

Abstract
To assess soil bacterial diversity, PCR systems consisting of several slightly different reverse primers together with forward primer F968-GC were used along with subsequent denaturing gradient gel electrophoresis (DGGE) or clone library analyses. In this study, a set of 13 previously used and novel reverse primers was tested with the canonical forward primer as to the DGGE fingerprints obtained from grassland soil. Analysis of these DGGE profiles by GelCompar showed that they all fell into two main clusters separated by a G/A alteration at position 14 in the reverse primer used. To assess differences between the dominant bacteria amplified, we then produced four (100-membered) 16S rRNA gene clone libraries by using reverse primers with either an A or a G at position 14, designated R1401-1a, R1401-1b, R1401-2a, and R1401-2b. Subsequent sequence analysis revealed that, on the basis of the about 410-bp sequence information, all four primers amplified similar, as well as different (including novel), bacterial groups from soil. Most of the clones fell into two main phyla, Firmicutes and Proteobacteria. Within Firmicutes, the majority of the clones belonged to the genus Bacillus. Within Proteobacteria, the majority of the clones fell into the alpha or gamma subgroup whereas a few were delta and beta proteobacteria. The other phyla found were Actinobacteria, Acidobacteria, Verrucomicrobia, Chloroflexi, Gemmatimonadetes, Chlorobi, Bacteroidetes, Chlamydiae, candidate division TM7, Ferribacter, Cyanobacteria, and Deinococcus. Statistical analysis of the data revealed that reverse primers R1401-1b and R1401-1a both produced libraries with the highest diversities yet amplified different types. Their concomitant use is recommended.
Denaturing gradient gel electrophoresis (DGGE) is a powerful molecular technique in which DNA fragments of the same length but with different sequences can be separated (7, 8, 23, 24, 32). When applied to 16S rRNA genes, the method allows the dissection of microbial communities at the level of the phylogeny of their constituents. PCR applied to regions of this gene with conserved primers allows the generation of a mixture of amplicons which can be separated by DGGE. The technique was initially introduced into microbial ecology by Muyzer et al. in 1993 and has been widely used since its inception (21). Whereas Muyzer and coworkers originally proposed a system based on the V3 region of the 16S rRNA gene, Heuer et al. (12) and Smalla et al. (34) described a PCR-DGGE system based on the V6 region of this gene. This region has the highest variability within the whole rRNA gene, thus theoretically allowing for the most nearly optimal dissection of bacterial communities. Since its concoction, several different reverse primers for region V6-based DGGE have been described in the literature, some of which show sequence differences (6, 15, 16, 25, 26). Moreover, some of the reverse primers carrying the same name had different nucleotide compositions whereas, on the other hand, primers were found to have the same nucleotide order but to have different names. Hence, it is legitimate to question the comparability of the soil bacterial DGGE patterns generated by these different reverse primers and, in addition, to interrogate which reverse primer can be best used for DGGE to characterize soil bacterial communities to their fullest breadth. To elucidate these questions, we analyzed, for a selected loamy sand soil, how primer sets composed of the existing reverse primers, as well as several novel different reverse primers, affect our picture of soil bacterial community structure by two methods, (i) PCR-DGGE fingerprinting followed by cluster analysis and (ii) analysis of the partial rRNA gene sequences of clones generated with different reverse primers. A species-rich grassland soil was used for this purpose, as previous research (9) has shown that this soil contains an elevated microbial diversity. It was thus of interest to optimize the molecular tools for the analysis of the bacterial diversity and community structure in this soil.
MATERIALS AND METHODS
Soil and soil sampling.
The soil samples used in this experiment were collected from a long-term ecological site at the Wildekamp field, located in Bennekom, The Netherlands. The soil in this field is a loamy sand rich in organic matter (2.5%) with a slightly acidic pH (5.5 to 6.5). The site sampled represented a long-term (>54 years) permanent grassland field. Samples were taken from replicate plots of the rhizosphere compartments (designated RG) and the grassland bulk soil (G). Specifically, 100 samples per plot (10 cm deep) were mixed to yield one composite sample per plot (10, 35, 37).
DNA extraction from soil.
DNA was extracted from soil with the UltraClean Soil DNA kit (Mo Bio Laboratories Inc.). DNA isolation was performed according to the manufacturer's instructions, modified as follows. Extra glass beads (0.15 to 0.30 g; bead size, 0.1 mm) were added to the soil samples, and the cells were disrupted by bead beating (mini-bead beater; Bio Spec Products) two times for 30 s. Final purification of the extracted DNA was performed with the Wizard DNA cleanup system (Promega).
Primers.
Primers for PCR amplification were designed to be specific for bacterial 16S rRNA gene targets. A 17-mer forward primer, designated F-968 (5′-AA CGC GAA GAA CCT TAC-3′), to which a 40-mer GC clamp (5′-CGC CCG GGG CGC GCC CCG GGC GGG GCG GGG GCA CGG GGG G) was attached at the 5′ end was combined with 13 different reverse primers situated roughly around position 1400 (Table 1) to amplify the bacterial 16S rRNA gene fragments (22).
TABLE 1.
Reverse primers used in PCR experiments in this study
| Original primera name | Primer code | 5′-3′ primer sequence (16S rRNA gene target)b | Proposed new name | % Match within RDP database relative to a 100% matchc | Reference(s) |
|---|---|---|---|---|---|
| R1378/R1401 | 1a | CGG TGT GTA CAA GGC CCG GGA ACG (1378-1401) | R1401-1a | 23 | 12, 34 |
| —d | 1b | CGG TGT GTA CAA GAC CCG GGA ACG (1378-1401) | R1401-1b | 7 | This study |
| — | 1c | CGG TGT GTA CAA GRC CCG GGA ACG (1378-1401) | 30 | This study | |
| — | 2a | CGG TGT GTA CAA GGC CC (1378-1394) | R1401-2a | 24 | This study |
| R1401/L1401 | 2b | CGG TGT GTA CAA GAC CC (1378-1394) | R1401-2b | 7 | 6, 25 |
| — | 2c | CGG TGT GTA CAA GRC CC (1378-1394) | 31 | This study | |
| — | 3a | GTA CAA GGC CCG GGA ACG (1384-1401) | 24 | This study | |
| — | 3b | GTA CAA GAC CCG GGA ACG (1384-1401) | 8 | This study | |
| — | 3c | GTA CAA GRC CCG GGA ACG (1384-1401) | 32 | This study | |
| — | 2*a | GCG TGT GTA CAA GGC CC (1378-1394) | <1e | This study | |
| R1401/L1401 | 2*b | GCG TGT GTA CAA GAC CC (1378-1394) | <1e | 16, 26 | |
| — | 2*c | GCG TGT GTA CAA GRC CC (1378-1394) | <1e | This study | |
| — | 1378* | GCG TGT GTA CAA GGC CCG GGA ACG (1378-1401) | <1e | This study |
Reverse primers.
Positions according to E. coli 16S rRNA gene sequence. Boldface type indicates nucleotides that differentiate primers.
Related to homologous 16S rRNA gene sequences in the Ribosomal Database Project II database.
—, own primers identified by primer code.
The match is <1% because of a GC mismatch at the 5′ end.
Theoretical primer match.
Ribosomal Database Collection II Release 9.50 (option Probe Match; http://rdp.cme.msu.edu/probematch/search.jsp) was used to collect the 16S rRNA gene sequences that matched the selected reverse primers. In particular, differences in the theoretical primer match were checked to allow selection of the best theoretical reverse primer for the amplification system (4).
PCR amplification.
PCR mixtures were composed as follows. Seven microliters of 10× Stoffel PCR buffer (Applera, Nieuwerkerk a/d IJssel, The Netherlands), 100 nmol MgCl2 (Applera), 0.5 μl formamide, 0.5 μg T4 gene 32 protein (Roche, Almere, The Netherlands), 10 nmol of each deoxyribonucleoside triphosphate, 10 pmol of each primer, and 3 U of 10 U/μl AmpliTaq DNA polymerase, Stoffel fragment (Applera), were combined with H2O (Applera) to 50 μl in a 0.2-ml Microfuge tube. After the addition of 5 ng of template DNA, the mixtures were incubated in a Gene Amp PCR system 9700 (Applera) programmed as follows: initial denaturation of double-stranded DNA for 5 min at 94°C; 10 (touchdown) cycles consisting of 1 min at 94°C, 1 min at 60°C, and 2 min at 72°C with a decrease in the annealing temperature of 0.5°C per cycle; 25 cycles consisting of 1 min at 94°C, 1 min at 55°C, and 2 min at 72°C; and extension for 30 min at 72°C. All amplification products were purified with the Wizard PCR DNA purification system (Promega, Madison, WI) and analyzed by electrophoresis in 1.0% (wt/vol) agarose gels, followed by ethidium bromide staining (1.2 mg/liter ethidium bromide in 1× Tris-acetate-EDTA) (19, 28).
DGGE.
Denaturing gradient gel electrophoresis (DGGE) was performed with the Ingeny Phor-U system (Ingeny International, Goes, The Netherlands). The PCR products were loaded onto a polyacrylamide gel (6% [wt/vol] acrylamide in 0.5× TAE buffer [2.42 g Tris base, 0.82 g sodium acetate, 0.185 g EDTA, 1 liter of H2O; pH adjusted to 7.8 with acetic acid]) with a 40 to 70% denaturant gradient (100% denaturant was 3.5 M urea and 32% [vol/vol] deionized formamide). The wells were loaded with roughly equal amounts of DNA (about 500 ng), and electrophoresis was carried out in 0.5× TAE buffer at 100 V for 16 h at 60°C. The gels were stained for 90 min in 0.5× TAE buffer with SYBR gold (final concentration, 0.5 μg/liter; Invitrogen, Breda, The Netherlands). Images of the gels were obtained by Imagemaster VDS (Amersham Biosciences, Buckinghamshire, United Kingdom) and stored as TIFF files. The DGGE patterns were compared by clustering the different lanes by Pearson's product-moment correlation coefficient with GelCompar II software (Applied Maths, Sint-Martens-Latem, Belgium) by the unweighted-pair group method with arithmetic mean, rolling-disk background subtraction, and no optimization (17, 27).
Preparation of clone libraries and sequencing.
Cleaned PCR products derived from the four reverse primers in conjunction with the forward primer with a GC clamp, F968-GC, were ligated into the pGEM-T easy vector (Promega, Madison, WI) and introduced into competent Escherichia coli MM294 cells (Sylphium Life Sciences, Groningen, The Netherlands) by transformation as described by the pGEM-T manufacturer's protocol. Blue-white screening was used, and white colonies were randomly picked. Colony PCR was then performed with pGEM-T primers T7F (5′-TAATACGACTCACTATAGGG-3′) and SP6R (5′-GATTTAGGTGACACTATAG-3′). Clones that had yielded PCR products of the correct sizes, as determined by gel electrophoresis, were selected, and inserts were sequenced with standard primer T7F. Sequencing was performed on an ABI 377 machine (Applied Biosystems) (28).
Sequence analysis, construction of trees, and statistical analyses.
Analysis of the sequences was done with Chromas (Technelysium, Tewantin, Australia). Chimera check with Bellerophon was used to check for chimeric sequences (http://greengenes.lbl.gov/cgi-bin/nph-bel3_interface.cgi) (13). Bellerophon is a program for detecting chimeric sequences in a multiple-sequence data set by comparative analysis. It was specifically developed to detect 16S rRNA gene chimeras in PCR clone libraries but can be applied to other gene data sets. Chimeric sequences were not detected (13). The partial 16S rRNA gene sequences were compared with sequences in GenBank with nucleotide-nucleotide BLAST (BLAST-N) to obtain the nearest phylogenetic neighbors (www.ncbi.nlm.nih.gov/BLAST/). Sequences showing more than 97% similarity were considered to belong to the same operational taxonomic unit (OTU) (1). Trees were constructed from libraries obtained with each reverse primer by neighbor joining within the program MEGA 3.1 (The Biodesign Institute) and bootstrapped with 500 repetitions. These trees were used to obtain broader groupings—supported by checks with the Ribosomal Database Project (RDP) database—which served to prepare histograms revealing the compositions of the bacterial communities detected by each of the four reverse primers combined with the forward primer (18).
The program DOTUR (distance-based OTU and richness determination) was used to create rarefaction curves and to determine the Shannon diversity index, as well as the bias-corrected Chao1 estimator of richness (http://www.plantpath.wisc.edu/fac/joh/dotur.html) (14, 29). The Shannon index (H′) was used to determine the diversity of bacteria present in the clone libraries created with the four reverse primers by the following equation:
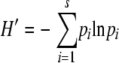 |
where s is the number of species (species richness) and pi is the proportion of the species i in sample i. The index measures diversity by incorporating both the richness and the distribution (evenness) of types (31). Chao1 is the species richness estimator and assesses the number of species present (3).
To compare the clone libraries for similarities, we used LIBSHUFF (30, 33). Differences between clone libraries were thus determined by calculating homologous and heterologous coverage curves and assessing the difference between the two curves, with the Cramér-von Mises statistic according to the method described by Singleton et al. (30, 33). For each comparison, if the lowest of the two P values calculated by LIBSHUFF is lower than or equal to the so-called critical P value (value for four libraries, 0.0043; relaxed, 0.0075) given by the program, a significant difference in the composition of the communities sampled in each library is indicated. The analyses were performed with the web-based LIBSHUFF program (version 0.96; J. R. Henriksen; http://libshuff.mib.uga.edu).
Nucleotide sequence accession numbers.
The nucleotide sequences obtained in this study have been submitted to the GenBank database and assigned accession numbers EU407826 to EU408221.
RESULTS
Theoretical primer match.
Table 1 shows the 13 different reverse primers that were designed between positions 1378 and 1401 of the rRNA gene (E. coli numbering) and their characteristics. All primers were checked versus the sequences within the RDP database to indicate the theoretical coverage of bacterial sequences within the database. These data were then converted to percentages of matching with the total 16S rRNA database (data not shown). When no mismatches were allowed, none of the primers gave matches to 100% of the database entries (Table 1), whereas at one or two mismatches, the theoretical match increased drastically to (close to) 100%. Of the four primers that were selected to create clone libraries (R1401-1a, -2a, -1b, and -2b), primer R1401-2a was found to match the largest fraction of the bacterial 16S rRNA gene sequences in the database (24%), followed by primers R1401-1a (23%), R1401-2b (7%), and R1401-1b (7%). Hence, the G at position 14 of the reverse primer (counted from the 5′ end), which corresponds to position 1391 of the small-subunit RNA gene (E. coli numbering system), apparently yielded a higher degree of matching with bacterial database sequences than the alternative base, an A, at this site (Table 1). The GC/CG shift (at position 1-2) had a dramatic influence on the percent matching of all primers with bacterial sequences (decreasing), whereas the GGGAACG extension at the 3′ end seemed to have little influence on the matching within the domain Bacteria.
DGGE fingerprints.
High-molecular-weight DNA was recovered from all replicates of the two grassland soil samples, RG and G. The mean molecular size was 25 kb, and an average of about 15 to 20 μg DNA was obtained per g of soil, as indicated by electrophoresis on agarose gels and comparison to a marker. After Wizard DNA purification, the soil DNA preparations were colorless and sufficiently pure for PCR amplification, as evidenced by ready amplification with the canonical 16S rRNA gene-based PCR system. The electrophoresis of the PCR products obtained with the 13 different reverse primers under nondenaturating conditions in all cases revealed the expected bands of approximately 450 bp (data not shown).
To validate the differences caused by using the different reverse primers, a within-experiment check was needed to assess the method's reproducibility. Therefore, the reproducibility of the DGGE method was tested prior to the actual assessment of the profiles generated with the 13 different reverse primers. This was done by performing PCR on the basis of the 13 reverse primers in triplicate assays, running DGGE, and analyzing the resulting patterns by GelCompar. Analysis of the DGGE patterns thus obtained yielded a clear clustering among reverse primer types, as the within-treatment replicates clustered tightly together and were actually indistinguishable (data not shown). Hence, the reproducibility of the PCR-DGGE technique was very high and there were clear effects of the type of reverse primer used. We then compared the DGGE profiles generated with the 13 primer sets (one replicate each) in a single gel to circumvent the problem of gel-to-gel variation. The clustering of the patterns on this gel revealed the appearance of two broad clusters, one cluster consisting of the patterns produced by all of the reverse primers containing a G at position 14 (designated cluster a) and one cluster of those produced by reverse primers with an A at this position (designated cluster b). Since four primers contained an R degeneracy (A or G) at position 14 (designated cluster c), these were taken out of further analysis to remove any confounding effects of this degeneracy on the clustering. Figure 1A shows the DGGE patterns of PCR products produced with the nine remaining reverse primers (1378*, 1a, 2a, 2*a, 3a, 1b, 2b, 2*b, and 3b) in combination with the forward GC-clamped primer on DNA from grass rhizosphere sample RG. Analysis by GelCompar produced two main clusters, designated I and II, at low levels of similarity (Fig. 1B). The patterns produced by all reverse primers with a G at position 14 clustered together, and so did those generated with the reverse primers with an A at position 14. No other conspicuous pattern variations were discerned. Thus, the G/A switch at position 14 was apparently more determinative than the GC/CG alteration at the 5′ end of the primer and/or the GGGAACG addition to the 3′ end. To ascertain whether this clustering was consistent across samples, a similar analysis was performed on soil DNA taken from the grassland bulk soil (G). Analysis with GelCompar indeed supported the clustering that was also seen for sample RG. Calculation of the Shannon diversity indices (GelCompar) showed that primer R1401-1a yielded patterns with the highest diversity index (3.23), followed by R1401-1b (3.18), R1401-2a (3.16), and R1401-2b (2.68).
FIG. 1.

Comparison of bacterial DGGE patterns obtained from soil sample RG by PCR with canonical primer F968 (joined to a GC clamp) and reverse primers R1401-1a, -2a, -2*a, -3a, -1378*, -1b, -2b, -2*b, and -3b (A) and the corresponding dendrogram (clustering by the unweighted-pair group method with arithmetic mean) (B). M, molecular size markers; 1a, 2a, 2*a, 3a, 1378*, 1b, 2b, 2*b, and 3b, reverse primers used.
To examine the nature of the dominant bacterial types that are targeted by the different primers used in the two clusters, two reverse primers from each cluster were taken (cluster I, R1401-1a and R1401-2a; cluster II, R1401-1b and R1401-2b) and clone libraries were produced.
Clone libraries produced with four selected primer sets.
Clone libraries were successfully produced from the same soil sample, RG, with each of the four reverse primers (R1401-1a, R1401-2a, R1401-1b, and R1401-2b) in combination with the canonical forward primer. The clones obtained in the pGEM-T easy vector system were then checked for the presence of inserts of the expected size (about 450 bp) and a random set of about 100 clones with inserts per reverse primer was sequenced on the ABI377 apparatus. Rarefaction analysis (Fig. 2) of the sequences obtained revealed that coverage, as expected, was not complete; we estimated that a >10- to 30-fold increased sampling effort would be required to achieve fair coverage. The coverage obtained with primer R1401-1b was conspicuously lower than that produced with the other three primers, whereas that obtained with primer R1401-2b was the highest. On the other hand, the Shannon diversity index determined on the basis of the sequences (threshold, 97%) revealed rather similar values between the primers. As expected, the value produced with primer R1401-1b (4.12) was slightly higher than those produced with the other primers (R1401-2a, 4.07; R1401-2b, 3.99; R1401-1a, 3.93). The ChaoI species richness estimator was the highest for primers R1401-2a (232) and R1401-2b (221) and lower for the other primers (R1401-1b, 195; R1401-1a, 192).
FIG. 2.

Rarefaction curves of bacterial clone libraries of soil sample RG generated with primer sets consisting of canonical primer U968f combined with four different reverse primers (R1401-1a, -2a, -1b, and -2b). Sequences were compared to GenBank rRNA gene-based entries. The number of different OTUs (defined at a 97% similarity cutoff) in each sample is plotted versus the number of sequences sampled.
The LIBSHUFF comparative analysis of the four clone libraries is presented in Table 2. This analysis revealed that the clone libraries produced with primers R1401-1a and -2b were different, per library, from those produced with the other three primer sets. Furthermore, the clone library produced with primer R1401-1b was different from those produced with primers R1401-1a and -2b and the one produced with primer R1401-2a differed from that produced with R1401-1a and -2b. Thus, considerable differences in library composition can be caused by primers differing at various positions, even at just a single site.
TABLE 2.
Comparison of four 16S rRNA gene libraries using LIBSHUFFa
| Homologous library (X) |
P value of ΔCxy heterologous library (Y)
|
|||
|---|---|---|---|---|
| 1401R-1a | 1401R-1b | 1401R-2a | 1401R-2b | |
| 1401R-1a | NAb | 0.073 | 0.091 | 0.014 |
| 1401R-1b | 0.016 | NA | 0.664 | 0.001 |
| 1401R-2a | 0.040 | 0.822 | NA | 0.190 |
| 1401R-2b | 0.003 | 0.012 | 0.063 | NA |
Libraries were constructed by using four different reverse primers (1401R-1a, n = 94; 1401R-2a, n = 103; 1401R-1b, n = 96; 1401R-2b, n = 103) in conjunction with the forward (clamped) F968 primer. Comparisons were made by using the integral form of the Cramér-von Mises statistic. The critical P value for the comparison of the four libraries was 0.0043.
NA, not applicable.
Analysis of bacterial types amplified by four selected primer sets.
The analysis of the clones generated per library is presented in the dendrograms in Fig. 3A to D. After analysis of these, a histogram was made to compare the relative abundances of the different groups revealed by each primer set (Fig. 4). Large fractions of the clones from each library had sequences that matched NCBI database sequences at ≥97% (R-1401-1a, 55.3%; R1401-1b, 42.7%; R1401-2a, 44.7%; R1401-2b, 50.5%). Moreover, 16.0% (R1401-1a), 15.6% (R1401-1b), 17.5% (R1401-2a), and 12.6% (R1401-2b) of the clones matched database entries between 95 and 97% and 25.5% (R1401-1a), 33.4% (R1401-1b), 34.0% (R1401-2a), and 32.0% (R1401-2b) of them matched database entries between 90 and 94%. Finally, low percentages of the clone libraries had matches in the database of 84 to 90% (R1401-1a, 3.2%; R1401-1b, 8.3%; R1401-2a, 3.8%; R1401-2b, 4.9%). The database entries with which matches were found at different similarity levels were often cultured organisms of defined taxonomic status, except for a few entries found in the phyla Acidobacteria, Verrucomicrobia, and Chloroflexi.
FIG. 3.


Phylogenetic trees constructed on the basis of partial 16S rRNA gene sequences (about 400 bp) generated from soil with canonical forward primer F968-GC and reverse primers R1401-1a (A, 94 sequences), R1401-2a (B, 103 sequences), R1401-1b (C, 96 sequences), and R1401-2b (D, 103 sequences). The trees were calculated with the neighbor-joining algorithm. To simplify the phylogenetic trees, clones are marked as follows: no symbol, sequences with ≥97% similarity; ▴, 95 to 96% similarity; •, 90 to 94% similarity; ○, <90% similarity. Grouping is according to phylum or class (Proteobacteria) or PN candidate divisions. Abbreviations: Acido, Acidobacteria; Actino, Actinobacteria; Bacter, Bacteroidetes; Chlam, Chlamydiae; Chloro, Chloroflexi; Chlorob, Chlorobi; Cyano, Cyanobacteria; Deino, Deinococcus; Ferri, Ferribacter; Firm, Firmicutes; Gemma, Gemmatimonadetes; α-, β-, γ-, and δ-Prot, Alpha-, Beta-, Gamma-, and Deltaproteobacteria, respectively; TM7, candidate division TM7; Verruco, Verrucomicrobia.
FIG. 4.

Frequency histogram of the clones found in this study on the basis of ≥97%, 95% to 96%, 90% to 94%, and <90% matching OTU. The x axis shows the major existing or possibly novel phylogenetic groups (phylum or class) found in the clone libraries with each of the reverse primers used (R1401-1a, -2a, R1401-1b, and -2b), whereas the y axis shows the number of clones found in each phylogenetic group. *, these groups matched unculturable bacteria in the NCBI database.
Overall, the sequences found in the four libraries fell into 14 known phylogenetic divisions, namely, Firmicutes, Proteobacteria, Actinobacteria, Acidobacteria, Verrucomicrobia, Chloroflexi, Gemmatimonadetes, Chlorobi, Bacteroidetes, Cyanobacteria, Chlamydiae, Ferribacter, Deinococcus, and candidate division TM7 (Fig. 4). The different primers amplified different numbers of these groups; i.e., R1401-1a yielded 7 groups, R1401-1b yielded 12 groups, R1401-2a yielded 10 groups, and R1401-2b yielded 8 groups.
The majority of the clones generated with all four primer sets fell into two main phyla, namely, Firmicutes (R1401-1a, 30.9%; R1401-1b, 18.8%; R1401-2a, 21.4%; R1401-2b, 32.0%) and the alpha, beta, gamma, and delta subgroups of Proteobacteria (R1401-1a, 27.6%; R1401-1b, 30.3%; R1401-2a, 28.1%; R1401-2b, 29.2%) (Table 3). The other phyla found with all four primers were Actinobacteria, Acidobacteria, and Chloroflexi. A main difference between the libraries generated with the four primer sets was the distribution of the Proteobacteria among the different subgroups. While 65.4% of the clones produced with primer R1401-1a were affiliated with the gamma subdivision of Proteobacteria, substantially lower frequencies of clones belonging to this subdivision were found with the other primers (R1401-1b, 24.1%; R1401-2a, 31.0%; R1401-2b, 43.3%). Also, while 37.9% of the clones obtained with primer R1401-1b fell within the delta subdivision of Proteobacteria, lower frequencies of this group were found with the other primers (R1401-1a, 3.8%; R1401-2a, 17.2%; R1401-2b, 16.7%). Members of Gemmatimonadetes and Verrucomicrobia were found with primers R1401-1a, R1401-1b, and R1401-2a, but primer R1401-2b did not yield any representative of these phyla. Further, primer R1401-1a stood out by having no clones related to the class Rubrobacteridae but having the highest number of clones associated with the class Actinobacteridae.
TABLE 3.
Relative abundances of major phylogenetic groups found in clone libraries constructed with forward primer F968-GC and four different reverse primers in a single soil samplea
| Phylogenetic group | Relative abundance (%) found with reverse primer:
|
|||
|---|---|---|---|---|
| R1401-1a | R1401-1b | R1401-2a | R1401-2b | |
| Firmicutes | 30.9 | 18.8 | 21.4 | 32.0 |
| Genus Bacillus | 75.9 | 83.3 | 86.4 | 81.8 |
| Actinobacteria | 14.9 | 15.6 | 19.4 | 15.5 |
| Class Actinobacteridae | 85.7 | 73.3 | 55.0 | 56.3 |
| Class Acidimicrobidae | 14.3 | —b | 5.0 | — |
| Class Rubrobacteridae | — | 26.7 | 35.0 | 43.7 |
| Class candidatus Microthrix | — | — | 5.0 | — |
| Acidobacteria | 14.9 | 11.5 | 18.4 | 12.6 |
| Proteobacteria | 27.6 | 30.3 | 28.1 | 29.2 |
| Class Alphaproteobacteria | 23.1 | 34.5 | 48.3 | 36.7 |
| Class Betaproteobacteria | 7.7 | 3.5 | 3.5 | 3.3 |
| Class Gammaproteobacteria | 65.4 | 24.1 | 31.0 | 43.3 |
| Class Deltaproteobacteria | 3.8 | 37.9 | 17.2 | 16.7 |
| Verrucomicrobia | 5.3 | 7.4 | 1.0 | — |
| Chloroflexi | 1.0 | 3.1 | 5.8 | 2.9 |
| Gemmatimonadetes | 3.2 | 3.1 | 1.0 | — |
| Chlamydiae | — | 1.0 | — | 2.9 |
| Chlorobi | — | 1.0 | 1.9 | — |
| Bacteroidetes | — | 2.1 | 1.0 | — |
| Candidate division TM7 | — | — | — | 1.9 |
| Cyanobacteria | — | 1.0 | — | — |
| Ferribacter | — | — | 1.0 | — |
| Deinococcus | — | — | — | 1.0 |
| PN-1 | 2.2 | — | — | — |
| PN-2 | — | 2.1 | — | — |
| PN-3 | — | 1.0 | — | — |
| PN-4 | — | 1.0 | — | — |
| PN-5 | — | 1.0 | — | 1.0 |
| PN-6 | — | — | 1.0 | — |
| PN-7 | — | — | — | 1.0 |
Based on the frequencies of occurrence in the 16S rRNA gene clone libraries (1401R-1a, n = 94; 1401R-2a, n = 103; 1401R-1b, n = 96; 1401R-2b, n = 103) (main entries) or on those within the indicated phylum (subentries).
—, not detected.
With all of the primers, rather low numbers of clones were found in the phyla Chlorobi, Bacteroidetes, Cyanobacteria, Chlamydiae, Ferribacter, Deinococcus, and candidate division TM7. Specifically, primer R1401-1b only detected members of Chlorobi, Bacteroidetes, Cyanobacteria, and Chlamydiae whereas primer R1401-2a had representatives in the phyla Chlorobi and Bacteroidetes and one representative in the phylum Ferribacter. Finally, Chlamydiae were also represented in the library produced by primer R1401-2b but this primer also yielded clones in the phyla Deinococcus and candidate division TM7.
Sequences with low matches to the database.
Some clones, loosely affiliated with the phyla Chlorobi and candidate division TM7, had lower than 90% matches with EMBL database entries and, when considering the phylogenetic tree, might define novel groups. All of the clones with lower than 90% matches to the database were also compared to the RDP database. This analysis confirmed that the clones affiliated with the phyla Chlorobi and candidate division TM7 actually belonged to these two groups and not to a group outside of these phyla. Finally, 10 clones (produced with any of the four reverse primers) had no specific association with any of the known divisions or candidate divisions. Hence, these were phylogenetically divided into seven putatively novel (PN) division level groups named PN-1 through PN-7. Group PN-1, which was represented by two clones (2.2%), was found in the library produced with primer R1401-1a. Groups PN-2, PN-3, and PN-4 (primer R1401-1b) represented, respectively, two clones (2.1%), one clone (1%), and one clone (1%). Both primers R1401-1b and R1401-2b yielded one representative (1%) of group PN-5, and primer R1401-2b produced one clone (1%) that represented group PN-7. Group PN-6, represented by one clone (1%), was found in the library created with R1401-2a.
DISCUSSION
The objective of this study was to compare different reverse primers used in the region V6-based bacterial PCR-DGGE fingerprinting system (22) with respect to their breadth of amplification of sequences from the bacterial communities in soil under grass. We thus compared the DGGE patterns produced by a suite of 13 different reverse primers in conjunction with a canonical forward primer by cluster analysis. Subsequently, we also analyzed the partial rRNA gene sequences of clones in libraries generated with four selected reverse primers. Overall, the DGGE and clone library-based data both quite consistently indicated that different parts of the bacterial community in a single soil sample were accessed by different reverse primers. This contention is worked out in detail below.
It has been shown before that bacterial community fingerprints obtained from soil are, to a great extent, reproducible, and so the levels of experimental variation due to methodological variation are often minimal (11). In this study, this high reproducibility of the PCR-DGGE method within methodological replicates was confirmed, providing the background for subsequent analyses in which the patterns generated with 13 primers (Table 1) were compared side by side on a single gel. Analysis of these patterns revealed the existence of two main clusters which grouped on the basis of the G/A alteration at position 14 of the reverse primers used. The existence of the two clusters was confirmed by excluding the reverse primers which carry an R (A or G) at position 14 of their sequences (R1401-1c, -2c, -3c, and -2*c) (Fig. 1A). Hence, the G-to-A switch at position 14 had a dramatic influence on the DGGE profiles obtained from the same soil sample. This was corroborated by the clear differences in the theoretical match percentages of the different primers with database sequences. In contrast, other changes in the reverse primers, such as the GGGAACG addition at the 3′ end and the GC/CG alteration at the 5′ end, had lesser effects. Statistical analyses of the DGGE profiles obtained showed that primer R1401-1a yielded patterns with the highest diversity, followed by primers R1401-1b, -2a, and -2b.
To further resolve the issue of reverse primer specificity effects, we produced clone libraries with primers R1401-1a, -1b, -2a, and -2b, which were examined with respect to the nature of the amplified sequences. First, rarefaction analysis indicated considerable incompleteness of sampling, an expected finding given the high bacterial diversity in most soils, including grassland (2, 5, 9). The difference in the rate of “exhaustion” of soil bacterial diversity between the different primers, notably, R1401-1b versus R1401-2b, was interesting, as it indicated that, indeed, different parts of the same soil bacterial community were being accessed. To further theoretically underpin these data, we analyzed the degree to which the four primers matched clones of the 139,340-member Waseca County farm soil metagenome available via the web. Primers R1401-1a, -1b, -2a, and -2b matched sequences present in seven, nine, two, and two clones in the library, respectively. These data are grossly consistent with an expected frequency of occurrence of the 16S rRNA operon in a soil DNA library of 1/104 to 105 clones but do not shed light on the exact prevalence because of statistical uncertainties.
Moreover, the analysis of the clone libraries by LIBSHUFF in several cases indicated a clear effect of the reverse primer type on the composition of the clone libraries, and hence we could confirm the initial observation—obtained by DGGE—that different parts of the bacterial community present in the same soil are targeted by different reverse primers, sometimes differing by only a single nucleotide.
However, the analysis of the different bacterial sequence types produced by the different primers, with a few exceptions, did not reveal major differences in the observed frequencies of specific sequence types (at >97% similarity). We attribute this finding to the diversity that is found within many bacterial taxa, allowing the amplification of apparently similar taxa with (slightly) different bacterial primers. An alternative explanation for this finding might lie in the nature of the primer-to-target annealing process, which is known to be determined, to a large extent, by the 3′-end sequence but may allow the production of amplicons resulting from sloppy annealing.
Overall and quite expectedly, the analysis of the sequences produced in the four libraries revealed the amplification, to different extents, of members of 14 different bacterial phyla from soil, namely, Firmicutes, Proteobacteria, Actinobacteria, Acidobacteria, Verrucomicrobia, Chloroflexi, Gemmatimonadetes, Chlorobi, Bacteroidetes, Cyanobacteria, Chlamydiae, Ferribacter, Deinococcus, and candidate division TM7, the dominant phyla being Firmicutes and Proteobacteria. The differences observed between the amplification rates of the different primers (Fig. 4) clearly indicate different annealing preferences. However, the finding of the 14 major groups is, overall, fairly consistent with data obtained in a range of other studies of different soils, and most of these phyla are now recognized as forming the numerically dominant parts of the bacterial communities in a majority of soils (36). For instance, Mummey et al. (20) recently found that members of Gemmatimonadetes, Actinobacteria (subdivision Rubrobacteridae), Acidobacteria, and the alpha subgroup of Proteobacteria were among the dominant members of the bacterial communities in two soils. In our study, the majority of the clones matched members of recognized bacterial divisions; however, a small fraction of the clones revealed the existence in the grassland soil of seven potential novel candidate divisions, PN-1 through PN-7, found to different extents in the libraries produced with all of the primers. This finding indicates that, in spite of the finding of a large fraction of sequences that match database entries with taxonomic status, our understanding of the extent of microbial diversity in soil is still incomplete, even at deeper phylogenetic levels. Moreover, even slightly different amplification systems may yield different phylogenetic novelty. However, given the scarcity of the current data (only a few clones found per putatively novel group), a more precise definition of the PN groups should await the production of more sequence information from other studies.
Although the data obtained from the clone libraries and the DGGE patterns indicated clear differences in the amplified bacterial communities, there was not an obvious connecting point between the data. On the basis of the matching diversity found within the RDP database (Table 1), the theoretically “best” reverse primers for use in amplifications would be R1401-1a or -2a, followed by R1401-1b and -2b. However, the numbers of matching RDP database entries do not necessarily represent the bacterial diversity typical for soil. Thus, the database does not (yet) contain categories of organisms such as the class “Spartobacteria” within Verrucomicrobia, the order “Solibacterales” within the phylum Acidobacteria, and the phylum Fibrobacteres. In our study, theoretically “suboptimal” primer R1401-1b gave high diversity indices on the basis of both DGGE patterns and clone libraries. Specifically, this primer yielded, among 100 clones, the highest numbers of different phyla (11) and novel candidate divisions (4) within the domain Bacteria. Thus, the G-to-A alteration at position 14 of the primer apparently strongly influenced the patterns generated, allowing primer R1401-1b to give high indices in both DGGE and clone libraries.
Considering the foregoing, we conclude that the present study, on the basis of the comparison of existing and newly designed primers, revealed a major effect of the sequence of the reverse primer used on the “apparent” (observed) soil bacterial diversity and community composition. Where the different primers thus provide different angles at the extant bacterial community in soil, if a single primer would have to be recommended on the basis of the observed levels of diversity, this might be R1401-1b. However, the concomitant use of primers R1401-1a and -1b might offer the best view of the diversity of bacterial communities in soil.
Acknowledgments
We thank Paolina Garbeva (University of Wageningen) for providing soil samples. Wouter Wanders (University of Nijmegen) is thanked for preliminary work on the DGGE reverse primers. Anouk Piquet (University of Groningen) is thanked for optimizing the Phor-U DGGE system. We thank Rodrigo Costa (University of Groningen) for support with GelCompar.
Footnotes
Published ahead of print on 29 February 2008.
REFERENCES
- 1.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burr, M. D., S. J. Clark, C. R. Spear, and A. K. Camper. 2006. Denaturing gradient gel electrophoresis can rapidly display the bacterial diversity contained in 16S rDNA clone libraries. Microb. Ecol. 51:479-486. [DOI] [PubMed] [Google Scholar]
- 3.Chao, A. 1984. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 11:265-270. [Google Scholar]
- 4.Cole, J. R., B. Chai, R. J. Farris, Q. Wang, S. A. Kulam, D. M. McGarrell, G. M. Garrity, and J. M. Tiedje. 2005. The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res. 33:D294-D296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunbar, J., S. M. Barns, L. O. Ticknor, and C. R. Kuske. 2002. Empirical and theoretical bacterial diversity in four Arizona soils. Appl. Environ. Microbiol. 68:3035-3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Felske, A., H. Rheims, A. Wolterink, E. Stackebrandt, and A. D. L. Akkermans. 1997. Ribosome analysis reveals prominent activity of an uncultured member of the class Actinobacteria in grassland soils. Microbiology 143:2983-2989. [DOI] [PubMed] [Google Scholar]
- 7.Fischer, S. G., and L. S. Lerman. 1979. Length-independent separation of DNA restriction fragments in two-dimensional gel electrophoresis. Cell 16:191-200. [DOI] [PubMed] [Google Scholar]
- 8.Fischer, S. G., and L. S. Lerman. 1983. DNA fragments differing by single base-pair substitutions are separated in denaturing gradient gels: correspondence with melting theory. Proc. Natl. Acad. Sci. USA 80:1579-1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garbeva, P. 2006. The significance of microbial diversity in agricultural soil for disease suppressiveness. Ph.D. thesis. University of Leiden, Leiden, The Netherlands.
- 10.Garbeva, P., J. A. van Veen, and J. D. van Elsas. 2003. Predominant Bacillus spp. in agricultural soil under different management regimes detected via PCR-DGGE. Microb. Ecol. 45:302-316. [DOI] [PubMed] [Google Scholar]
- 11.Gelsomino, A., A. C. Keijzer-Wolters, G. Cacco, and J. D. van Elsas. 1999. Assessment of bacterial community structure in soil by polymerase chain reaction and denaturing gradient gel electrophoresis. J. Microbiol. Methods 38:1-15. [DOI] [PubMed] [Google Scholar]
- 12.Heuer, H., M. Krsek, P. Baker, K. Smalla, and E. M. H. Wellington. 1997. Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl. Environ. Microbiol. 63:3233-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huber, T., G. Faulkner, and P. Hugenholtz. 2004. Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20:2317-2319. [DOI] [PubMed] [Google Scholar]
- 14.Hughes, J. B., J. J. Hellmann, T. H. Ricketts, and B. J. Bohannan. 2001. Counting the uncountable: statistical approaches to estimating microbial diversity. Appl. Environ. Microbiol. 67:4399-4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kowalchuk, G. A., Z. S. Naoumenko, P. J. Derikx, A. Felske, J. R. Stephen, and I. A. Arkhipchenko. 1999. Molecular analysis of ammonia-oxidizing bacteria of the beta subdivision of the class Proteobacteria in compost and composted materials. Appl. Environ. Microbiol. 65:396-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozdrój, J., and J. D. van Elsas. 2000. Application of polymerase chain reaction-denaturing gradient gel electrophoresis for comparison of direct and indirect extraction methods of soil DNA used for microbial community fingerprinting. Biol. Fertil. Soils 31:372-378. [Google Scholar]
- 17.Kropf, S., H. Heuer, M. Gruning, and K. Smalla. 2004. Significance test for comparing complex microbial community fingerprints using pairwise similarity measures. J. Microbiol. Methods 57:187-195. [DOI] [PubMed] [Google Scholar]
- 18.Kumar, S., K. Tamura, and M. Nei. 2004. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform. 5:150-163. [DOI] [PubMed] [Google Scholar]
- 19.Mullis, K. B. 1990. The unusual origin of the polymerase chain reaction. Sci. Am. 262:56-61, 64-65. [DOI] [PubMed] [Google Scholar]
- 20.Mummey, D., W. Holben, J. Six, and P. Stahl. 2006. Spatial stratification of soil bacterial populations in aggregates of diverse soils. Microb. Ecol. 51:404-411. [DOI] [PubMed] [Google Scholar]
- 21.Muyzer, G., E. C. de Waal, and A. G. Uitterlinden. 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59:695-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muyzer, G., and K. Smalla. 1998. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie van Leeuwenhoek 73:127-141. [DOI] [PubMed] [Google Scholar]
- 23.Myers, R. M., S. G. Fischer, L. S. Lerman, and T. Maniatis. 1985. Nearly all single base substitutions in DNA fragments joined to a GC-clamp can be detected by denaturing gradient gel electrophoresis. Nucleic Acids Res. 13:3131-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Myers, R. M., T. Maniatis, and L. S. Lerman. 1987. Detection and localization of single base changes by denaturing gradient gel electrophoresis. Methods Enzymol. 155:501-527. [DOI] [PubMed] [Google Scholar]
- 25.Nübel, U., B. Engelen, A. Felske, J. Snaidr, A. Wieshuber, R. I. Amann, W. Ludwig, and H. Backhaus. 1996. Sequence heterogeneities of genes encoding 16S rRNAs in Paenibacillus polymyxa detected by temperature gradient gel electrophoresis. J. Bacteriol. 178:5636-5643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Postma, J., J. E. I. M. Willemsen-de Klein, and J. D. van Elsas. 1999. Effect of the indigenous microflora on the development of root and crown rot caused by Pythium aphanidermatum in cucumber grown on rockwool. Phytopathology 90:125-133. [DOI] [PubMed] [Google Scholar]
- 27.Rademaker, J. L. W., F. J. Louws, U. Rossbach, P. Vinuesa, and F. J. de Bruijn. 1999. Computer-assisted pattern analysis of molecular fingerprints and database construction, p. 1-33. In A. D. L. Akkermans, J. D. van Elsas, and F. J. de Bruijn (ed.), Molecular microbial ecology manual. Kluwer Academic Publishers, Dordrecht, The Netherlands.
- 28.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 29.Schloss, P. D., and J. Handelsman. 2005. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 71:1501-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schloss, P. D., B. R. Larget, and J. Handelsman. 2004. Integration of microbial ecology and statistics: a test to compare gene libraries. Appl. Environ. Microbiol. 70:5485-5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shannon, C. E. 1997. The mathematical theory of communication. MD Comput. 14:306-317. [Reprint from 1943.] [PubMed] [Google Scholar]
- 32.Sheffield, V. C., D. R. Cox, L. S. Lerman, and R. M. Myers. 1989. Attachment of a 40-base-pair G+C-rich sequence (GC-clamp) to genomic DNA fragments by the polymerase chain reaction results in improved detection of single-base changes. Proc. Natl. Acad. Sci. USA 86:232-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singleton, D. R., M. A. Furlong, S. L. Rathbun, and W. B. Whitman. 2001. Quantitative comparisons of 16S rRNA gene sequence libraries from environmental samples. Appl. Environ. Microbiol. 67:4374-4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smalla, K., U. Wachtendorf, H. Heuer, W. T. Liu, and L. Forney. 1998. Analysis of BIOLOG GN substrate utilization patterns by microbial communities. Appl. Environ. Microbiol. 64:1220-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sørensen, J., and A. Sessitsch. 2006. Plant-associated bacteria—lifestyle and molecular interactions, p. 211-236. In J. D. van Elsas, J. K. Jansson, J. T. Trevors (ed.), Modern soil microbiology II. CRC Press, Boca Raton, FL.
- 36.van Elsas, J. D., V. Torsvik, A. Hartmann, L. Øureås, and J. K. Jansson. 2006. The bacteria and archaea in soil, p. 83-106. In J. D. van Elsas, J. K. Jansson, and J. T. Trevors (ed.), Modern soil microbiology II. CRC Press, Boca Raton, FL.
- 37.van Veen, J. A., L. S. van Overbeek, and J. D. van Elsas. 1997. Fate and activity of microorganisms introduced into soil. Microbiol. Mol. Biol. Rev. 61:121-135. [DOI] [PMC free article] [PubMed] [Google Scholar]