

Abstract
Purpose
Bevacizumab is a recombinant IgG1humanized monoclonal antibody against vascular endothelial growth factor (VEGF). Its proposed mechanism of action is independent of immune effector functions. Many human carcinomas not only secrete VEGF but also express membrane-bound VEGF. In addition, VEGF receptors are expressed on tumor cells. It is hypothesized that bevacizumab could bind membrane-bound VEGF or VEGF-VEGF receptor complexes on tumors, thereby initiating potential immunologic consequences. We previously showed that yeast-derived β-glucan functions with antitumor antibodies that activate complement to recruit complement receptor 3– expressing leukocytes capable of mediating complement receptor 3– dependent cellular cytotoxicity of tumors opsonized with iC3b. In the current study, the therapeutic efficacy mediated by combining bevacizumab with yeast-derived β-glucan was studied in human carcinoma xenograft models.
Experimental Design
Human tumor cell lines were screened for membrane-bound VEGF expression both in vitro and in vivo. Complement activation mediated by bevacizumab was examined. Tumor cell lines positive or negative for membrane-bound VEGF expression were implanted in severe combined immunodeficient mice to establish xenograft models. Tumor-bearing mice were treated with different regimens. Tumor regression and long-term survival were recorded.
Results
Human ovarian carcinoma SKOV-3 cells expressed membrane-bound VEGF both in vitro and in vivo. Bevacizumab was bound to membrane-bound VEGF, activated complement, and synergized with β-glucan to elicit cellular cytotoxicity in vitro. In vivo study showed that β-glucan could significantly augment the therapeutic efficacy mediated by bevacizumab.
Conclusions
Yeast-derived β-glucan can synergize with anti-VEGF monoclonal antibody bevacizumab for the treatment of cancer with membrane-bound VEGF expression.
Vascular endothelial growth factor (VEGF) stimulates abundant angiogenesis, which allows exponential tumor growth and provides the hematogenous route for metastasis (1, 2). VEGF-A is the member of the VEGF family that seems to exercise the greatest control of angiogenesis during tumor and metastatic development (3, 4). The human VEGF-A gene is structured in eight exons that give rise to four main isoforms by alternative splicing (5). The isoform VEGF165, the most physiologically relevant, is secreted by both cancerous and noncancerous cells. However, a significant fraction remains bound to the cell surface and the extracellular matrix, which is mediated by its heparin-binding properties (6). It has been shown that most human tumors overexpress VEGF, which is associated with tumor progression and poor prognosis in colorectal, lung, breast, pancreatic, and gastrointestinal carcinomas and melanoma (7–12). Human VEGF exerts its functions through binding to two related receptors, VEGF receptor (VEGFR) 1 (Flt-1) and VEGFR2 (Flk-1 or KDR), which are expressed mostly on endothelial cells (13, 14). VEGFR2 is the primary receptor for transmitting VEGF signals and is a transmembrane protein with an intracellular tyrosine kinase–active end. The induction of this tyrosine kinase by VEGF fastening initiates a cascade of phosphorylation of other signaling molecules, resulting in microvascular permeability, endothelial cell proliferation, invasion, migration, and survival (15, 16). Interestingly, VEGFRs are also expressed on tumor cells, including those from non–small cell lung carcinoma, leukemia, prostate carcinoma, and breast carcinoma (17–20). Although the significance of this observed expression pattern is still under investigation, it is intriguing to hypothesize that circulating VEGF could bind to its receptor on tumor cells to form VEGF-VEGFR complex, thereby stimulating tumor growth and metastasis.
Anti-VEGF monoclonal antibody (mAb; bevacizumab, Avastin) is a murine-derived recombinant mAb with a human IgG1 framework. It is capable of binding and neutralizing all biologically active isoforms of VEGF, thus potently blocking VEGF (21, 22). Bevacizumab was shown as having no direct effect on the proliferation of tumor cell lines. Rather, it was concluded that its target is the endothelial cells and the tumor blood supply (23). Thus, the proposed mechanism of action of bevacizumab is the blocking of secreted VEGF, resulting in regression to tumor microvessels, normalization of surviving mature vasculature, and inhibition of vessel growth and neovascularization (22, 24). Although bevacizumab uses the human IgG1 framework, which itself is capable of activating complement, it has not been shown to activate complement or to be cytotoxic to tumor cells, neither in vitro nor in vivo. This tendency to attack tumor cells is a mechanism exhibited by other antitumor antibodies, such as rituximab (anti-CD20 mAb; ref. 25).
β-Glucan, a pathogen-associated molecular pattern, has shown therapeutic benefits in a variety of animal disease models (26–29). Yeast-derived β-glucan or barley β-glucan has shown significant therapeutic efficacy in murine breast, liver metastasis, lung, and lymphoma tumor models as well as in human neuroblastoma, lymphoma, and melanoma xenograft models when used in combination with antitumor mAbs or naturally occurring antitumor antibodies (30–35). Our previous in vitro studies have shown that the small molecular mass of yeast-derived β-glucan binds a lectin-like domain within the COOH-terminal region of the CD11b subunit of leukocyte complement receptor 3 (CR3; CD11b/CD18, αMβ2 integrin, Mac-1; refs. 36, 37). β-Glucans prime CR3 of neutrophils, macrophages, and natural killer cells for cytotoxicity against tumors opsonized with iC3b as a result of complement activation by antitumor mAbs or natural antibodies. Dual occupancy of leukocyte CR3 by the I-domain ligand iC3b and the lectin-like domain ligand β-glucan leads to degranulation and cytotoxic responses (38, 39). Further studies have shown that successful β-glucan–mediated tumor immunotherapy requires tumor-reactive antibodies that activate complement and deposit iC3b on tumor cells and CR3 on leukocytes (30, 31). In addition, neutrophils have been identified as the predominate effector cells for β-glucan–mediated tumor therapy (31, 40).
In the current study, we hypothesized that bevacizumab, in addition to its conventional effects on circulating VEGF, also binds membrane-bound VEGF on tumor cells, leading to complement activation and iC3b deposition on tumors. This effect can be augmented by coadministration of yeast-derived β-glucan, which results in the synergistic and heightened antitumor effects for tumor therapy. This study has a double therapeutic consequence. First, we established the need for testing membrane-bound VEGF expression in tumors. Second, we revealed an augmented therapeutic efficacy against tumors mediated by the combination of bevacizumab and β-glucan therapy, one that allows the clinical use of this therapy without added chemotherapy or adverse effects.
Materials and Methods
Cell lines
The human breast tumor cell line HBL-100, human melanoma cell line Colo38, and human ovarian carcinoma SKOV-3 were obtained from the American Type Culture Collection. Human breast carcinoma MDA-MB-483 was obtained from Dr. R.L. Ceriani (Cancer Research Fund of Contra Costa, Walnut Creek, CA). The cell lines were cultured in DMEM with 10% newborn calf serum (Hyclone), MEM nonessential amino acids (Life Technologies), 100 units/mL penicillin (Sigma), 100 μg/mL streptomycin (Sigma), and 2 mmol/L L-glutamine (Sigma).
Antibodies and other reagents
Anti-mouse C3-FITC and anti-human C3-FITC were purchased from Cappel. Anti-mouse Gr-1-phycoerythrin (PE), anti-mouse CD31-biotin mAbs, and relevant isotype controls were purchased from eBioscience. Streptavidin and peroxidase substrate kit 3-amino-9-ethylcarbazole were purchased from Vector Laboratories. The humanized mAb against VEGF (bevacizumab) and humanized mAb against Her-2/neu (trastuzumab) were produced by Genentech. Therapeutic soluble poly-(1,6)- β-D-glucopyranosyl-(1,3)- β-D-glucopyranose (PGG) β-glucan, a pharmaceutical-grade β-glucan with an average molecular mass of 150 kDa, was obtained from Biothera.
VEGF expression on tumor cells
To detect the expression of membrane-bound VEGF on human tumor cell lines both in vitro and in vivo, flow cytometry and fluorescence microscopy were done. For in vitro staining, tumor cells were harvested and Fc receptors were blocked by incubation with anti-CD32/CD16 mAb. Cells were then stained with PE-labeled anti-VEGF mAb or isotype control and analyzed by flow cytometry. For in vivo staining, solid tumors were excised and snap frozen in tissue freezing medium (OCT, Sakura Finetechnical Co. Ltd.). Tumor sections were first blocked with 3% bovine serum albumin/PBS and then stained with PE-labeled anti-VEGF mAb and FITC-labeled anti–Her-2/neu mAb. Images were acquired by fluorescence microscopy (Nikon Eclipse TE300 confocal cell images).
Detection of complement activation
To determine whether anti-VEGF mAb can activate mouse or human complement, mouse or human serum was freshly collected and kept on ice. For every million tumor cells, a 100 μL volume of diluted mouse (1:4) or human (1:10) serum containing 10 μg/mL working dilution of anti-VEGF mAb was used. Tumor cells were mixed and incubated at 37°C for 30 min. Cells were washed in ice-cold flow cytometry staining buffer and the tumor cell pellet was resuspended in 100 μL of diluted detecting antibody (goat anti-mouse or human C3-FITC). Cells were incubated on ice for 30 min and washed twice as above. Propidium iodide was used to exclude the dead cells.
In vitro β-glucan–mediated cellular cytotoxicity
In vitro cytotoxicity of tumor cells by β-glucan–primed human neutrophils was analyzed using a real-time measure of the impedance of electrical current by viable target cells adhered to a conductor on the bottom of wells in a 16-well plate (Acea Biosciences) according to the manufacturer’s instruction and our previous publication (38). Briefly, 5 × 103 tumor cells were placed into the wells of the Acea 16-well plates for 24 h. Following this, fresh human serum and sufficient anti-VEGF mAb were added to the adherent tumor cells. The cells were incubated for 30 min at 37°C to permit complement activation and deposition of human iC3b. Human neutrophils were added to achieve E:T cell ratios of 20:1 with or without the 25-kDa active moiety of β-glucan (38). Cells were incubated at 37°C in a humidified 5% CO2 incubator for 12 h. Cytotoxicity was calculated by measuring the relative decrease in current impedance among wells containing iC3b-opsonized tumor cells and β-glucan–primed neutrophils and wells containing iC3b-opsonized tumor cells and non-β-glucan–primed neutrophils or wells containing iC3b-opsonized tumor cells only.
Mice and tumor models
Fox Chase ICR severe combined immunodeficient (SCID) mice were purchased from Taconic. The murine tumor therapy protocols were done in compliance with all guidelines and were approved by the Institutional Animal Care and Use Committee of the University of Louisville For the SKOV-3 or Colo38 cell xenograft model, 6- to 8-week-old SCID mice were implanted s.c. in a mammary fat pad with 10 × 106 SKOV-3 or Colo38 cells. When tumor volume reached ~300 mm3, animals were divided into groups (n = 9) and received the anti-VEGF mAb bevacizumab (0.2 mg i.v. twice weekly) with or without soluble PGG β-glucan (1.2 mg i.v. twice weekly). PBS-treated, PGG β-glucan–treated only, and bevacizumab-treated only animals served as controls. Therapy was conducted for up to 4 weeks, during which time tumor volumes were recorded twice weekly. Tumor volume (mm3) was calculated by caliper measurements of the largest diameter (a) and its perpendicular (b) according to the following formula: volume = [(a + b)/2]3. Mice were sacrificed when tumors reached 2,744 mm3 as recommended by Institutional Animal Care and Use Committee guidelines. In some experiments, survival was monitored up to 100 days beyond tumor implantation.
Immunofluorescence staining for neutrophil infiltration and complement activation in tumors and immunohistochemical staining for tumor microvascularization
Tumors were excised and snap frozen in tissue freezing medium (OCT). Tissue blocks were cut and fixed with cold acetone. To detect intratumor neutrophil infiltration or complement activation, the sections were first blocked with 3% bovine serum albumin/PBS and then stained with PE-labeled anti–Gr-1 mAb or FITC-labeled anti-mouse C3 antibody. Images were acquired by fluorescence microscopy (Nikon Eclipse TE300 confocal cell images). The number of infiltrating neutrophils was calculated as the mean of the number of Gr-1–positive cells in 10 representative high-power fields (×400 total magnification). For immunohistochemical staining for tumor microvascularization, tumor sections were blocked with TBS plus 3% bovine serum albumin buffer and then incubated with an avidin/biotin blocking kit (Vector Laboratories) and stained with anti–CD31-biotin for 1 h at room temperature. After three 10-min washes with blocking buffer, the sections were stained with streptavidin-horseradish peroxidase (Southern Biotechnology Associates) for 1 h at room temperature. After additional washes, horseradish peroxidase substrate (Vector Laboratories) was added for 30 min at room temperature. Following three additional washes, the sections were counterstained with hematoxylin to provide morphologic detail. Microvessel density was determined by counting the number of CD31+ blood vessels (four fields per slide; magnification, ×200).
Statistical analysis
Data from mouse therapy protocols were entered into Prism 4.0 (GraphPad Software) to generate graphs of tumor regression or survival and to determine the significance of differences between data sets. One-way ANOVA with Fisher’s least significant difference was used to compare tumor sizes at the end of treatment and the numbers of infiltrating neutrophils in tumors with different treatment as well as microvessel density within tumors, whereas the log-rank test was used to determine the significance of differences between two survival curves.
Results
Expression of membrane-bound VEGF and complement activation on human carcinomas
Although most tumors are VEGF producers, little is known about whether some tumors indeed express membrane-bound VEGF. We first detected membrane-bound VEGF expression on human tumor cell lines. Human breast carcinomas MDA-MB-483 and HBL-100, human ovarian carcinoma SKOV-3, and human melanoma Colo38 cells were stained with fluorescein-labeled anti-VEGF mAb and assessed by flow cytometry. As indicated in Fig. 1A, only SKOV-3 cells, but not other tumor cell lines, expressed high levels of membrane-bound VEGF. To further confirm that SKOV-3 tumors express membrane-bound VEGF in vivo, human ovarian carcinoma SKOV-3 cells were implanted into SCID mice. SKOV-3 cells were previously shown to express high levels of Her-2/neu oncoprotein. Therefore, we can use anti–Her-2 mAb to track tumor cells. After tumors reached 7 to 8 mm in diameter, mice were sacrificed and tumors were removed and snap frozen. Tumor sections were stained with anti–VEGF-PE mAb (red) or anti–Her-2/neu-FITC (green) or both. As indicated in Fig. 1B, SKOV-3 tumors expressed a high density of Her-2/neu (green) and colocalized with anti-VEGF mAb staining, indicating that SKOV-3 tumors indeed express membrane-bound VEGF.
Fig. 1.

The expression of membrane-bound VEGF and complement activation on human tumor cell lines. A, human tumor cell lines were stained with anti-VEGF mAb (bold line) or isotype control mAb (filled gray) and then assessed by flow cytometry. B, human ovarian carcinoma SKOV-3 tumors were sectioned and stained with anti – Her-2/neu-FITC (green) or anti – VEGF-PE (red) or both. The images were acquired by fluorescent microscopy. Representative tumor sections of five total tumor specimens. Original magnification, ×400. C, human tumor cell lines were incubated with anti-VEGF mAb in the presence of mouse complement. Bold line, cells were then stained with anti-mouse iC3b-FITC mAb. Tumor cells incubated with mouse complement (dotted line) or without mouse complement (filled gray) and then stained with anti-mouse iC3b-FITC mAb were used as controls.
Next, we examined whether anti-VEGF mAb is capable of binding surface-bound VEGF, thereby activating complement leading to iC3b deposition on tumor cells. We mixed tumor cells with anti-VEGF mAb in the presence of mouse complement and then stained with anti–iC3b-FITC mAb. As shown in Fig. 1C, anti-VEGF mAb could bind membrane-bound VEGF on SKOV-3 cells and efficiently activate complement as assessed by flow cytometry. All other tumor cell lines that were negative for membrane-bound VEGF expression were also negative for complement activation.
β-Glucan synergizes with anti-VEGF mAb to elicit CR3-dependent cellular cytotoxicity
Yeast-derived β-glucan functions with antitumor antibodies to activate CR3 and to recruit neutrophils that mediate CR3-dependent cytotoxicity of tumors coated with iC3b. Because anti-VEGF mAb bevacizumab is an IgG1 isotype antibody and thus is capable of both binding membrane-bound VEGF on tumor cells and efficiently activating complement as indicated in Fig. 1C, it is hypothesized that bevacizumab could synergize with β-glucan to elicit CR3-dependent cellular cytotoxicity. To this end, tumor cells were mixed with anti-VEGF mAb in the presence of human complement to achieve iC3b opsonization on tumor cells and then cocultured with neutrophils primed with or without β-glucan. As shown in Fig. 2, the complement-dependent cytotoxicity mediated by bevacizumab was ~3%, with antibody-dependent cellular cytotoxicity being similar to complement-dependent cytotoxicity. These data reaffirm that the mechanism of action of bevacizumab is independent of immune effector functions, including complement-dependent cytotoxicity and antibody-dependent cellular cytotoxicity. Strikingly, bevacizumab plus β-glucan resulted in ~18% cytotoxicity against iC3b-opsonized SKOV-3 tumor cells but not other tumor cells, which are negative for membrane-bound VEGF expression (Fig. 2).
Fig. 2.
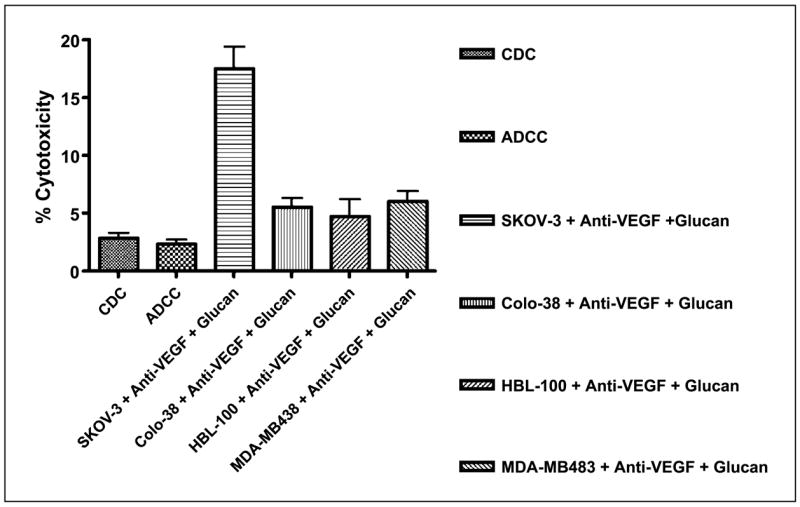
Anti-VEGF mAb bevacizumab synergizes with β-glucan to elicit CR3-dependent cellular cytotoxicity. In vitro cytotoxicity experiments suggested that bevacizumab in conjunction with β-glucan elicits cellular cytotoxicity of SKOV-3 tumors expressing membrane-bound VEGF but not other tumor cells. Minimal complement-dependent cytotoxicity (CDC ; SKOV-3 cells with anti-VEGF mAb in the presence of mouse complement) and antibody-dependent cellular cytotoxicity (ADCC ; SKOV-3 cells with anti-VEGF mAb plus mouse complement plus neutrophils without β-glucan) were observed.
Combined β-glucan with anti-VEGF mAb therapy in vivo on SKOV-3 tumors
To determine if the combined PGG β-glucan with bevacizumab therapy has an augmented antitumor effect compared with the bevacizumab only therapy in vivo, human carcinoma xenografts on SCID mice were produced. Because in vitro study showed that only human ovarian carcinoma SKOV-3 cells express membrane-bound VEGF, SKOV-3 xenografts on SCID mice were used to assess therapeutic efficacy with different therapeutic regimens. In addition, human melanoma Colo38 cells, which do not express membrane-bound VEGF, were used as a control. After SKOV-3 or Colo38 cell implantation, tumors were allowed to grow until they were 300 mm3. The mice were then assigned to four different groups and received (a) no treatment (PBS injections), (b) PGG β-glucan only, (c) anti-VEGF mAb bevacizumab alone, or (d) PGG β-glucan plus anti-VEGF mAb bevacizumab. As shown in Fig. 3A, in SKOV-3 xenograft model, mice receiving PGG β-glucan only did not show any significant tumor regression compared with untreated animals (P = 0.14). Indeed, mice treated with anti-VEGF mAb alone exhibited a significantly reduced tumor burden compared with untreated or treated with PGG β-glucan only (P < 0.0001 with respect to β-glucan only–treated animal). Strikingly, mice receiving PGG β-glucan in combination with anti-VEGF mAb therapy had significantly smaller tumors compared with anti-VEGF mAb treatment alone (P < 0.05). Most of these tumors still retained their sizes before therapy. More importantly, 86% of these mice achieved long-term survival compared with 43% of tumor-bearing mice treated with anti-VEGF mAb only (Fig. 3B). In Colo38 xenograft model, mice treated with anti-VEGF mAb alone also showed a significantly reduced tumor burden compared with untreated or treated with PGG β-glucan only (P < 0.01). However, no significant difference was observed in tumor-bearing mice treated with anti-VEGF mAb alone or anti-VEGF mAb in combination with PGG β-glucan (P = 0.67; Fig. 3C). These data suggest that the addition of PGG β-glucan to anti-VEGF mAb therapy significantly enhances the regression of the membrane-bound VEGF-positive SKOV-3 tumors and long-term survival.
Fig. 3.
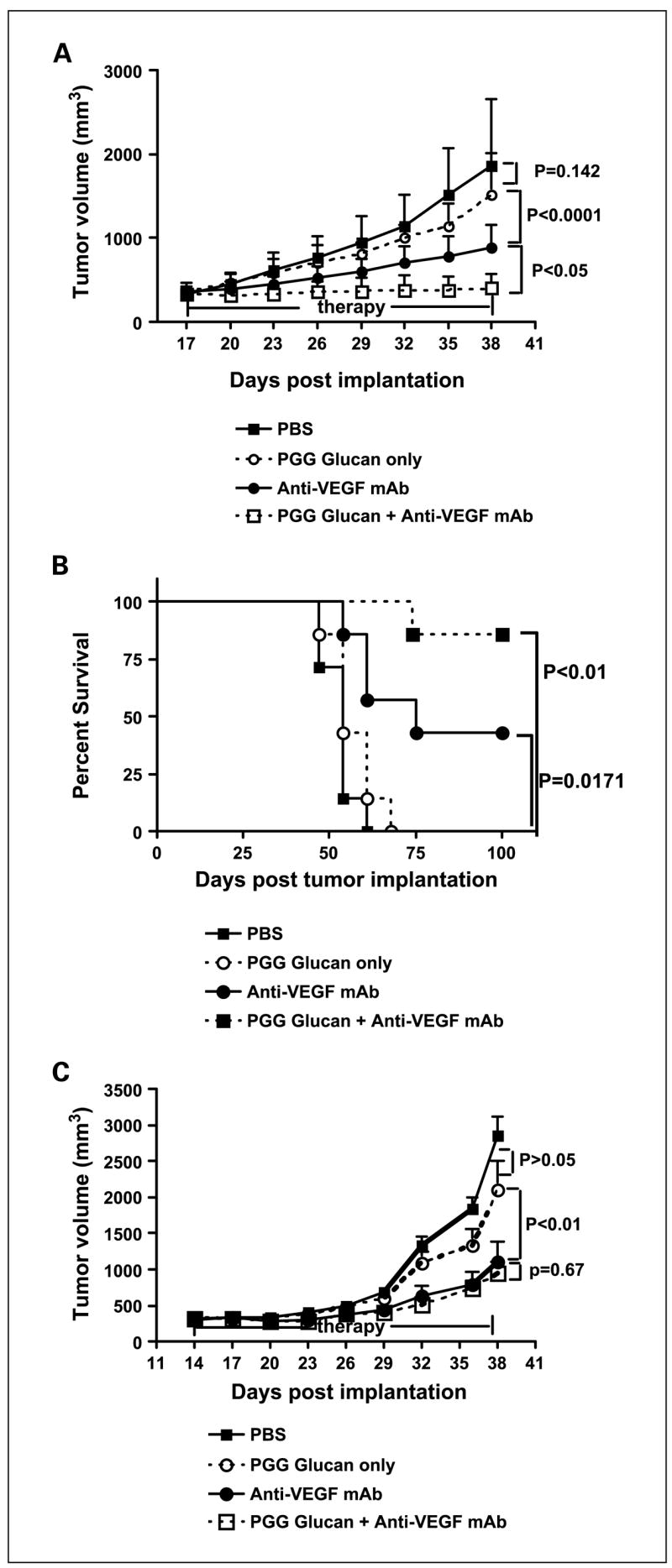
The tumoricidal activity of immunotherapy with PGG β-glucanin combination with bevacizumab. A and B, ICRSCID mice (n = 9) were implanted s.c. with SKOV-3 cells and tumors were allowed to form ~300 mm3 in volume before therapy. Mice received PBS, humanized anti-VEGF mAb(0.2mg every 3rd day)with or without PGG β-glucan (1.2 mg every 3rd day), or β-glucan only for 4 wk. Both tumor growth (A) and survival (B) were monitored. C, ICRSCID mice (n = 10) were implanted s.c. with Colo38 cells and tumors were allowed to form ~300 mm3 in volume before therapy.
Tumor-bearing mice received four different therapies as described above.
Tumor measurements were made at the indicated time. Mice were sacrificed when the tumor sizes reached 2,744 mm3 in volume. Points, mean; bars, SE.
Intratumor neutrophil infiltration and complement deposition within SKOV-3 tumors
We have previously shown that complement activation and neutrophil trafficking within tumors are critical for successful therapy using β-glucan combined with antitumor mAb therapy (40). Therefore, immunofluorescence staining was carried out to examine complement activation and neutrophil infiltration within tumors after treatment with different regimens. The tumor sections from four groups were stained with anti–iC3b-FITC (green) and anti–VEGF-PE (red) mAbs. As indicated in Fig. 4A, massive iC3b deposition was observed in tumors treated with anti-VEGF mAb with or without β-glucan, indicating that anti-VEGF mAb is capable of activating complement in vivo. There was no iC3b deposition on the tumors from mice treated with β-glucan alone or untreated with PBS injections. Furthermore, the tumor sections were stained with anti–Gr-1-PE (red) mAb to detect neutrophil infiltration. As indicated in Fig. 4B and C, the tumors from mice receiving β-glucan plus anti-VEGF mAb bevacizumab therapy had a significantly increased number of infiltrating neutrophils than the mice receiving PBS injection or β-glucan treatment only. Interestingly, tumors treated with anti-VEGF mAb alone also had a significant neutrophil infiltration, suggesting that anti-VEGF mAb has a unique function. It alters the tumor microenvironment that subsequently chemoattracts inflammatory neutrophil infiltration within tumors.
Fig. 4.

Complement activation and neutrophil infiltration within SKOV-3 tumors. A and B, SKOV-3 tumors from animals receiving different treatment regimens as described in Materials and Methods were sectioned and stained with anti – VEGF-PE (red) and anti – iC3b-FITC (green; A) or stained with anti – Gr-1-PE to reveal neutrophil infiltration (B). Original magnifications, ×100 (A) and ×400 (B). C, quantitative summary of the neutrophil infiltration measured as the mean number of Gr-1+ cells in 10 representative high-power fields. Magnification, ×400. Columns, mean; bars, SE.
Tumor microvascularization after anti-VEGF mAb therapy
To observe tumor blood vessel development after anti-VEGF mAb therapy, the tumor sections were stained with anti–CD31-biotin mAb. As shown in Fig. 5, a decreased tumor microvessel density was observed in the tumors from mice treated with PGG β-glucan plus anti-VEGF mAb or anti-VEGF mAb alone when compared with the groups treated with either PBS or β-glucan alone. These data further suggest that the mechanism of action of anti-VEGF mAb is to block angiogenesis.
Fig. 5.
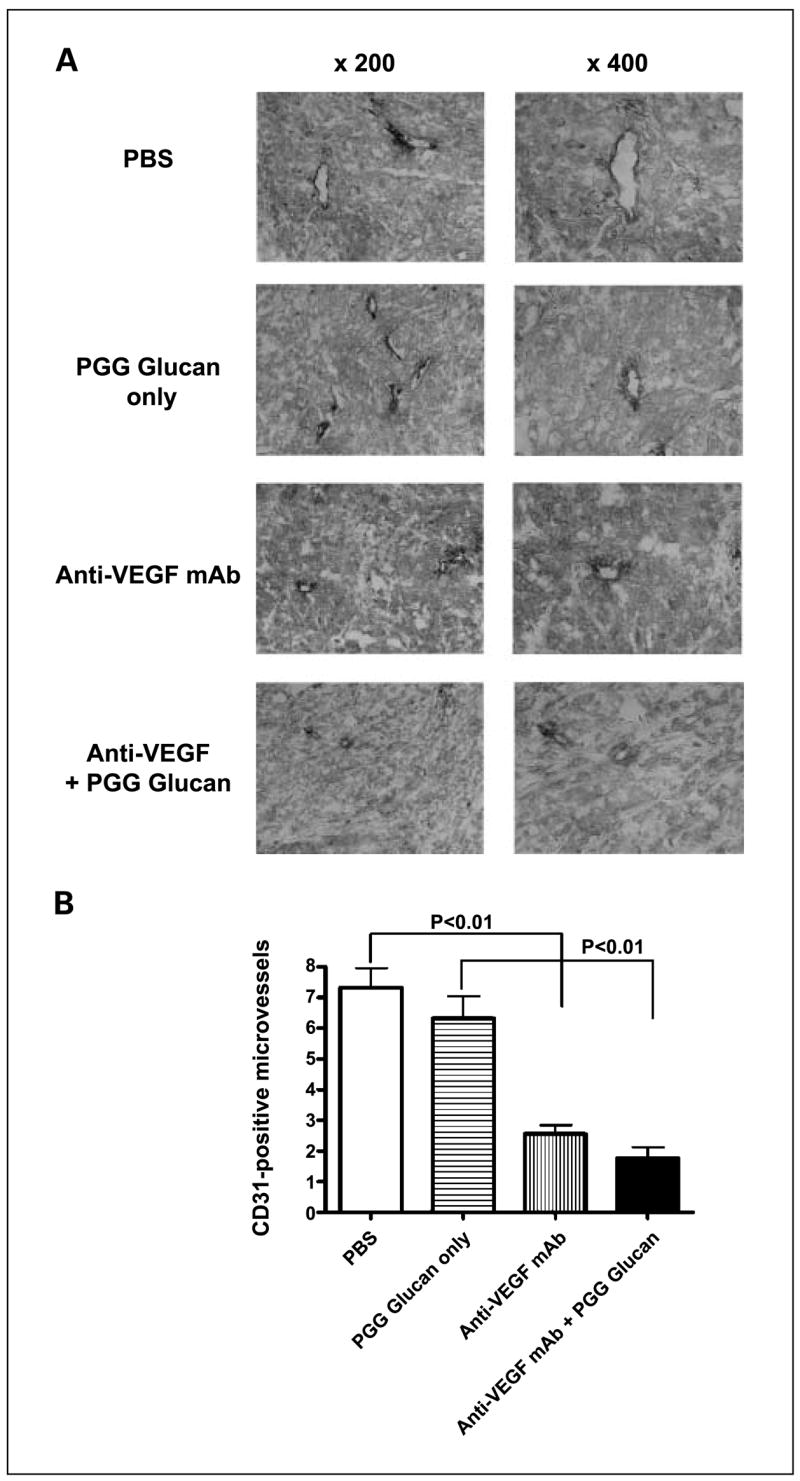
Bevacizumab treatment impairs tumor microvessel formation. A, SKOV-3 tumors from animals treated with different regimens were sectioned and stained with anti – CD31-biotin mAb. Left, low-power fields. Magnification, ×200. Right, representative high-power field. Magnification, ×400. B, quantification of immunohistochemical staining for microvessel density in SKOV-3 tumors. Columns, mean; bars, SE.
Discussion
In the current study, we showed that human ovarian tumor cells express membrane-bound VEGF both in vitro and in vivo. This unique expression pattern leads to the complement activation and iC3b opsonization on tumor cells mediated by anti-VEGF mAb bevacizumab. More importantly, the augmented therapeutic efficacy is achieved by combined PGG β-glucan with anti-VEGF mAb therapy. The enhanced anti-tumor responses and long-term survival for tumor-bearing mice treated with the combined therapy seem to be associated with intratumor massive complement activation and neutrophil infiltration. Therefore, this strategy may offer its clinical benefits for tumor patients with membrane-bound VEGF expression.
Bevacizumab in combination with chemotherapy has been approved by the Food and Drug Administration for use in colorectal and lung cancer treatments (41). Although the mechanism of action of bevacizumab has not been fully elucidated, the proposed mechanism of action is via blockade of circulating VEGF secreted by cancer cells or cancer stromal cells and is independent of immune effector mechanisms, such as complement-dependent cytotoxicity or antibody-dependent cellular cytotoxicity. Indeed, our in vitro cytotoxicity experiments supported this notion (Fig. 2). However, some tumors not only secrete soluble VEGF into the extracellular matrix but also express a membrane-bound form of VEGF. Here, we showed that human ovarian carcinoma SKOV-3 expresses membrane-bound VEGF both in vitro and in vivo. Additionally, VEGFRs are also expressed on tumor cells (17–20). It is possible that the secreted VEGF may bind to its receptor on tumor cells forming a ligand-receptor complex. Indeed, studies have shown that functional VEGF/VEGFR2 autocrine loops are present in human leukemia and support leukemic cell survival and migration (42, 43). Therefore, anti-VEGF mAb may bind surface-bound VEGF or VEGF-VEGFR complex and initiate potential immunologic consequences. Although anti-VEGF mAb bevacizumab itself does not have significant direct cytotoxicity against tumor cells, in vitro study showed that yeast-derived β-glucan is able to synergize with anti-VEGF mAb to elicit leukocyte-mediated CR3-dependent cellular cytotoxicity (Fig. 2). These data suggest that the anti-VEGF mAb bevacizumab can be manipulated in such a way as to elicit effective antitumor immune responses. Indeed, our in vivo study showed that PGG β-glucan in addition to anti-VEGF mAb therapy achieved a significantly smaller tumor burden and long-term survival with respect to anti-VEGF mAb–treated only animals (Fig. 3). However, this synergy did not occur in Colo38 tumors, which do not express membrane-bound VEGF. The augmented therapeutic efficacy offers potential clinical benefits for cancer patients who are subject to anti-VEGF mAb therapy. This may also pose a question of whether membrane-bound VEGF expression should be included in the clinical pathologic report. Although anti-VEGF mAb therapy has been approved for clinical use, the response rate mediated by anti-VEGF mAb as an autonomous monotherapy is limited. In clinical practice, anti-VEGF mAb has been used in combination with chemotherapy, which significantly increases the therapeutic efficacy (44–46). However, those combination therapies also have more severe adverse effects, which limit general use for most patients. Yeast-derived PGG β-glucan is a polysaccharide and has a minimal toxicity (28, 47). Our in vitro and in vivo data clearly suggest that PGG β-glucan can significantly augment the therapeutic efficacy mediated by anti-VEGF mAb bevacizumab in membrane-bound VEGF-positive SKOV-3 tumors but not in membrane-bound VEGF-negative Colo38 tumors. The premise for this combination therapy requires tumors expressing membrane-bound VEGF. Therefore, it seems necessary to detect membrane-bound VEGF expression for patients who potentially undergo this combination therapy. Nevertheless, the combined PGG β-glucan with anti-VEGF mAb therapy offers an alternative strategy for cancer therapy and suggests that bevacizumab therapy can be incorporated with other immune effector functions, such as β-glucan–mediated CR3-dependent cellular cytotoxicity.
Our previous studies in murine syngeneic tumor models have shown that the successful β-glucan–mediated tumor therapy requires glucan-primed neutrophils traffic into tumors and iC3b opsonization on tumor cells (31, 40). The dual ligation of leukocyte CR3 leads to degranulation and cytotoxic responses to iC3b-coated tumor cells (38). This seems to be the case in the current study. Massive complement activation and intratumor neutrophil infiltration occurred in tumors treated with anti-VEGF mAb with or without PGG β-glucan (Fig. 4). This process is independent of PGG β-glucan because tumor-bearing mice treated with PGG β-glucan alone also showed the paucity of iC3b deposition and neutrophil infiltration within the tumors similar to mice treated with PBS injection. This may suggest that SKOV-3 tumors have established an immune-suppressive mechanism to escape immune surveillance. Interestingly, tumors treated with anti-VEGF mAb alone exhibited massive iC3b deposition and neutrophil infiltration. Our previous study showed that SKOV-3 tumors express high levels of membrane complement regulatory proteins, such as CD46, CD55, and CD59 (48). Particularly, up-regulation of CD55 (decay-accelerating factor) on SKOV-3 cells protected SKOV-3 cells from complement-mediated lysis (49). Although SKOV-3 tumors express a high density of Her-2/neu oncoprotein, SKOV-3 tumors were resistant to the anti–Her-2/neu mAb treatment, even in conjunction with PGG β-glucan when a few neutrophils were infiltrated within the SKOV-3 tumors. However, anti-VEGF mAb treatment alone led to the alteration of such a suppressive microenvironment. This seems to be independent of complement activation because both mAbs have human IgG1 frameworks and potently activate complement. It is possible that anti-VEGF mAb uses its unique antiangiogenic activity, which disrupts tumor blood vessel supply and therefore modulates the tumor microenvironment to favor immunotherapy. A recent study has shown that VEGF secreted by carcinoma cells either directly or indirectly participates in maintaining an inflammatory microenvironment (50). Bevacizumab reduced the density of macrophages, MHC class II antigen expression by macrophages, and interleukin-1βmRNA expression. Interestingly, VEGF also stimulates CD55 production on tumor cells (51). Therefore, anti-VEGF mAb bevacizumab treatment could down-regulate CD55 expression, thereby leading to potent neutrophil chemoattractant C5a release within the tumor.
In summary, our study is the first to prove that anti-VEGF mAb bevacizumab can be used in concert with other immune effector functions when it is coadministered with yeast-derived PGG β-glucan. This strategy provides a novel mechanism of action of bevacizumab and has a significant clinical implication for combination therapies. We believe that the combination of bevacizumab and β-glucan has great potential to become part of the armament against cancer and deserves further investigation with the goal of translation into clinical practice.
Acknowledgments
NIH/National Cancer Institute grant RO1CA86412, Kentucky Lung Cancer Research Board, James Graham Brown Cancer Center Pilot Project Program, and Biothera, Inc. (Eagan, MN).
References
- 1.Rini BI, Small EJ. Biology and clinical development of vascular endothelial growth factor-targeted therapy in renal cell carcinoma. J Clin Oncol. 2005;23:1028–43. doi: 10.1200/JCO.2005.01.186. [DOI] [PubMed] [Google Scholar]
- 2.Fidler IJ, Ellis LM. The implications of angiogenesis for the biology and therapy of cancer metastasis. Cell. 1994;79:185–8. doi: 10.1016/0092-8674(94)90187-2. [DOI] [PubMed] [Google Scholar]
- 3.Senger DR, Galli SJ, Dvorak AM, et al. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–5. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- 4.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–9. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- 5.Tischer E, Mitchell R, Hartman T, et al. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J Biol Chem. 1991;266:11947–54. [PubMed] [Google Scholar]
- 6.Park JE, Keller GA, Ferrara N. The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell. 1993;4:1317–26. doi: 10.1091/mbc.4.12.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi Y, Kitadai Y, Bucana CD, Cleary KR, Ellis LM. Expression of vascular endothelial growth factor and its receptor, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer. Cancer Res. 1995;55:3964–8. [PubMed] [Google Scholar]
- 8.Fontanini G, Lucchi M, Vignati S, et al. Angiogenesis as a prognostic indicator of survival in non-small-cell lung carcinoma: a prospective study. J Natl Cancer Inst. 1997;89:881–6. doi: 10.1093/jnci/89.12.881. [DOI] [PubMed] [Google Scholar]
- 9.Berns EM, Klijn JG, Look MP, et al. Combined vascular endothelial growth factor and TP53 status predicts poor response to tamoxifen therapy in estrogen receptor-positive advanced breast cancer. Clin Cancer Res. 2003;9:1253–8. [PubMed] [Google Scholar]
- 10.Ikeda N, Adachi M, Taki T, et al. Prognostic significance of angiogenesis in human pancreatic cancer. Br J Cancer. 1999;79:1553–63. doi: 10.1038/sj.bjc.6690248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takahashi Y, Cleary KR, Mai M, et al. Significance of vessel count and vascular endothelial growth factor and its receptor (KDR) in intestinal-type gastric cancer. Clin Cancer Res. 1996;2:1679–84. [PubMed] [Google Scholar]
- 12.Goydos JS, Gorski DH. Vascular endothelial growth factor C mRNA expression correlates with stage of progression in patients with melanoma. Clin Cancer Res. 2003;9:5962–7. [PubMed] [Google Scholar]
- 13.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 14.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- 15.Millauer B, Wizigmann-Voos S, Schnurch H, et al. High affinity VEGF binding and developmental expression suggest Flk-1as a major regulator of vasculogenesis and angiogenesis. Cell. 1993;72:835–46. doi: 10.1016/0092-8674(93)90573-9. [DOI] [PubMed] [Google Scholar]
- 16.Zeng H, Dvorak HF, Mukhopadhyay D. Vascular permeability factor (VPF)/vascular endothelial growth factor (VEGF) receptor-1 down-modulates VPF/VEGF receptor-2-mediated endothelial cell proliferation, but not migration, through phosphatidylinositol 3-kinase-dependent pathways. J Biol Chem. 2001;276:26969–79. doi: 10.1074/jbc.M103213200. [DOI] [PubMed] [Google Scholar]
- 17.Decaussin M, Sartelet H, Robert C, et al. Expression of vascular endothelial growth factor (VEGF) and its two receptors (VEGF-R1-Flt1 and VEGF-R2-Flk1/KDR) in non-small cell lung carcinomas (NSCLCs): correlation with angiogenesis and survival. J Pathol. 1999;188:369–77. doi: 10.1002/(SICI)1096-9896(199908)188:4<369::AID-PATH381>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 18.Bellamy WT, Richter L, Frutiger Y, Grogan TM. Expression of vascular endothelial growth factor and its receptors in hematopoietic malignancies. Cancer Res. 1999;59:728–33. [PubMed] [Google Scholar]
- 19.Ferrer FA, Miller LJ, Lindquist R, et al. Expression of vascular endothelial growth factor receptors in human prostate cancer. Urology. 1999;54:567–72. doi: 10.1016/s0090-4295(99)00156-9. [DOI] [PubMed] [Google Scholar]
- 20.Price DJ, Miralem T, Jiang S, Steinberg R, Avraham H. Role of vascular endothelial growth factor in the stimulation of cellular invasion and signaling of breast cancer cells. Cell Growth Differ. 2001;12:129–35. [PubMed] [Google Scholar]
- 21.Kim KJ, Li B, Houck K, Winer J, Ferrara N. The vascular endothelial growth factor proteins: identification of biologically relevant regions by neutralizing monoclonal antibodies. Growth Factors. 1992;7:53–64. doi: 10.3109/08977199209023937. [DOI] [PubMed] [Google Scholar]
- 22.Presta LG, Chen H, O’Connor SJ, et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57:4593–9. [PubMed] [Google Scholar]
- 23.Kim KJ, Li B, Winer J, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362:841–4. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 24.Willett CG, Boucher Y, di Tomaso E, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004;10:145–7. doi: 10.1038/nm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gelderman KA, Tomlinson S, Ross GD, Gorter A. Complement function in mAb-mediated cancer immunotherapy. Trends Immunol. 2004;25:158–64. doi: 10.1016/j.it.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 26.Williams DL, Browder IW, Di Luzio NR. Immunotherapeutic modification of Escherichia coli-induced experimental peritonitis and bacteremia by glucan. Surgery. 1983;93:448–54. [PubMed] [Google Scholar]
- 27.Lavigne LM, Albina JE, Reichner JS. β-Glucan is a fungal determinant for adhesion-dependent human neutrophil functions. J Immunol. 2006;177:8667–75. doi: 10.4049/jimmunol.177.12.8667. [DOI] [PubMed] [Google Scholar]
- 28.Yan J, Allendorf DJ, Brandley B. Yeast whole glucan particle (WGP) β-glucan in conjunction with antitumour monoclonal antibodies to treat cancer. Expert Opin Biol Ther. 2005;5:691–702. doi: 10.1517/14712598.5.5.691. [DOI] [PubMed] [Google Scholar]
- 29.Kournikakis B, Mandeville R, Brousseau P, Ostroff G. Anthrax-protective effects of yeast β1,3 glucans. Med Gen Med. 2003;5:1. [PubMed] [Google Scholar]
- 30.Yan J, Vetvicka V, Xia Y, et al. β-Glucan, a “specific” biologic response modifier that uses antibodies to target tumors for cytotoxic recognition by leukocyte complement receptor type 3 (CD11b/CD18) J Immunol. 1999;163:3045–52. [PubMed] [Google Scholar]
- 31.Hong F, Hansen RD, Yan J, et al. β-Glucan functions as an adjuvant for monoclonal antibody immunotherapy by recruiting tumoricidal granulocytes as killer cells. Cancer Res. 2003;63:9023–31. [PubMed] [Google Scholar]
- 32.Hong F, Yan J, Baran JT, et al. Mechanism by which orally administered β-1,3-glucans enhance the tumoricidal activity of antitumor monoclonal antibodies in murine tumor models. J Immunol. 2004;173:797–806. doi: 10.4049/jimmunol.173.2.797. [DOI] [PubMed] [Google Scholar]
- 33.Cheung NK, Modak S. Oral (1→3),(1→4)-β-D-glucan synergizes with antiganglioside GD2 monoclonal antibody 3F8 in the therapy of neuroblastoma. Clin Cancer Res. 2002;8:1217–23. [PubMed] [Google Scholar]
- 34.Cheung NK, Modak S, Vickers A, Knuckles B. Orally administered β-glucans enhance anti-tumor effects of monoclonal antibodies. Cancer Immunol Immuno ther. 2002;51:557–64. doi: 10.1007/s00262-002-0321-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Modak S, Koehne G, Vickers A, O’Reilly RJ, Cheung NK. Rituximab therapy of lymphoma is enhanced by orally administered (1→3),(1→4)-D-β-glucan. Leuk Res. 2005;29:679–83. doi: 10.1016/j.leukres.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 36.Thornton BP, Vetvicka V, Pitman M, Goldman RC, Ross GD. Analysis of the sugar specificity and molecular location of the β-glucan-binding lectin site of complement receptor type 3 (CD11b/CD18) J Immunol. 1996;156:1235–46. [PubMed] [Google Scholar]
- 37.Xia Y, Vetvicka V, Yan J, et al. The β-glucan-binding lectin site of mouse CR3 (CD11b/CD18) and its function in generating a primed state of the receptor that mediates cytotoxic activation in response to iC3b-opsonized target cells. J Immunol. 1999;162:2281–90. [PubMed] [Google Scholar]
- 38.Li B, Allendorf DJ, Hansen R, et al. Yeast β-glucan amplifies phagocyte killing of iC3b-opsonized tumor cells via complement receptor 3-Syk-phosphatidylinositol 3-kinase pathway. J Immunol. 2006;177:1661–9. doi: 10.4049/jimmunol.177.3.1661. [DOI] [PubMed] [Google Scholar]
- 39.Tsikitis VL, Morin NA, Harrington EO, Albina JE, Reichner JS. The lectin-like domain of complement receptor 3 protects endothelial barrier function from activated neutrophils. J Immunol. 2004;173:1284–91. doi: 10.4049/jimmunol.173.2.1284. [DOI] [PubMed] [Google Scholar]
- 40.Allendorf DJ, Yan J, Ross GD, et al. C5a-mediated leukotriene B4-amplified neutrophil chemotaxis is essential in tumor immunotherapy facilitated by anti-tumor monoclonal antibody and β-glucan. J Immunol. 2005;174:7050–6. doi: 10.4049/jimmunol.174.11.7050. [DOI] [PubMed] [Google Scholar]
- 41.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23:1011–27. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 42.Dias S, Hattori K, Heissig B, et al. Inhibition of both paracrine and autocrine VEGF/VEGFR-2 signaling pathways is essential to induce long-term remission of xenotransplanted human leukemias. Proc Natl Acad Sci U S A. 2001;98:10857–62. doi: 10.1073/pnas.191117498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dias S, Hattori K, Zhu Z, et al. Autocrine stimulation ofVEGFR-2 activates human leukemic cell growth and migration. J Clin Invest. 2000;106:511–21. doi: 10.1172/JCI8978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sandler A, Gray R, Perry MC, et al. Paclitaxelcarboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–50. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 45.Johnson DH, Fehrenbacher L, Novotny WF, et al. Randomized phase II trial comparing be vacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxelalonein previously untreated locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol. 2004;22:2184–91. doi: 10.1200/JCO.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 46.Ellis LM. Bevacizumab. Nat Rev Drug Discov. 2005;(Suppl):S8–9. doi: 10.1038/nrd1727. [DOI] [PubMed] [Google Scholar]
- 47.Zimmerman JW, Lindermuth J, Fish PA, et al. A novel carbohydrate-glycosphingolipid interaction between a β-(1-3)-glucan immunomodulator, PGG-glucan, and lactosylceramide of human leukocytes. J Biol Chem. 1998;273:22014–20. doi: 10.1074/jbc.273.34.22014. [DOI] [PubMed] [Google Scholar]
- 48.Li B, Allendorf DJ, Hansen R, et al. Combined yeast β-glucan and antitumor monoclonal antibody therapy requires CSA-mediated neatrophil chemotaxis via regulation of decay-accelerating factor CD55. Cancer Research. 2007;67:7421–30. doi: 10.1158/0008-5472.CAN-07-1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bjorge L, Hakulinen J, Wahlstrom T, Matre R, Meri S. Complement-regulatory proteins in ovarian malignancies. Int J Cancer. 1997;70:14–25. doi: 10.1002/(sici)1097-0215(19970106)70:1<14::aid-ijc3>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 50.Salnikov AV, Heldin NE, Stuhr LB, et al. Inhibition of carcinoma cell-derived VEGF reduces inflammatory characteristics in xenograft carcinoma. Int J Cancer. 2006;119:2795–802. doi: 10.1002/ijc.22217. [DOI] [PubMed] [Google Scholar]
- 51.Mason JC, Steinberg R, Lidington EA, et al. Decay-accelerating factor induction on vascular endothelium by vascular endothelial growth factor (VEGF) is mediated via a VEGF receptor-2 (VEGF-R2)- and protein kinase C-α/ε (PKCα/ε)-dependent cytoprotective signaling pathway and is inhibited by cyclosporin A. J Biol Chem. 2004;279:41611–8. doi: 10.1074/jbc.M407981200. [DOI] [PubMed] [Google Scholar]