treated groups are 0.0002 for experiment depicted in A and 0.11 for experiment in B. (C.) Severity of intra-islet infiltration in mice that remain free of overt diabetes at the end of the experiment in B was evaluated histologically and the percentage of islets with no infiltration (0), peri-insulitis (1), moderate insulitis with less than 50% islet area infiltrated (2), and severe insulitis with greater than 50% islet area infiltrated (3) were determined and plotted. Data for the Saline group represents 40 islets from 4 mice and data from IL-2 treatment group represents 341 islets from 8 mice.
Discussion
In this study, we demonstrated that T1D progression in the NOD mouse was associated with a progressive loss of Treg:Teff balance in the inflamed islets, but not in the PLN. Intra-islet Treg cells expressed reduced amount of CD25 and Bcl-2 relative to the Treg cells in the PLN, suggesting that the Treg:Teff imbalance was due a defect in intra-islet Treg survival. We further demonstrated that IL-2 treatment of NOD mice restored CD25 expression on intra-islet Treg cells and led to diabetes prevention.
It is well established that Treg cells control the progression of T1D in the NOD mouse model (Chen et al., 2005; Salomon et al., 2000). However, the precise temporal and anatomical basis of Treg cell control is not clear, especially in regards to the relevant role of the PLN versus inflamed islets as the major site of immune regulation (Bour-Jordan et al., 2004; Chen et al., 2005; Tang and Bluestone, 2006). In this study, we demonstrate that NOD Treg cells mount an appropriate response to tissue destruction in the PLN by expanding, acquiring an activated phenotype, and infiltrating inflamed islets. This attempt to “control” disease was also manifested within the islets, as a high fraction of the Treg cells continue to divide within the pancreatic islet tissue, suggesting that further activation occurred at the site of inflammation. In fact, quantitative in vivo analysis of the Treg:Teff balance in individual islets revealed a high frequency of Treg cells in early islet infiltration in young prediabetic mice similar to what has been observed in other settings including models of tumor, infectious disease and other autoimmune diseases (Aluvihare and Betz, 2006; Belkaid et al., 2006; Munn and Mellor, 2006; Rouse et al., 2006; Sakaguchi et al., 2006; Waldmann et al., 2006). However, in NOD mice, Treg cells failed to survive in the tissue long term likely due to the limited availability of IL-2.
The importance of IL-2 in the maintenance of Treg cell homeostasis and suppression of T1D has been suggested by IL-2 neutralization studies (Setoguchi et al., 2005). Genetic mapping studies have demonstrated the NOD Idd3 allele contributes to IL-2 defect in the NOD mice (Denny et al., 1997). More recently, an elegant study further demonstrated the NOD idd3 allele led to systemic reduction in Treg frequency and higher mortality due to diabetes in a CD8+ T cell receptor transgenic NOD model (Yamanouchi et al., 2007). Our findings in this study further illustrate that in untreated NOD mice, there is a selective defect in Treg cell survival in inflamed tissues. The survival defect was observed in mice as young as 6 weeks of age, supporting the notion that this phenotype is most likely genetically encoded. The reduced expression of IL-2 and IL-2-regulated genes such as CD25 and Bcl-2 on Treg cells suggests that the Treg cell survival defect was secondary to a IL-2 deficiency. The local inflammatory milieu in the islets may exacerbate the IL-2 shortage by further inhibiting IL-2 expression (Villarino et al., 2007), competing for IL-2 by activated Teff cells, and cleaving of CD25 by matrix metalloproteases induced by local inflammation (Sheu et al., 2001). Thus, the genetically-encoded inborn IL-2 deficiency in the NOD mice may be more pronounced in inflamed tissues compromising Treg cell survival locally. Common γ chain binding cytokines such as IL-4, IL-7, and IL-15 can help to sustain Treg cell survival in vitro through provision of anti-apoptosis signals (Pandiyan et al., 2007). It is thus possible that deficiency in these cytokines may also contribute to the demise of Treg cells in the inflamed islets. Finally, it should be noted that the deficiency in Treg survival may be compounded by an independent genetic defect in Bcl-2 expression in NOD mice (Garchon et al., 1994). In fact, over-expression of Bcl-2 in T cells and B cells alleviated insulitis and conferred diabetes protection (Rietz et al., 2003).
Similar to intra-islet Treg cells, Treg cells isolated from inflamed lachrymal and salivary glands in the NOD mice also expressed markedly reduced CD25. Several published reports in various disease models demonstrated that tissue infiltrating Treg cells could be readily identified by their surface expression of CD25 and the cells were functional in controlling local inflammation (Belkaid et al., 2002; Yu et al., 2005). Thus, loss of CD25 is not a general characteristic of Treg cells in inflamed tissues. It remains to be determined whether the local IL-2 deficiency and Treg cell imbalance observed in this study were restricted to sites of autoimmune inflammation or reflected a more general defect of Treg cells in the NOD mouse. Examining CD25 expression on Treg cells in other inflammatory settings in the NOD mice such as microbial infection would help to clarify this issue.
The polarized effect of high and low dose IL-2 therapy on autoimmune response observed in this study is striking and highlights the pleiotropic effect of this cytokine. Although IL-2 has an indispensable role in Treg cell homeostasis, it was originally discovered as a T cell growth factor and activator of cytotoxic lymphocytes (Taniguchi et al., 1983). IL-2 has been used in the clinic since mid-1980s as an immune-boosting cancer therapy with limited success. Part of the limitations in IL-2 cancer therapy be due to the expansion of Treg cells (Wei et al., 2007). We observed that a regimen of multiple, low dose injections IL-2 favored Treg over Teff cells while a high dose regimen led to rapid Teff expansion and disease onset. In light of this result, it is interesting to point out that a high bolus dose of IL-2 was more efficacious than low dose in treating renal cell carcinomas (Fisher et al., 2000; Yang et al., 2003). Together, the results suggest that the in vivo effect of IL-2 can vary widely depending on the dosing regimen, amount of endogenous IL-2, and the numbers of activated CD4 and CD8 Teff cells, NK cells, and Treg cells in the host. Thus, optimal IL-2 treatment regimen may be difficult to predict for a heterogeneous patient population and adjunct therapy will be needed to ensure desired outcome. For example, combining IL-2 with Teff cell-depleting treatments such as anti-CD3 or Rapamycin may prevent potential disease exacerbation and help to restore long-term self-tolerance in an autoimmune setting (Chatenoud, 2003; Rabinovitch et al., 2002). In contrast, in cancer setting, IL-2 treatment in conjunction with Treg cell depletion may be more effective than IL-2 monotherapy in inducing tumor regression.
Three independent studies in the NOD mice demonstrated that diabetes progression is associated with the acquisition of regulation resistance by Teff cells over time (Gregori et al., 2003; Pop et al., 2005; You et al., 2005). Our results extend these findings suggesting that the development of diabetes in the face of increasing frequencies of Treg cells may reflect this increased Teff cell resistance. The protracted disease course in the NOD mice may reflect the time needed for regulation-resistant Teff cells to emerge and accumulate to sufficient numbers under the constant control of Treg cells. Furthermore, the regulation-resistant phenotype may be linked to reduced IL-2 production by these Teff cells. We previously demonstrated that Treg cells expanded in the presence of strong co-stimulation through CD28 and large doses of IL-2 can effectively prevent and even reverse diabetes (Tang et al., 2004). These Treg cells survive long-term (greater than 50 days) in recipient mice and are less dependent on B7 co-stimulation and IL-2 from the hosts (QT and JAB unpublished observations). Thus, the heightened regulation resistance and unfavorable survival environment for Treg cells found in the NOD mice can be overcome by Treg cell therapies if appropriate Treg cell preparative regimens are used.
Immune deficiency is often associated with autoimmunity in mice and man (Arkwright et al., 2002; Dupuis-Girod et al., 2003; Horak et al., 1995; Mombaerts et al., 1993), and in the NOD mouse, immune stimulation can protect mice against diabetes (Qin and Singh, 1997; Sharif et al., 2001). Our findings provide one possible explanation for these paradoxical observations suggesting that normal Treg cell homeostasis and a healthy balanced immune system depend ultimately on a robust Teff cell response. IL-2 production by activated Teff cells expand and sustain Treg cells, which in turn feed back to suppress the Teff cell response and maintain normal immune homeostasis. Disruption of this cross-talk can lead to the dysregulation of the Treg:Teff balance and contribute to the development of autoimmune diseases in the NOD mice. The effective control of diabetes with low dose IL-2 treatment leads to an intriguing suggestion that, in some instances, immune stimulation rather than immunosuppression may be an effective approach for the treatment of autoimmune diseases.
Methods
Mice
Female NOD (Taconic, Germantown, NY) and NOD.BDC2.5 T cell receptor transgenic mice were housed and bred under specific pathogen-free conditions at the UCSF Animal Barrier Facility.
Treatment with IL-2
Eight to 10-week-old female NOD mice were treated with an IL-2 and anti-IL-2 complex. A 100 μL PBS solution containing 0.5 to 5 μg recombinant mouse IL-2 (eBioscience, San Diego, CA) and 5 to 50 μg rat anti-mouse IL-2 (clone JES6-12A1, R & D Systems, Minneapolis, MN) were injected into the peritoneal cavity every day for 5 days. Control mice were injected with either saline or 50 μg irrelevant mAb (rat anti-human HLA-Bw6). For diabetes prevention studies, female NOD mice were treated for five consecutive days followed by twice a week maintenance treatments for a total duration of 10 weeks. Dosings for specific experiment are indicated in the result section. In separate experiments, human recombinant IL-2 was used at a dose of 25,000 international units three times a week for the duration as indicated in the results section.
Flow Cytometry
Islets were purified following standard collagenase protocols as described (Tang et al., 2004) and dissociated by incubating with a non-enzymatic solution (Sigma, St, Lois, MO) followed by trituration per the manufacturer’s instructions. LN cells were made into a single cell suspension by mechanic disruption or as described for islet cells. The following antibodies were used to stain the cells: FITC- or PerCP-labeled anti-CD45 (LCA, BD Pharmingen, San Diego, CA), Phycoerythrin (PE)- or FITC-labeled anti-CD25 (clone PC61, eBiosciences), PerCP- or Alexa 700-labeled anti-CD4 (BD Pharmingen), and FITC- or Alexa 700-conjugated anti-CD8, biotinylated anti-NKG2D followed by Quantumdot™ 605 labeled streptavidin (Invitrogen). The cells were then fixed, permeabilized, and stained with APC-labeled Foxp3 antibody using a Foxp3 labeling kit per manufacturer’s instructions (eBioscience). Cell surface staining with anti-CD25 was omitted in some experiments stained intracellularly with PE anti-Bcl-2 (BD) and/or FITC anti-Ki67 (BD). Flow cytometric analyses were performed on a LSRII™ flow cytometer with FACS Diva™ software (both Becton Dickenson, San Jose, CA).
Multicolor immunofluorescent labeling and confocal microscopy
The PLN and pancreata were harvested and frozen in OCT™. Six micron cryosections were fixed in ethanol and incubated with rabbit anti-mouse Foxp3 antisera (provided by Dr. Roli Khattri), biotinylated mouse monoclonal antibody anti-Ki67, followed by goat anti-rabbit Alexa 555 (Invitrogen, Carlsbad, CA), streptavidin FITC, and anti-CD4 Alexa 647 (eBioscience, San Diego, CA). The resulting fluorescent staining pattern was detected and acquired on an SP2 laser scanning confocal microscope (Leica, Wetzlar, Germany) and post-acquisition analyses were performed with the aid of the Metamorph software (Universal Imaging Corp, Downingtown, PA). To quantify the numbers of CD4+Foxp3+, CD4+Foxp3-, and Ki67+ cells within each of the individual islets, a manual counting method was used with the aid of Metamorph software. High resolution digitally zoomed images were used to ensure accuracy in counting. Numbers obtained from contiguous non-overlapping regions of a single islet were added to derive a total number of Teff cells and Treg cells in each islet. To compare the number of Treg cells and proliferating Teff cells in the PLN of 6-week old and recently diabetic NOD mice, the numbers of CD4+Foxp3+ cells and CD4+Foxp3-Ki67+ cells in non-overlapping 105 pixel (one objective field) areas in the T cell zones (indicated by continuous CD4 staining) were determined by manual counting with the aid of Metamorph software.
Two-photon laser scanning microscopy analysis
FACS-purified CD4+CD62LhiCD25- cells from NOD.BDC2.5 TCR transgenic mice were labeled with 5 μM carboxyfluorescein diacetate succinimidyl ester (CFSE, Invitrogen) before retro-orbital injection into 6-week old prediabetic or recently diabetic NOD recipients (3.5-5 × 106/mouse). Two-photon microscopy was performed on a custom resonant scanning instrument as described (Tang et al., 2006). Briefly, PLN were excised, immobilized to cover slips with the hilum of the LN facing away from the objective. During imaging, the LNs were maintained in 36°C RPMI medium bubbled with 95% O2/5% CO2, and imaged through the capsule in a region distal to the hilum. For time-lapse image acquisition, images of up to 50 xy-planes with 2-5 μm z spacing were acquired every 30 seconds for 30 to 60 minutes. Post-acquisition data analyses were performed using Metamorph software (Universal Imaging Corp.).
Quantification of IL-2 mRNA by real-time RT-PCR
Islet single cell suspension was made as described in the section for flow cytometry. CD4+ T cells from islets, ILN, PLN and spleen were purified by positive selection using autoMACS. Samples were kept separate for individual mice and two thirds of the purified CD4+ cells from each sample were lysed immediately after purification in Trizol (Invitrogen) for RNA isolation. The remaining one third were stimulated in vitro with plate bound anti-CD3 and anti-CD28 (1 μg/ml each) for 16 hours before RNA isolation by Trizol. Same amount of RNA was used to make cDNA using oligo dT primers and a reverse transcription kit per manufacture’s instructions (SuperScript III Reverse Transcription kit, Invitrogen). Then 10% of each of the cDNA reactions was used as a template for real-time PCR. The sequences for PCR primers are: IL-2 forward: 5′ AAA AGC TTT CAA TTG GAA GAT GCT G; IL-2 reverse: 5′ TTG AGG GCT TGT TGA GAT GA; β-actin forward, 5′ AAG TGT GAC GTT GAC ATC CGT AA; and β-actin reverse, 5′ TGC CTG GGT ACA TGG TGG TA. PCR was carried out on a Bio-Rad iQ5 Real-Time PCR Detection System (Hercules, CA) and Cyber Green (Invitrogen) was used for quantitative detection of amplified DNA. Signals from the β-actin reactions were used to normalize the signals in the IL-2 PCR reactions of the same cDNA sample to derive the relative IL-2 mRNA level for each sample.
Statistical analysis
To determine the relationship between the size of intra-islet infiltrates and the percentages of Treg cells in individual islets, correlation analysis was performed with the aid of GraphPad Prism software (San Diego, CA). Spearman r value was calculated to determine the types of correlation (0 = no correlation, 0 to 1 = positive correlation, and -1 to 0 = inversion correlation) and two tailed p value was used to determine the significance of the correlation. To determine the effect of IL-2 treatment on diabetes progression, diabetes-free fractions in control and IL-2 treated groups over time were calculated using the Kaplan-Meier method and the Log-rank test was used to compare the difference between the treatment groups. Two tailed student t test was used to determine the significance of two different treatment groups as specified in the result section.
Supplementary Material
Acknowledgements
We thank S. Jiang, C. McArthur, P. Wegfahrt, P. Koudria and J. Liang for technical assistance and Drs. Abul Abbas and Samantha Bailey for helpful discussions. This study was supported by a Juvenile Diabetes Research Foundation Center Grant and grants from the National Institutes of Health, Canadian Institutes for Health Research (CIHR) and Canadian Diabetes Association. J.Y.A. was supported by a Howard Hughes Medical Institute fellowship. E.S. is a recipient of a fellowship from the CIHR training grant in neuroinflammation. C.A.P is the recipient of the Canada Research Chair.
References
- Aluvihare VR, Betz AG. The role of regulatory T cells in alloantigen tolerance. Immunol Rev. 2006;212:330–343. doi: 10.1111/j.0105-2896.2006.00408.x. [DOI] [PubMed] [Google Scholar]
- Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- Arkwright PD, Abinun M, Cant AJ. Autoimmunity in human primary immunodeficiency diseases. Blood. 2002;99:2694–2702. doi: 10.1182/blood.v99.8.2694. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, Blank RB, Suffia I. Natural regulatory T cells and parasites: a common quest for host homeostasis. Immunol Rev. 2006;212:287–300. doi: 10.1111/j.0105-2896.2006.00409.x. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- Bour-Jordan H, Salomon BL, Thompson HL, Szot GL, Bernhard MR, Bluestone JA. Costimulation controls diabetes by altering the balance of pathogenic and regulatory T cells. J Clin Invest. 2004;114:979–987. doi: 10.1172/JCI20483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311:1924–1927. doi: 10.1126/science.1122927. [DOI] [PubMed] [Google Scholar]
- Brusko TM, Wasserfall CH, Clare-Salzler MJ, Schatz DA, Atkinson MA. Functional defects and the influence of age on the frequency of CD4+ CD25+ T-cells in type 1 diabetes. Diabetes. 2005;54:1407–1414. doi: 10.2337/diabetes.54.5.1407. [DOI] [PubMed] [Google Scholar]
- Chatenoud L. CD3 antibody treatment stimulates the functional capability of regulatory T cells. Novartis Found Symp. 2003;252:279–286. discussion 286-290. [PubMed] [Google Scholar]
- Chen Z, Herman AE, Matos M, Mathis D, Benoist C. Where CD4+CD25+ T reg cells impinge on autoimmune diabetes. J Exp Med. 2005;202:1387–1397. doi: 10.1084/jem.20051409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chentoufi AA, Polychronakos C. Insulin expression levels in the thymus modulate insulin-specific autoreactive T-cell tolerance: the mechanism by which the IDDM2 locus may predispose to diabetes. Diabetes. 2002;51:1383–1390. doi: 10.2337/diabetes.51.5.1383. [DOI] [PubMed] [Google Scholar]
- D’Cruz LM, Klein L. Development and function of agonist-induced CD25+Foxp3+ regulatory T cells in the absence of interleukin 2 signaling. Nat Immunol. 2005;6:1152–1159. doi: 10.1038/ni1264. [DOI] [PubMed] [Google Scholar]
- Denny P, Lord CJ, Hill NJ, Goy JV, Levy ER, Podolin PL, Peterson LB, Wicker LS, Todd JA, Lyons PA. Mapping of the IDDM locus Idd3 to a 0.35-cM interval containing the interleukin-2 gene. Diabetes. 1997;46:695–700. doi: 10.2337/diab.46.4.695. [DOI] [PubMed] [Google Scholar]
- Dupuis-Girod S, Medioni J, Haddad E, Quartier P, Cavazzana-Calvo M, Le Deist F, de Saint Basile G, Delaunay J, Schwarz K, Casanova JL, et al. Autoimmunity in Wiskott-Aldrich syndrome: risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics. 2003;111:e622–627. doi: 10.1542/peds.111.5.e622. [DOI] [PubMed] [Google Scholar]
- Fisher RI, Rosenberg SA, Fyfe G. Long-term survival update for high-dose recombinant interleukin-2 in patients with renal cell carcinoma. Cancer J Sci Am. 2000;6(Suppl 1):S55–57. [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005a;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005b;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Furtado GC, Curotto de Lafaille MA, Kutchukhidze N, Lafaille JJ. Interleukin 2 signaling is required for CD4(+) regulatory T cell function. J Exp Med. 2002;196:851–857. doi: 10.1084/jem.20020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garchon HJ, Luan JJ, Eloy L, Bedossa P, Bach JF. Genetic analysis immune dysfunction in non-obese diabetic (NOD) mice: mapping of a susceptibility locus close to the Bcl-2 gene correlates with increased resistance of NOD T cells to apoptosis induction. Eur J Immunol. 1994;24:380–384. doi: 10.1002/eji.1830240217. [DOI] [PubMed] [Google Scholar]
- Gregori S, Giarratana N, Smiroldo S, Adorini L. Dynamics of pathogenic and suppressor T cells in autoimmune diabetes development. J Immunol. 2003;171:4040–4047. doi: 10.4049/jimmunol.171.8.4040. [DOI] [PubMed] [Google Scholar]
- Horak I, Lohler J, Ma A, Smith KA. Interleukin-2 deficient mice: a new model to study autoimmunity and self-tolerance. Immunol Rev. 1995;148:35–44. doi: 10.1111/j.1600-065x.1995.tb00092.x. [DOI] [PubMed] [Google Scholar]
- Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, Debes GF, Lauber J, Frey O, Przybylski GK, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med. 2004;199:303–313. doi: 10.1084/jem.20031562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoechel B, Lohr J, Kahn E, Bluestone JA, Abbas AK. Sequential development of interleukin 2-dependent effector and regulatory T cells in response to endogenous systemic antigen. J Exp Med. 2005;202:1375–1386. doi: 10.1084/jem.20050855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Backstrom BT, Sobel RA, Wucherpfennig KW, Strom TB, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko DV, D’Herde K, Vandenabeele P. Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis. 2006;11:1709–1726. doi: 10.1007/s10495-006-9527-8. [DOI] [PubMed] [Google Scholar]
- Kukreja A, Cost G, Marker J, Zhang C, Sun Z, Lin-Su K, Ten S, Sanz M, Exley M, Wilson B, et al. Multiple immuno-regulatory defects in type-1 diabetes. J Clin Invest. 2002;109:131–140. doi: 10.1172/JCI13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes. 2005;54:92–99. doi: 10.2337/diabetes.54.1.92. [DOI] [PubMed] [Google Scholar]
- Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu. 1980;29:1–13. doi: 10.1538/expanim1978.29.1_1. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Liu ZJ, Kawahara A, Minami Y, Yamada K, Tsujimoto Y, Barsoumian EL, Permutter RM, Taniguchi T. Three distinct IL-2 signaling pathways mediated by bcl-2, c-myc, and lck cooperate in hematopoietic cell proliferation. Cell. 1995;81:223–231. doi: 10.1016/0092-8674(95)90332-1. [DOI] [PubMed] [Google Scholar]
- Mombaerts P, Mizoguchi E, Grusby MJ, Glimcher LH, Bhan AK, Tonegawa S. Spontaneous development of inflammatory bowel disease in T cell receptor mutant mice. Cell. 1993;75:274–282. doi: 10.1016/0092-8674(93)80069-q. [DOI] [PubMed] [Google Scholar]
- Munn DH, Mellor AL. The tumor-draining lymph node as an immune-privileged site. Immunol Rev. 2006;213:146–158. doi: 10.1111/j.1600-065X.2006.00444.x. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, Wegmann DR, Hutton JC, Elliott JF, Eisenbarth GS. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature. 2005;435:220–223. doi: 10.1038/nature03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4(+)CD25(+)Foxp3(+) regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4(+) T cells. Nat Immunol. 2007;8:1353–1362. doi: 10.1038/ni1536. [DOI] [PubMed] [Google Scholar]
- Pop SM, Wong CP, Culton DA, Clarke SH, Tisch R. Single cell analysis shows decreasing FoxP3 and TGFbeta1 coexpressing CD4+CD25+ regulatory T cells during autoimmune diabetes. J Exp Med. 2005;201:1333–1346. doi: 10.1084/jem.20042398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochazka M, Leiter EH, Serreze DV, Coleman DL. Three recessive loci required for insulin-dependent diabetes in nonobese diabetic mice. Science. 1987;237:286–289. doi: 10.1126/science.2885918. [DOI] [PubMed] [Google Scholar]
- Putnam AL, Vendrame F, Dotta F, Gottlieb PA. CD4+CD25high regulatory T cells in human autoimmune diabetes. J Autoimmun. 2005;24:55–62. doi: 10.1016/j.jaut.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Qin HY, Singh B. BCG vaccination prevents insulin-dependent diabetes mellitus (IDDM) in NOD mice after disease acceleration with cyclophosphamide. J Autoimmun. 1997;10:271–278. doi: 10.1006/jaut.1997.0136. [DOI] [PubMed] [Google Scholar]
- Rabinovitch A, Suarez-Pinzon WL, Shapiro AM, Rajotte RV, Power R. Combination therapy with sirolimus and interleukin-2 prevents spontaneous and recurrent autoimmune diabetes in NOD mice. Diabetes. 2002;51:638–645. doi: 10.2337/diabetes.51.3.638. [DOI] [PubMed] [Google Scholar]
- Rietz C, Screpanti V, Brenden N, Bohme J, Fernandez C. Overexpression of bcl-2 in T cells affects insulitis in the nonobese diabetic mouse. Scand J Immunol. 2003;57:342–349. doi: 10.1046/j.1365-3083.2003.01244.x. [DOI] [PubMed] [Google Scholar]
- Rouse BT, Sarangi PP, Suvas S. Regulatory T cells in virus infections. Immunol Rev. 2006;212:272–286. doi: 10.1111/j.0105-2896.2006.00412.x. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, Shimizu J, Takahashi T, Nomura T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. 2005;201:723–735. doi: 10.1084/jem.20041982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif S, Arreaza GA, Zucker P, Mi QS, Sondhi J, Naidenko OV, Kronenberg M, Koezuka Y, Delovitch TL, Gombert JM, et al. Activation of natural killer T cells by alpha-galactosylceramide treatment prevents the onset and recurrence of autoimmune Type 1 diabetes. Nat Med. 2001;7:1057–1062. doi: 10.1038/nm0901-1057. [DOI] [PubMed] [Google Scholar]
- Sheu BC, Hsu SM, Ho HN, Lien HC, Huang SC, Lin RH. A novel role of metalloproteinase in cancer-mediated immunosuppression. Cancer Res. 2001;61:237–242. [PubMed] [Google Scholar]
- Shi FD, Flodstrom M, Balasa B, Kim SH, Van Gunst K, Strominger JL, Wilson SB, Sarvetnick N. Germ line deletion of the CD1 locus exacerbates diabetes in the NOD mouse. Proc Natl Acad Sci U S A. 2001;98:6777–6782. doi: 10.1073/pnas.121169698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, Santamaria P, Locksley RM, Krummel MF, Bluestone JA. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol. 2006;7:83–92. doi: 10.1038/ni1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Bluestone JA. Regulatory T-cell physiology and application to treat autoimmunity. Immunol Rev. 2006;212:217–237. doi: 10.1111/j.0105-2896.2006.00421.x. [DOI] [PubMed] [Google Scholar]
- Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, Bluestone JA. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Henriksen KJ, Boden EK, Tooley AJ, Ye J, Subudhi SK, Zheng XX, Strom TB, Bluestone JA. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J Immunol. 2003;171:3348–3352. doi: 10.4049/jimmunol.171.7.3348. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Matsui H, Fujita T, Takaoka C, Kashima N, Yoshimoto R, Hamuro J. Structure and expression of a cloned cDNA for human interleukin-2. Nature. 1983;302:305–310. doi: 10.1038/302305a0. [DOI] [PubMed] [Google Scholar]
- Todd JA, Bell JI, McDevitt HO. HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature. 1987;329:599–604. doi: 10.1038/329599a0. [DOI] [PubMed] [Google Scholar]
- Todd JA, Wicker LS. Genetic protection from the inflammatory disease type 1 diabetes in humans and animal models. Immunity. 2001;15:387–395. doi: 10.1016/s1074-7613(01)00202-3. [DOI] [PubMed] [Google Scholar]
- Turley S, Poirot L, Hattori M, Benoist C, Mathis D. Physiological beta cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J Exp Med. 2003;198:1527–1537. doi: 10.1084/jem.20030966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, Rainbow DB, Hunter KM, Smith AN, Di Genova G, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–511. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- Villarino AV, Tato CM, Stumhofer JS, Yao Z, Cui YK, Hennighausen L, O’Shea JJ, Hunter CA. Helper T cell IL-2 production is limited by negative feedback and STAT-dependent cytokine signals. J Exp Med. 2007;204:65–71. doi: 10.1084/jem.20061198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann H, Adams E, Fairchild P, Cobbold S. Infectious tolerance and the long-term acceptance of transplanted tissue. Immunol Rev. 2006;212:301–313. doi: 10.1111/j.0105-2896.2006.00406.x. [DOI] [PubMed] [Google Scholar]
- Wei S, Kryczek I, Edwards RP, Zou L, Szeliga W, Banerjee M, Cost M, Cheng P, Chang A, Redman B, et al. Interleukin-2 administration alters the CD4+FOXP3+ T-cell pool and tumor trafficking in patients with ovarian carcinoma. Cancer Res. 2007;67:7487–7494. doi: 10.1158/0008-5472.CAN-07-0565. [DOI] [PubMed] [Google Scholar]
- Yamanouchi J, Rainbow D, Serra P, Howlett S, Hunter K, Garner VE, Gonzalez-Munoz A, Clark J, Veijola R, Cubbon R, et al. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat Genet. 2007;39:329–337. doi: 10.1038/ng1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JC, Sherry RM, Steinberg SM, Topalian SL, Schwartzentruber DJ, Hwu P, Seipp CA, Rogers-Freezer L, Morton KE, White DE, et al. Randomized study of high-dose and low-dose interleukin-2 in patients with metastatic renal cancer. J Clin Oncol. 2003;21:3127–3132. doi: 10.1200/JCO.2003.02.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You S, Belghith M, Cobbold S, Alyanakian MA, Gouarin C, Barriot S, Garcia C, Waldmann H, Bach JF, Chatenoud L. Autoimmune diabetes onset results from qualitative rather than quantitative age-dependent changes in pathogenic T-cells. Diabetes. 2005;54:1415–1422. doi: 10.2337/diabetes.54.5.1415. [DOI] [PubMed] [Google Scholar]
- Yu P, Lee Y, Liu W, Krausz T, Chong A, Schreiber H, Fu YX. Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. J Exp Med. 2005;201:779–791. doi: 10.1084/jem.20041684. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.