

Abstract
A molecular method, termed hierarchical oligonucleotide primer extension (HOPE), was used to determine the relative abundances of predominant Bacteroides spp. present in fecal microbiota and wastewaters. For this analysis, genomic DNA in feces of healthy human adults, bovines, and swine and in wastewaters was extracted and total bacterial 16S rRNA genes were PCR amplified and used as the DNA templates for HOPE. Nineteen oligonucleotide primers were designed to detect 14 Bacteroides spp. at different hierarchical levels (domain, order, cluster, and species) and were arranged into and used in six multiplex HOPE reaction mixtures. Results showed that species like B. vulgatus, B. thetaiotaomicron, B. caccae, B. uniformis, B. fragilis, B. eggerthii, and B. massiliensis could be individually detected in human feces at abundances corresponding to as little as 0.1% of PCR-amplified 16S rRNA genes. Minor species like B. pyogenes, B. salyersiae, and B. nordii were detected only collectively using a primer that targeted the B. fragilis subgroup (corresponding to ∼0.2% of PCR-amplified 16S rRNA genes). Furthermore, Bac303-related targets (i.e., most Bacteroidales) were observed to account for 28 to 44% of PCR-amplified 16S rRNA genes from human fecal microbiota, and their abundances were higher than those detected in the bovine and swine fecal microbiota and in wastewaters by factors of five and two, respectively. These results were comparable to those obtained by quantitative PCR and to those reported previously from studies using whole-cell fluorescence hybridization and 16S rRNA clone library methods, supporting the conclusion that HOPE can be a sensitive, specific, and rapid method to determine the relative abundances of Bacteroides spp. predominant in fecal samples.
Approximately 1014 microbial cells reside in the human intestine and form close symbiotic associations with the host (18). Members of the genera Bacteroides and Parabacteroides in the order Bacteroidales are known to make up 14 to 40% of cultivable microorganisms in human feces (21). Species including Bacteroides caccae, B. fragilis, B. eggerthii, B. uniformis, B. thetaiotaomicron, B. vulgatus, B. nordii, B. salyersiae, B. coprocola, B. massiliensis, B. intestinalis, Parabacteroides distasonis, P. merdae, and P. goldsteinii have been successfully isolated from the human gut or human feces (3, 12, 23, 26, 38) and constitute up to 80% of the fecal bacterial isolates from the genus Bacteroides documented in Ribosomal Database Project II (http://rdp.cme.msu.edu).
Due to their high prevalence in the gut and fecal microbiota, Bacteroides spp. and Parabacteroides spp. have been considered to be clinically important anaerobes that are related to gastrointestinal well-being (13). For example, species like B. vulgatus and B. caccae can colonize the surface of the intestinal mucosa and inhibit the adherence of enteroinvasive pathogens (8, 18). Others like B. thetaiotaomicron and B. ovatus can break down a wide variety of indigestible polysaccharides to supply the host with 10 to 15% of the daily metabolic needs (35, 42). Besides relating to gastrointestinal functioning, certain Bacteroides spp. and Parabacteroides spp. are also suspected to be human gut specific and can act as biomarkers for identifying fecal pollutants from human and nonhuman sources (9, 27, 29). For instance, P. distasonis, B. fragilis, B. thetaiotaomicron, and B. vulgatus are reported to be 100- to 1,000-fold more abundant in human feces than in animal feces (27), while species in uncultivated animal-specific Bacteroidales clusters (Fig. 1) are seldom detected in human feces. Given the importance of Bacteroides spp. and Parabacteroides spp., determining the abundances of all or specific strains in the microbiota has become important for establishing a correlation to the health status of the host and for performing microbial source tracking.
FIG. 1.

Phylogenetic representation of different cultivated and uncultivated Bacteroides spp. In this study, a total of 19 primers were designed to target 14 functionally important Bacteroides spp. and Parabacteroides spp. at various taxonomical levels. The associated coverage of each primer is indicated on the right. Primers were arranged into six reaction tubes, with those targeting at the species level distributed across reaction tubes 1 to 5. Within reaction tubes 1 to 5, an associated higher-level primer, usually targeting not beyond the family level, was placed alongside. In the last reaction tube, all higher-level primers that were used in reaction tubes 1 to 5 (i.e., Bth274 and Bfrg602) were placed along with primers targeting at even higher taxonomical levels, like Bac303 and U1390.
At present, qualitative and quantitative analyses of Bacteroidales in feces-related samples are achieved mainly through cell cultivation and the use of conventional molecular tools (2, 4, 10, 19, 21, 33). Both approaches, however, have their strengths and weaknesses. The former can be time-consuming and favor the growth of selective species (40). The latter methods (e.g., the use of whole-cell fluorescence in situ hybridization [FISH], quantitative PCR [Q-PCR], and 16S rRNA gene clone libraries) can circumvent the issues related to cultivability but may also present other technical challenges. For example, FISH can be affected by the in situ accessibility of labeled probes through the cell membranes and the copies of rRNA inside inactive cells (7, 17). Clone library construction, with its resolution dependent on the total number of clones selected for analysis, can be time-consuming (15). Q-PCR can provide excellent quantitative detection sensitivity (down to a few copies of target fragments) but requires standard curves to be established for individual targets (24). Therefore, there is still a need to develop a direct and simple-to-use technique which can complement the existing molecular methods to rapidly evaluate the relative abundances of Bacteroides spp. and Parabacteroides spp. present in complex microbial communities.
Recently, a molecular method termed hierarchical oligonucleotide primer extension (HOPE) has been developed to measure the relative abundances of multiple bacterial 16S rRNA genes among a total set of PCR-amplified 16S rRNA genes (41). In this method, multiple oligonucleotide primers that are designed with different lengths by the addition of poly(A) tails are used to target specific 16S rRNA gene sequences at different taxonomical levels (i.e., domain, phylum, order, and so on down to species). During the primer extension reaction, these primers anneal to their complementary target sequences and are subsequently extended with a fluorescently labeled dideoxynucleoside triphosphate (ddNTP) by DNA polymerase. The extended primers are identified by a DNA sequencer based on their fragment sizes and the type of the extended fluorescent ddNTP. Based on the fluorescence intensity detected, peak areas for extended specific primers are then determined, and the ratios of the areas to that for a common reference primer are calculated and used to determine the relative abundances of the bacterial targets of interest (Fig. 2).
FIG. 2.

Schematic illustration of HOPE. PCR amplicons, fluorophore-labeled ddNTPs, DNA polymerase, and oligonucleotide primers were mixed together to form a HOPE reaction mixture. The mixture underwent a thermal program involving 20 to 25 cycles of denaturation (96°C), annealing (60 to 64°C), and single-base extension (72°C). The products were purified and the excess ddNTP residue was removed by shrimp alkaline phosphatase (SAP) digestion. An appropriate amount of the product mixture was mixed with synthetic oligonucleotide standards and then subjected to capillary electrophoresis. From the electropherograms, the extended primers were recognized by means of consistent fragment sizes and the colors of the extended ddNTPs. PA to PD, primers A to D.
In a previous study, a 10-plex HOPE reaction protocol targeting Bacteroides spp. at domain, group, and species levels was developed and applied to detect the relative abundances of Bacteroides spp. in the influent and effluent of a domestic wastewater treatment plant (41). It was successfully shown that HOPE could achieve single-mismatch discrimination and had the sensitivity to detect targets accounting for as little as 0.01 to 0.05% of the total set of PCR-amplified 16S rRNA genes (41). These promising results have further encouraged us to develop HOPE as a robust method to rapidly detect and measure the relative abundances of a total of 14 functionally important Bacteroides and Parabacteroides spp. at species, group, and domain levels in feces and wastewaters. To do so, we have added 12 primers specific for species in the genera Bacteroides and Parabacteroides (Fig. 1 and Table 1). To further validate the ability of HOPE to determine the relative abundances of bacterial targets, we have compared the HOPE results with results from conventional molecular methods like Q-PCR.
TABLE 1.
HOPE primers designed to target human-specific Bacteroides spp. at various hierarchical levels (species, group, order, and domain levels)
| Primer | Target | Sequence (5′-3′) | Length of poly(A) tail (nt) | Type of ddNTP added | HOPE reaction tube no. | Reference |
|---|---|---|---|---|---|---|
| Bnd136 | B. nordii (JCM12987) | AGC CTA TCC CCG AGT AAA A | 0 | G | 1 | This study |
| Bsls1016 | B. salyersiae (JCM12988) | GCC TTG CGG CTA TGC CG | 10 | T | 1 | This study |
| Pg_dtc732 | B. pyogenes (JCM6294) | GTT CCG GCC CGG TGA GCT | 20 | G | 1 | This study |
| Bth584 | B. thetaiotaomicron (BCRC10624) | CAA CTG ACT TAA CTG TCC AC | 16 | C | 1 | 41 |
| Bfrg1026 | B. fragilis (BCRC10619) | TCA CAG CGG TGA TTG CTC | 20 | A | 2 | This study |
| Bcc1066 | B. caccae (JCM9498) | CGT ATG GGT TTC CCC ATA A | 15 | T | 3 | This study |
| Badf1009 | B. acidifaciens (JCM10556) | CGG CTA ACA TGT TTC CAC | 0 | A | 3 | This study |
| Bitt141 | B. intestinalis (JCM13266) | CGA AAG GCT ATC CCG GAA | 0 | T | 3 | 41 |
| Begt999 | B. eggerthii (JCM12986) | GTT TCC ACT ACA TTC CGC | 0 | T | 4 | This study |
| Bhcg171 | B. helcogenes (JCM6297) | TTT CAG TGC CAT CGG GCA T | 8 | T | 4 | This study |
| Bufm1018 | B. uniformis (JCM5828) | CTG CCT TGC GGC TGA CA | 20 | T | 4 | 41 |
| Bvg1016 | B. vulgatus (BCRC12903) | ATG CCT TGC GGC TTA CGG C | 0 | T | 5 | This study |
| Bcpc1015 | B. coprocola (JCM12979) | CGC CTT GCG GCT TAC AAG T | 24 | T | 5 | This study |
| Bmsl1000 | B. massiliensis (JCM12982) | GCG TTT CCG CCA TAT TCG G | 19 | T | 5 | This study |
| Bdts_gp980 | P. distasonis (JCM5825) | CGT TCA AAC CCG GGT AA | 12 | G | 6 | This study |
| P. merdae (JCM9497) | ||||||
| P. goldsteinii (JCM13446) | ||||||
| Bth274 | B. fragilis subgroups | CCC CTA TCC ATC GAA GG | 15 | C or T | 6, 2, 1 | 41 |
| Bfrg602 | B. fragilis cluster | GAG CCG CAA ACT TTC ACA A | 19 | C | 6, 5, 4, 3, 2 | 19 |
| Bac303 | Most Bacteroidales | CCA ATG TGG GGG ACC TT | 5 | C | 6 | 31 |
| U1390 | Most Bacteria | YGA CGG GCG GTG TGT | 17 | A | 6 | 43 |
MATERIALS AND METHODS
Bacterial strains and environmental samples.
Nineteen bacterial reference strains obtained from the Japan Collection of Microorganisms (Wako, Japan) or the Bioresources Collection and Research Centre (Hsinchu, Taiwan) were used (Table 1). These bacterial strains are classified into the B. fragilis group (i.e., B. uniformis, B. helcogenes, B. stercoris, B. caccae, B. acidifaciens, B. fragilis, B. thetaiotaomicron, B. vulgatus, B. coprocola, B. massiliensis, B. eggerthii, B. nordii, B. salyersiae, B. intestinalis, and B. ovatus) and the P. distasonis group (i.e., P. distasonis, P. merdae, and P. goldsteinii). Within the B. fragilis subgroup, bacterial strains can be further classified into the subcluster for which primer Bth274 extended with nucleotide T (the Bth274 [T]-related subcluster, i.e., B. pyogenes, B. tectus, B. nordii, B. salyersiae, and B. thetaiotaomicron) and the subcluster for which primer Bth274 extended with nucleotide C (the Bth274 [C]-related subcluster, i.e., B. uniformis and B. fragilis). Fecal samples were collected from (i) three healthy donors aged 26 to 32 years old, (ii) three healthy pigs, and (iii) three healthy cows. Environmental samples were collected from the influents of a municipal treatment plant located in Singapore and a swine wastewater treatment plant in Tainan, Taiwan. Samples were collected and kept at −20°C prior to DNA extraction.
DNA extraction.
Total DNA from pure cultures and influent samples was extracted according to a previously described protocol with minor modifications (36). Total DNA from fecal samples was extracted using a QIAamp DNA stool mini kit (Qiagen) (32).
PCR amplification of 16S rRNA genes.
Each PCR mixture (50 μl in volume) contained 50 to 100 ng of genomic DNA in 1× TaKaRa Ex Taq buffer, 200 nM (each) forward primer (11F; 5′-GTT TGA TCC TGG CTC AG-3′ [25]) and reverse primer (1512R; 5′-GGC TAC CTT GTT ACG ACT T-3′ [28]), 200 mM deoxynucleoside triphosphate, and 0.5 U of Ex Taq DNA polymerase (TaKaRa). The optimal number of thermal cycles required to obtain a proportional amplification of the DNA of the microbial community was examined by varying the cycle number of the thermal program (denaturation, 95°C for 30 s; annealing, 55°C for 45 s; and extension, 72°C for 60 s) from 10 to 35 cycles at an increment of 5 cycles. Amplicons obtained at each cycle were concentrated and purified using a QIAquick PCR purification kit (Qiagen), and the concentrations were determined by UV absorbance measurement using a DU730 spectrophotometer (Beckman Coulter).
Cloning and sequencing.
To ensure culture purity, 16S rRNA genes of reference bacterial strains were separately cloned into the pCRII vector by using the TA cloning kit (Invitrogen Corporation, Carlsbad, CA). For each clone library, 20 colonies were selected, and the presence of DNA insertion was confirmed by direct PCR amplification with M13R and M13F primers. The inserted DNA fragment was then sequenced using an ABI PRISM 3130 genetic analyzer (Applied Biosystems) and a BigDye Terminator sequencing kit (Applied Biosystems). The sequences were compared against the entries in the 16S rRNA gene database by using BLAST software (1).
Species-specific primer design.
A total of 12 new primers specifically targeting different human-specific Bacteroides spp. were designed using the probe design function of ARB software (30). An updated ssu_jan04.arb database (http://www.arb-home.de), which contained 28,289 nearly complete 16S rRNA sequences (with lengths of >1,450 nucleotides [nt]), was used together with 189 aligned sequences of cultivated Bacteroides spp. obtained from Ribosomal Database Project II and nearly complete 16S rRNA sequences of the 19 reference bacterial strains. To enhance the specificity of HOPE primers, each primer was designed with mismatch bases corresponding to nontargets located at the 3′ terminus. The specificity of the designed primers was verified first in silico against entries in the Ribosomal Database Project II database and subsequently in HOPE reactions against DNA from all reference strains to ensure that primers did not extend when DNA from nontarget bacterial strains was used as the DNA template.
Hierarchical primer arrangement.
Table 1 lists the names, sequences, and specificities of the 19 different HOPE primers used in this study. These primers were assigned to six different multiplex HOPE reaction mixtures based on the phylogenetic affiliations of the bacterial targets (Table 1 and Fig. 1). Reaction mixtures 1 to 5 contained one of two higher-level primers that targeted both the B. fragilis group and its subgroups (i.e., primers Bfrg602 and Bth274), together with 14 species-specific primers. Thus, the abundance of the individual bacterial target targeted by each species-specific primer relative to those of targets targeted by higher-level primers could be determined. With reaction mixture 6, the relative abundances of Bacteroidales, the B. fragilis group and its subgroups, and the Parabacteroides group as represented by Bac303, Bfrg602, Bth274, and Bdts_gp980 relative to the total community as represented by all the 16S rRNA gene amplicons could be determined. To clearly differentiate extended primers in the same HOPE reaction mixture, primers that extended with the same ddNTP were further modified with poly(A) tails of different lengths (5 to 24 nt) at the 5′ ends.
Internal standard oligonucleotide mixture.
The internal standard oligonucleotide mixture contained four oligonucleotides of different lengths [5′-(GT)x-3′, where x equals 18, 20, 22, and 24], which were synthesized and labeled at the 5′ ends with four different fluorophores (i.e., dR110, dR6G, deoxy-6-carboxytetramethylrhodamine [dTAMRA], and deoxy-6-carboxy-X-rhodamine [dROX]; Applied Biosystems). The concentrations of individual oligonucleotides at 260 nm were measured and subsequently diluted to 3 nM for 5′-dR110-(GT)20-3′, 15 nM for 5′-dTAMRA-(GT)22-3′, 18 nM for 5′-dROX-(GT)24-3′, and 5 nM for 5′-dR6G-(GT)18-3′.
HOPE reactions.
Unless stated otherwise, each HOPE reaction mixture (in total, 5 μl) contained 2.5 μl of SNaPshot premix, 5 to 30 fmol of the DNA template, 10 to 60 pmol of oligonucleotide primers (Sigma-Proligo, Singapore, and Mission Biotech, Taiwan), and various amounts of deionized water. The SNaPshot premix consisted of DNA polymerase, ionic buffer, and fluorescently labeled dideoxynucleotides (dROX-ddTTP, dTAMRA-ddCTP, dR110-ddGTP, and dR6G-ddATP). The HOPE thermal program consisted of 20 cycles of denaturation (96°C for 10 s), annealing (64°C for 30 s), and extension (72°C for 15 s). After the primer extension reaction, 1 U of shrimp alkaline phosphatase (Roche Applied Science, Penzberg, Germany) was added and the mixture was incubated at 37°C for 60 min in order to remove unincorporated dye terminators. The reaction was terminated by heating at 75°C for 10 min.
Capillary electrophoresis.
HOPE products (0.5 to 1 μl) were mixed with 0.125 μl of GeneScan Liz 120 standard (Applied Biosystems), 0.25 μl of the standard oligonucleotide mixture, and 12 μl of Hi-Di formamide (Applied Biosystems). The electrophoresis program included a denaturation step (60°C), an injection step with an applied voltage of 1.0 to 2.1 kV for 12 to 40 s, and a separation step. Fluorescence data were subsequently analyzed by the fragment analysis software GeneMapper, in which the fragment sizes and peak areas for the extended primers and internal standard oligonucleotides were recorded.
Calculation of relative abundances of HOPE products.
The concentrations of extended primers were quantified as follows:
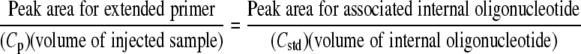 |
(1) |
where Cp represents the molar concentration of a specific primer extended in the HOPE reactions for the samples and Cstd represents the molar concentration of the associated standard oligonucleotide.
Calibration factors (CFs) for each specific primer with respect to higher-level primers could be obtained using the M13 amplicons from associated reference strains as templates and calculating as follows:
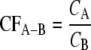 |
(2) |
where primer B targets a higher hierarchical level than primer A, CFA−B is the calibration factor for primer A with respect to primer B, CA is the concentration of extended primer A, and CB is the concentration of extended primer B. The relative abundances of 16S rRNA gene amplicons targeted by primer A with respect to those of amplicons targeted by primer B can then be calculated as follows:
 |
(3) |
Quantification of Bacteroides spp. with Q-PCR.
Standard curves used for the detection of Bacteroides spp. at different taxonomical levels were obtained using replicates with 400 nM of forward and reverse primers (Table 2), 1× iQ SYBR green mastermix (Bio-Rad), and various concentrations (1 to 10,000 pg per 25 μl of reaction mixture) of DNA from two independent reference strains (B. fragilis BCRC10619 and B. uniformis JCM5825). Forty cycles of the thermal program (denaturation at 96°C for 10 s, annealing at 64°C for 30 s, and extension at 72°C for 30 s) were performed with iCycler (Bio-Rad). Standard curves were generated by plotting the concentration of the genomic DNA template against mean numbers of threshold cycles. Similarly, real-time quantitative measurement of Bacteroides spp. present in human fecal samples was subsequently performed using replicates with 1,000 pg of genomic DNA.
TABLE 2.
Primers targeting Bacteroides spp. at various taxonomical levels that were used in Q-PCR to validate HOPE resultsa
| Target | Primer | Primer sequence (5′-3′) | Standard curve | Amplification efficiency (%) |
|---|---|---|---|---|
| Total Bacteria | 1390F | ACA CAC CGC CCG TCR | y = −3.309x + 26.069 | 100.5 |
| 1512R | GGC TAC CTT GTT ACG ACT T | |||
| Bacteroides-Prevotella | Bac303F | AAG GTC CCC CAC ATT GG | y = −3.306x + 24.345 | 100.7 |
| 530R | CCG CGG CKG CTG GCA C | |||
| B. fragilis cluster | Bfrg602F | TTG TGA AAG TTT GCG GCT C | y = −3.4043x + 25.999 | 96.7 |
| 797R | GGA CTA CCA GGG TAT CTA ATC CTG TT | |||
| P. distasonis cluster | Bdts_gp980F | TTA CCC GGG TTT GAA CG | y = −3.375x + 29.742 | 97.8 |
| 1055R | CAR CCA TGC AGC ACC | |||
| Bth274[C]- and Bth274[T]- | Bth274F | CCT TCG ATG GAT AGG GG | y = −3.565x + 28.925 | 90.8 |
| related subclusters | 530R | CCG CGG CKG CTG GCA C |
Appropriate reverse primers were chosen to result in an average amplicon size of less than 250 bp.
Nucleotide sequence accession numbers.
The sequences determined in this study have been deposited in the GenBank database under the accession numbers EU136678 to EU136697.
RESULTS
PCR amplification.
PCR amplification was first examined at various cycle intervals to determine the appropriate number of thermal cycles needed to generate amplicons that were representative of the actual microbial community. With 2 ng of genomic DNA/μl as the starting template in PCR, it was observed that after 15 to 20 thermal cycles, the amplicons obtained were proportionally amplified and the use of these amplicons for downstream analyses could minimize the effect of PCR bias. After 20 to 35 cycles of PCR, the amplification curve deviated from the exponential shape and underwent a quasilinear phase. Amplification eventually reached a plateau beyond 35 cycles, as the amount of accumulated amplicons could not be increased further (Fig. 3). Thus, amplicons that underwent 20 thermal amplification cycles were subsequently used as the DNA templates for all HOPE reactions in this study.
FIG. 3.

Graphical representation of PCR amplicon masses obtained with different numbers of thermal cycles. Exponential amplification was achieved from cycles 10 to 20, and a plateau was reached after 20 cycles. To minimize PCR bias that can affect the determination of the relative abundances of bacterial targets, the total set of PCR-amplified 16S rRNA genes was obtained after 20 cycles of PCR amplification, purified, and then used for HOPE. Lanes 1 and 7 represent the DNA size ladder. Lanes 2 to 6 show the PCR amplicons obtained from the piggery wastewater influent after 10, 15, 20, 25, and 30 cycles, respectively. Lanes 8 to 12 show the PCR amplicons obtained from the municipal wastewater after 10, 15, 20, 25, and 30 cycles, respectively.
Specificity of primer extension.
The specificities of primer extension were validated against reference strains. Results showed that all species-specific primers correctly extended with the same nucleotide as the in silico analysis predicted when DNA from their targeted species was used as the DNA template. Likewise, no species-specific primers were extended when nontargets were used. Group-specific primer Bth274 extended with nucleotide C in the presence of B. uniformis and B. fragilis and with nucleotide T in the presence of B. tectus, B. pyogenes, B. nordii, B. salyersiae, and B. thetaiotaomicron. Bfrg602, which was designed to target Bacteroides spp. in the B. fragilis cluster, extended with nucleotide C, and Bdts_gp980, which was designed to target the P. distasonis group, correctly extended with nucleotide G. Bac303 and U1390 were designed to target all reference bacterial strains at the order and domain levels, respectively, and extended with C and A, respectively (Table 1). However, minimal extension of the Bac303 primer in the presence of P. goldsteinii JCM13446 was observed. Resequencing of the 16S rRNA gene of P. goldsteinii revealed that the target sequence region had a single mismatch relative to Bac303 at the fifth position from the 3′ end of the primer and that this mismatch could significantly reduce the primer extension efficiency in comparison to that obtained with the perfect-match sequence (37). Since P. goldsteinii is not a predominant member in the fecal microbiota, its mismatch to Bac303 should not affect the measurement of the relative abundances of Bacteroidales in the fecal microbiota as represented by the total set of PCR-amplified 16S rRNA genes.
Calculation of CFs.
CFs were established in the presence of 16S RNA gene amplicons derived from the bacterial reference clone. For instance, when the B. thetaiotaomicron 16S rRNA gene amplicon was used as the HOPE template, only primers in reactions mixtures 1 and 6 were extended, and they were correctly identified by size and the type of the extended fluorescent ddNTP (Fig. 4A). Peak areas for individual primers Bth584 and Bth274 [T] in reaction 1 and Bth274, Bfrg602, Bac303, and U1390 in reaction 6 were noted, and the individual extended-primer concentrations were calculated. The concentration of the extended B. thetaiotaomicron-specific primer (i.e., primer Bth584) was normalized against that of the higher-level primer in the same reaction mixture (i.e., Bth274 [T]) to obtain the CF (i.e., CFBth584−Bth274 [T]). By repeating this procedure for all tested reference bacterial strains, CFs for all primers were obtained (Table 3). These values were subsequently used for calculating the relative abundances of bacterial targets present in the fecal and wastewater samples.
FIG. 4.

Actual electropherograms for HOPE products obtained in the presence of the total set of PCR-amplified 16S rRNA genes from the B. thetaiotaomicron BCRC10624 clone (A) and human feces sample H3 (B). Black, red, and green peaks represent primers that were extended with dTAMRA-ddCTP, dROX-ddTTP, and dR6G-ddATP, respectively. rflu, relative fluorescence units.
TABLE 3.
Relative abundances of bacterial targetsa
| Group | Species | Relative abundance (%) of bacteria of indicated species in samplee:
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H1 | H2 | H3 | P1 | P2 | P3 | C1 | C2 | C3 | Municipal WW | Pig WW | ||
| Bfrg602-related | B. fragilis | NDd | 3.1 ± 0.1 | 2.9 ± 0.2 | ND | ND | ND | ND | ND | ND | 3.6 ± 0.3 | ND |
| group | B. caccae | 8.9 ± 1.5 | 1.3 ± 0.8 | 1.6 ± 0.2 | ND | ND | ND | ND | ND | ND | 2.7 ± 0.4 | ND |
| B. acidifaciens | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| B. intestinalis | ND | ND | 1.2 ± 0.3 | ND | ND | ND | ND | ND | ND | ND | ND | |
| B. uniformis | 7.5 ± 0.5 | 3.9 ± 0.4 | 7.1 ± 0.3 | ND | ND | ND | ND | ND | ND | 18.3 ± 0.7 | ND | |
| B. eggerthii | ND | 0.6 ± 0.1 | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| B. helcogenes | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| B. vulgatus | 28.4 ± 0.9 | 14.8 ± 0.3 | 49.8 ± 2.2 | ND | ND | ND | ND | ND | ND | 31.5 ± 1.2 | ND | |
| B. massiliensis | ND | 2.9 ± 1.0 | ND | ND | ND | ND | ND | ND | ND | 5.2 ± 0.2 | ND | |
| B. coprocola | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| Bth274 [T]- | B. nordii | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| related | B. salyersiae | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| group | B. pyogenes | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| B. thetaiotaomicron | 72.2 ± 0.3 | 58.9 ± 0.7 | 97.5 ± 0.4 | ND | ND | ND | ND | ND | ND | 95.2 ± 2.0 | ND | |
| U1390-related domain | Bac303-related species | 28.1 ± 0.3 | 44.4 ± 2.0 | 29.9 ± 3.0 | 5.2 ± 0.4 | 4.6 ± 0.9 | 5.1 ± 0.7 | 2.3 ± 0.3 | 2.9 ± 0.2 | 1.8 ± 0.2 | 10.4 ± 1.6 | 4.6 ± 0.6 |
| Bfrg602-related species | 5.0 ± 0.9 | 11.8 ± 1.0 | 12.1 ± 1.2 | ND | ND | ND | ND | ND | ND | 6.2 ± 0.2 | ND | |
| Bdts_gp980-related species | 2.4 ± 0.1 | 2.5 ± 0.2 | 4.1 ± 0.2 | 1.3 ± 0.1 | 1.4 ± 0.1 | 1.7 ± 0.1 | 0.7 ± 0.2 | 0.7 ± 0.2 | 0.5 ± 0.1 | 5.1 ± 0.2 | 1.8 ± 0.1 | |
| Bth274 [C]-related speciesb | 0.8 ± 0.1 | 1.2 ± 0.2 | 1.3 ± 0.2 | ND | ND | ND | ND | ND | ND | 1.5 ± 0.8 | ND | |
| Bth274 [T]-related speciesc | 1.1 ± 0.3 | 0.5 ± 0.1 | 0.2 ± 0.1 | ND | ND | ND | ND | ND | ND | 0.3 ± 0.2 | ND | |
Relative abundances of bacterial targets in the human (H1 to H3), pig (P1 to P3), and cow (C1 to C3) feces, together with those present in the influents of municipal and swine wastewater treatment plants (municipal WW and pig WW, respectively), were determined individually by using a multiplex HOPE method. CFs obtained for primers Bth274 [T]_15dA, Bnd136, Bsls1016_10dA, Pg_dtc732_20dA, and Bth584_16dA in multiplex tube 1 were as follows: 1:2.05:0.39:0.36:0.19. CFs were obtained using clones from bacterial strains that included B. nordii, B. salyersiae, B. pyogenes, and B. thetaiotaomicron strains. CFs obtained for primers Bfrg602_19dA, Bfrg1026_20dA, Bcc1066_15dA, Badf1009, Bitt141, Bufm1018_20dA, Begt999, Bhcg171_8dA, Bvg1016, Bmsl1000_19dA, and Bcpc1015_24dA in multiplex tubes 2 to 5 were as follows: 1:0.17:2.96:0.36:16.51:4.61:8.17:13.86:8.37:3.13:3.06. CFs were obtained using clones from bacterial strains that included B. fragilis, B. caccae, B. acidifaciens, B. intestinalis, B. uniformis, B. eggerthii, B. helcogenes, B. vulgatus, B. massiliensis, and B. coprocola strains. CFs obtained for primers U1390_17dA, Bac303_5dA, Bfrg602_19dA, Bdts_gp980_12dA, Bth274 [C]_15dA, and Bth274 [T]_15dA in multiplex tube 6 were as follows: 1:1.206:0.091:1.998:0.158:0.432. CFs were obtained using clones from bacterial strains that included B. fragilis, B. thetaiotaomicron, B. tectus, B. uniformis, B. nordii, P. distasonis, and P. merdae strains.
Primer Bth274 [C]_15dA extends with ddCTP when annealed to bacterial targets in this group.
Primer Bth274 [T]_15dA extends with ddTTP when annealed to bacterial targets in this group.
ND, not detected.
The relative abundances of all bacterial targets were averaged from triplicate measurements and are expressed as means ± standard deviations (n = 3).
Electropherograms for tested samples.
Figure 4B shows the electropherograms obtained after HOPE was carried out in the presence of the total set of 16S rRNA gene amplicons derived from human stool sample H3. As higher-level primers had more targets for priming, the peak heights and areas were considerably larger than those corresponding to species-specific primers. Furthermore, as primers should be extended at a specific fragment size, all other peaks that did not coincide with the anticipated size could be treated as background noise and removed from subsequent analyses.
Relative abundances of Bacteroides spp. in feces as determined by HOPE.
Table 3 and Fig. 5 describe the relative abundances of the 14 tested Bacteroides spp. present in the three tested human feces samples at various taxonomical levels. At the higher taxonomical levels, the relative abundances of the Bacteroidales group targeted by the Bac303 primer corresponded to a range from 28.1 to 44.4% of PCR-amplified 16S rRNA genes. The B. fragilis Bth274 [C]-related subcluster (including B. uniformis and B. fragilis) accounted for 0.8 to 1.3% of amplified genes, while the B. fragilis Bth274 [T]-related subcluster (including B. thetaiotaomicron, B. tectus, B. caccae, B. pyogenes, B. nordii, and B. salyersiae) accounted for 0.2 to 1.1% of amplified genes. The abundance of the B. fragilis cluster, which varied across human samples, corresponded to a range from 5.0 to 12.1% of PCR-amplified 16S rRNA genes.
FIG. 5.

Statistical interpretation of the HOPE results. (A) Abundances of the B. fragilis cluster, the B. fragilis subclusters, and the P. distasonis cluster relative to the total set of amplified 16S rRNA genes were averaged for the individual sample types. WW, wastewater. (B) B. massiliensis, B. vulgatus, B. eggerthii, B. uniformis, B. intestinalis, B. caccae, and B. fragilis were detected, and their average relative abundances within the B. fragilis cluster are illustrated. A predominant portion within the B. fragilis cluster remains unidentified. (C) Multidimensional scaling plot obtained by first performing a Euclidean distance square-root transformation of the raw data to generate a similarity matrix. The matrix then underwent a 100-iteration multidimensional scaling analysis. C1 to C3, cow fecal samples; P1 to P3, pig fecal samples.
Within the B. fragilis group, B. vulgatus was observed to be the predominant species (accounting for 14.8 to 49.8% of amplified 16S rRNA genes from the B. fragilis group), and B. caccae and B. uniformis were present at various relative abundances (corresponding to 1.3 to 8.9% of amplified genes) in microbiota from different human fecal samples. Other species, like B. fragilis, B. eggerthii, B. intestinalis, and B. massiliensis, were detected to account for between 0.6 and 3.1% of amplified genes from samples H2 and H3, but their levels were too low to be detected in sample H1. In total, these targeted Bacteroides spp. accounted for 26.6 to 62.6% of amplified 16S rRNA genes from the B. fragilis group. These observations further suggest that other important and unknown Bacteroides spp. from this group were present in the human fecal microbiota (Fig. 5B). Within the B. fragilis Bth274 [T]-related subcluster, B. thetaiotaomicron was detected as the predominant and major species (58.9 to 97.5%) (Table 3).
In contrast, with the swine and bovine fecal microbiota, none of the 14 species-specific primers extended, suggesting that the Bacteroides spp. examined in this study were present at abundances below the detection sensitivity of HOPE. These results further suggest that the Bacteroides spp. examined in this study are probably host-specific to human fecal microbiota. In the animal fecal microbiota, most Bacteroidales could account for only 4.6 to 5.2% and 1.8 to 2.9% of PCR-amplified 16S rRNA genes from the swine and bovine feces, respectively. These abundances were lower than those in human fecal microbiota by factors of 5 to 25 and were clustered significantly apart from the latter. Furthermore, the interindividual differences among the animal fecal microbiota were less apparent than those among the human fecal microbiota (Fig. 5C).
Abundances of Bacteroides spp. as quantified by Q-PCR.
To validate the relative abundances of Bacteroides spp. as determined by HOPE, Q-PCR was also employed to determine the abundances of Bacteroidales, the B. fragilis group and its subgroups, and the Parabacteroides group that were present in the human fecal samples H1 to H3. As the 16S rRNA operon copy numbers of different Bacteroides spp. are not well defined yet, the quantitative expression of Q-PCR calculations was based on the genomic mass (in picograms). Standard curves generated showed a linear relationship between mean numbers of threshold cycles and the logarithmic mass over the range of 1 to 10,000 picograms, and the amplification efficiency at an annealing temperature of 64°C was 90.8 to 100.7%. The relative abundances of most Bacteroidales in the feces of all three human subjects as targeted by primer Bac303 were 30.1 to 50.9%. Likewise, the B. fragilis group, the Parabacteroides group, and the B. fragilis subgroups accounted for 4.7 to 14.8%, 3.1 to 5.4%, and 1.7 to 2.0% of amplified genes, respectively (Table 4). These results were in close proximity to those obtained using HOPE.
TABLE 4.
Comparison of the abundances of Bacteroides spp. at different hierarchical levels as reported in the published literature
| Reference | Method | Sampling size | Relative abundance (%) of bacteria classified as:
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Bacteroides-Prevotella | B. fragilis subgroup | P. distasonis subgroup | Bth274-related subgroup | B. vulgatus | B. thetaiotaomicron | B. uniformis | B. caccae | |||
| Franks et al. (16) | FISHa | Fecal samples from 9 subjects | NAd | 21 (Bfra602 and Bdis656 related) | 21 (Bfra602 and Bdis656 related) | NDe | ND | ND | ND | ND |
| Harmsen et al. (19) | FISH | Fecal samples from 11 subjects | 27.7 (Bac303 related) | ND | ND | ND | ND | ND | ND | ND |
| Rigottier-Gois et al. (34) | FISH | Fecal samples from 20 subjects | 14.5 (Bac303 related) | 3.9 (Bfra998 related) | 0.4 (Bdist1025 related) | ND | 4.2 (Bvg1017 related) | ND | ND | ND |
| Suau et al. (39) | 16S rRNA clone library analysisb | Fecal samples from 1 subject; 284 clones | 31 | 13.3 | 2.3 | NA | 2.1 | 1.4 | 4.9 | 1.1 |
| Eckburg et al. (11) | 16S rRNA clone library analysis | Mucosa and fecal samples from 3 subjects; total of 395 bacterial phylotypes | 48 | NA | NA | NA | 15 | 6.2 | NA | NA |
| This study | HOPEc | Fecal samples from 3 subjects | 28.1-44.4 (Bac303 related) | 5.0-12.1 (Bfrg602 related) | 2.4-4.1 (Bdts_gp980 related) | 1.5-1.9 | 1.4-6.0 (Bvg1016 related) | 0.2-0.8 (Bth584 related) | 0.4-0.9 (Bufm1018 related) | 0.2-0.4 (Bcc1066 related) |
| This study | Q-PCRc | Fecal samples from 3 subjects | 30.1-50.9 (Bac303 related) | 4.7-14.8 (Bfrg602 related) | 3.1-5.4 (Bdts_gp980 related) | 1.7-2.0 | ND | ND | ND | ND |
Results from FISH analyses are expressed in terms of hybridized cell counts normalized against Eub338.
Results from clone library analyses are expressed with respect to the total numbers of clones selected.
Results from HOPE and Q-PCR are expressed with respect to the total set of PCR-amplified 16S rRNA genes.
NA, information not available in the literature.
ND, not determined.
Relative abundances of Bacteroides spp. in wastewaters.
Table 3 and Fig. 5 indicate the relative abundances of Bacteroides spp. detected in the influents of swine and municipal wastewater treatment plants. Bacteroides spp. that were previously detected in the human feces were also present in municipal wastewater, with B. fragilis, B. caccae, B. uniformis, B. vulgatus, and B. massiliensis accounting for 3.6, 2.7, 18.3, 31.5, and 5.2% of the B. fragilis group, respectively. B. thetaiotaomicron remained the predominant species in the B. fragilis Bth274 [T]-related subcluster (95.2%). The relative abundances of these Bacteroides spp. in the municipal wastewater therefore approximate those found in human fecal microbiota. However, the relative abundances of most Bacteroidales decreased by more than twofold compared to those in human fecal samples, possibly due to a die-off effect for these Bacteroides spp. and the proliferation of other microbial populations during the transportation in the sewage system.
With the influent of the swine wastewater treatment plant, none of the primers targeting Bacteroides spp. at the species level could be detected. The relative abundances of targets in the order Bacteroidales were also about twofold lower than those in municipal wastewater.
DISCUSSION
In this study, HOPE, with its advantages in detection sensitivity, specificity, rapidity, and throughput, has been successfully demonstrated to be a method that can improve understanding of the relative abundances of Bacteroides and Parabacteroides spp. present in fecal microbiota. Findings from the analysis of human feces suggested that the fecal microbiota contain a relatively stable profile of Bacteroides spp., with major species like B. vulgatus, B. fragilis, B. thetaiotaomicron, B. uniformis, and B. caccae consistently detected in all tested samples. Clear interindividual differences in the diversity of Bacteroides spp. were observed, presumably due to differences in living habits, age, and diet (20, 22). The total abundances of the Bacteroides spp. within the B. fragilis cluster targeted by the species-specific primers were 27 to 63%, further suggesting the existence of other Bacteroides spp. in human fecal microbiota. It is possible that a small fraction of these unidentified Bacteroides spp. may be B. ovatus and B. stercoris, which were not detected in this study. However, these two species are unlikely to be predominant, as previous FISH studies have reported a low frequency of detection of these species (34). The remaining, largely unidentified fraction may represent the yet-to-be-cultured species in the B. fragilis cluster as described by Eckburg and coworkers (11). Based on a 16S rRNA gene clone library, they detected 65 Bacteroidetes-related phylotypes, and as many as 65% are classified as novel.
Likewise, when HOPE was applied to animal feces and wastewaters, all of the species-specific primers were undetected and a reduction in the abundances of Bacteroidales in the wastewater treatment influent compared to those in human fecal samples was observed. These results reconfirmed previous reports that those Bacteroides spp. predominant in human fecal microbiota are present in much lesser abundance in animal stools (27). Therefore, certain Bacteroides 16S rRNA genes have been suggested and used as effective biomarkers to differentiate the origin and duration of fecal contamination, thereby achieving microbial source tracking (5, 9, 14).
We further compared the HOPE results on the diversity and abundances of those fecal bacterial populations with results obtained previously using 16S rRNA clone libraries and FISH (11, 16, 19, 34, 39) (Table 4). Though the abundances of bacterial targets as determined by 16S rRNA clone library and FISH analyses were expressed based on different normalization units (i.e., the percentage of the total number of clones selected and the percentage of the total number of cells hybridized with a domain Bacteria-specific probe) and interindividual variation in the composition of the Bacteroides flora exists, both molecular methods showed ranges of abundances of Bacteroides-Prevotella bacteria (14.5 to 48%) in the human fecal microbiota similar to those shown by HOPE (28.1 to 44.4%). FISH results showed that the B. fragilis cluster and the P. distasonis group together accounted for 4.3 to 21%, respectively, of the total Bacteria and that B. vulgatus accounted for 4.2% of the total Bacteria (16, 19, 34). Similarly, after the selection and sequencing of 300 to 700 clones to construct a clone library, the B. fragilis group and the P. distasonis group were found to account for 13.3 and 2.3% of total Bacteria, respectively, while predominant species like B. vulgatus, B. thetaiotaomicron, B. uniformis, and B. caccae accounted for 2.1 to 15%, 1.4 to 6.2%, 4.9%, and 1.1% of total Bacteria (11, 39).
To further validate the reliability of HOPE results obtained in this study, we used Q-PCR analysis of the same set of human fecal samples analyzed by HOPE to determine the abundances of selected Bacteroides spp. The relative abundances of Bacteroidales, the B. fragilis cluster and its subclusters, and the P. distasonis cluster as determined by Q-PCR and HOPE were in close approximation (Table 4). This outcome suggests that when intersample differences in the composition of the Bacteroides spp. are minimal, the two PCR-based molecular methods complement each other in determining the abundances of specific bacterial targets.
It is clear through this comparison that HOPE, with its advantages in detection sensitivity, specificity, and throughput, can complement other available molecular methods and aid microbial studies. It has been demonstrated previously that HOPE is able to detect bacterial targets at different phylogenetic levels (i.e., species, genus, and so on up to domain) and at a sensitivity corresponding to 0.1% of the total set of PCR-amplified bacterial targets (41). This sensitivity approximates that obtained by constructing a 300-clone library (39), and thus, HOPE can easily detect bacterial targets like Bacteroides spp. in the human feces at the species level. Unlike clone library analysis, in which the abundance of a particular bacterial target is dependent on the ultimate number of clones picked, HOPE analysis examines the entire set of PCR-amplified products from the microbial community and can better reveal the relative abundances of bacterial targets. The comparability of the results obtained by HOPE to results obtained by other molecular tools (Table 4) suggests that the specificity and reliability of HOPE were also not compromised. The entire HOPE procedure for the identification and quantification of those 14 Bacteroides and Parabacteroides spp. after primary DNA extraction and PCR amplification took less than 120 min and was significantly shorter than that required for FISH or clone library construction. Furthermore, the total number of bacterial targets can be easily increased simply by adding HOPE reactions or by adding primer(s) to individual reactions (Fig. 1).
At present, HOPE has been demonstrated to determine the relative abundances of only known bacterial targets and, therefore, does not aid in further understanding the diversity of the microbial community. The complexity of making multiplex tube arrangements and analyzing the electropherograms can also increase with an increase in the number of applied primers. Thus, there is a strong need to automate both processes. It should be noted that the determination of the relative abundances of bacterial targets by HOPE can be subject to PCR bias. To minimize this bias, one can apply amplicons obtained at the exponential phase of PCR amplification as DNA templates in HOPE reactions. The sensitivity of the method may also be compromised by an excessively long poly(A) tail (41) and by possible interference from the nontarget DNA in complex environmental samples. To circumvent these effects, it is therefore essential to perform a systematic study that looks into an optimal poly(A) tail length and to perform thorough purification of the HOPE products prior to capillary electrophoresis.
In summary, this study has demonstrated the potential of HOPE as a rapid and high-throughput detection method that is able to examine the relative abundances of bacterial targets at various taxonomical levels. It can be used to identify disease-related bacterial populations that are present in the gut or feces over temporal and spatial intervals. Likewise, the present set of human-specific Bacteroides HOPE primers can be improved to include primers targeting animal-specific uncultivated Bacteroidales (5, 6, 9, 10, 14, 29) and be used to detect the presence of host-specific Bacteroidales in water bodies that may be contaminated with feces from multiple sources. The simplicity and versatility of this method can further facilitate the attempts to understand microbial diversity in different ecological systems when HOPE is used to complement other existing molecular tools. For example, one can use fingerprinting methods to quickly evaluate the microbial diversity of an environmental sample. Once the key microbial populations are identified using the fingerprinting methods, HOPE can be subsequently applied to effectively monitor the dynamics of those key populations at a much-refined resolution.
Footnotes
Published ahead of print on 14 March 2008.
REFERENCES
- 1.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 2.Amann, R., B. M. Fuchs, and S. Behrens. 2001. The identification of microorganisms by fluorescence in situ hybridisation. Curr. Opin. Biotechnol. 12:231-236. [DOI] [PubMed] [Google Scholar]
- 3.Bakir, M. A., M. Kitahara, M. Sakamoto, M. Matsumoto, and Y. Benno. 2006. Bacteroides intestinalis sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 56:151-154. [DOI] [PubMed] [Google Scholar]
- 4.Bartosch, S., A. Fite, G. T. Macfarlane, and M. E. McMurdo. 2004. Characterization of bacterial communities in feces from healthy elderly volunteers and hospitalized elderly patients by using real-time PCR and effects of antibiotic treatment on the fecal microbiota. Appl. Environ. Microbiol. 70:3575-3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernhard, A. E., and K. G. Field. 2000. Identification of nonpoint sources of fecal pollution in coastal waters by using host-specific 16S ribosomal DNA genetic markers from fecal anaerobes. Appl. Environ. Microbiol. 66:1587-1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernhard, A. E., and K. G. Field. 2000. A PCR assay to discriminate human and ruminant feces on the basis of host differences in Bacteroides-Prevotella genes encoding 16S rRNA. Appl. Environ. Microbiol. 66:4571-4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crosby, L. D., and C. S. Criddle. 2003. Understanding bias in microbial community analysis techniques due to rrn operon copy number heterogeneity. BioTechniques 34:790-803. [DOI] [PubMed] [Google Scholar]
- 8.Croucher, S. C., A. P. Houston, C. E. Bayliss, and R. J. Turner. 1983. Bacterial populations associated with different regions of the human colon wall. Appl. Environ. Microbiol. 45:1025-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dick, L. K., A. E. Bernhard, T. J. Brodeur, J. W. Santo Domingo, J. M. Simpson, S. P. Walters, and K. G. Field. 2005. Host distributions of uncultivated fecal Bacteroidales bacteria reveal genetic markers for fecal source identification. Appl. Environ. Microbiol. 71:3184-3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dick, L. K., and K. G. Field. 2004. Rapid estimation of numbers of fecal Bacteroidetes by use of a quantitative PCR assay for 16S rRNA genes. Appl. Environ. Microbiol. 70:5695-5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eckburg, P. B., E. M. Bik, C. N. Bernstein, E. Purdom, L. Dethlefsen, M. Sargent, S. R. Gill, K. E. Nelson, and D. A. Relman. 2005. Diversity of the human intestinal microbial flora. Science 308:1635-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fenner, L., V. Roux, M.-N. Mallet, and D. Raoult. 2005. Bacteroides massiliensis sp. nov., isolated from blood culture of a newborn. Int. J. Syst. Evol. Microbiol. 55:1335-1337. [DOI] [PubMed] [Google Scholar]
- 13.Finegold, S. M., and H. Jousimies-Somer. 1997. Recently described clinically important anaerobic bacteria: medical aspects. Clin. Infect. Dis. 25(Suppl. 2):S88-S93. [DOI] [PubMed] [Google Scholar]
- 14.Fogarty, L. R., and M. A. Voytek. 2005. Comparison of Bacteroides-Prevotella 16S rRNA genetic markers for fecal samples from different animal species. Appl. Environ. Microbiol. 71:5999-6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forney, L. J., X. Zhou, and C. J. Brown. 2004. Molecular microbial ecology: land of the one-eyed king. Curr. Opin. Microbiol. 7:210-220. [DOI] [PubMed] [Google Scholar]
- 16.Franks, A. H., H. J. Harmsen, G. C. Raangs, G. J. Jansen, F. Schut, and G. W. Welling. 1998. Variations of bacterial populations in human feces measured by fluorescent in situ hybridization with group-specific 16S rRNA-targeted oligonucleotide probes. Appl. Environ. Microbiol. 64:3336-3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuchs, B. M., G. Wallner, W. Beisker, I. Schwippl, W. Ludwig, and R. Amann. 1998. Flow cytometric analysis of the in situ accessibility of Escherichia coli 16S rRNA for fluorescently labeled oligonucleotide probes. Appl. Environ. Microbiol. 64:4973-4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guarner, F., and J. R. Malagelada. 2003. Gut flora in health and disease. Lancet 361:512-519. [DOI] [PubMed] [Google Scholar]
- 19.Harmsen, H. J., G. C. Raangs, T. He, J. E. Degener, and G. W. Welling. 2002. Extensive set of 16S rRNA-based probes for detection of bacteria in human feces. Appl. Environ. Microbiol. 68:2982-2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayashi, H., M. Sakamoto, and Y. Benno. 2002. Fecal microbial diversity in a strict vegetarian as determined by molecular analysis and cultivation. Microbiol. Immunol. 46:819-831. [DOI] [PubMed] [Google Scholar]
- 21.Hayashi, H., M. Sakamoto, and Y. Benno. 2002. Phylogenetic analysis of the human gut microbiota using 16S rDNA clone libraries and strictly anaerobic culture-based methods. Microbiol. Immunol. 46:535-548. [DOI] [PubMed] [Google Scholar]
- 22.Hopkins, M. J., and G. T. Macfarlane. 2002. Changes in predominant bacterial populations in human faeces with age and with Clostridium difficile infection. J. Med. Microbiol. 51:448-454. [DOI] [PubMed] [Google Scholar]
- 23.Johnson, J. L., W. E. C. Moore, and L. V. H. Moore. 1986. Bacteroides caccae sp. nov., Bacteroides merdae sp. nov., and Bacteroides stercoris sp. nov. isolated from human feces. Int. J. Syst. Bacteriol. 36:499-501. [Google Scholar]
- 24.Jung, R., K. Soondrum, and M. Neumaier. 2000. Quantitative PCR. Clin. Chem. Lab. Med. 38:833-836. [DOI] [PubMed] [Google Scholar]
- 25.Kane, M. D., L. K. Poulsen, and D. A. Stahl. 1993. Monitoring the enrichment and isolation of sulfate-reducing bacteria by using oligonucleotide hybridization probes designed from environmentally derived 16S rRNA sequences. Appl. Environ. Microbiol. 59:682-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kitahara, M., M. Sakamoto, M. Ike, S. Sakata, and Y. Benno. 2005. Bacteroides plebeius sp. nov. and Bacteroides coprocola sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 55:2143-2147. [DOI] [PubMed] [Google Scholar]
- 27.Kreader, C. A. 1995. Design and evaluation of Bacteroides DNA probes for the specific detection of human fecal pollution. Appl. Environ. Microbiol. 61:1171-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lane, D. J. 1991. 16S/23S rRNA sequencing, p. 115-175. In E. Stackebrandt and M. Goodfellow (ed.), Nucleic acids techniques in bacterial systematics. John Wiley & Sons, Inc., New York, NY.
- 29.Layton, A., L. McKay, D. Williams, V. Garrett, R. Gentry, and G. Sayler. 2006. Development of Bacteroides 16S rRNA gene TaqMan-based real-time PCR assays for estimation of total, human, and bovine fecal pollution in water. Appl. Environ. Microbiol. 72:4214-4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ludwig, W., O. Strunk, R. Westram, L. Richter, H. Meier, Yadhukumar, A. Buchner, T. Lai, S. Steppi, G. Jobb, W. Forster, I. Brettske, S. Gerber, A. W. Ginhart, O. Gross, S. Grumann, S. Hermann, R. Jost, A. Konig, T. Liss, R. Lussmann, M. May, B. Nonhoff, B. Reichel, R. Strehlow, A. Stamatakis, N. Stuckmann, A. Vilbig, M. Lenke, T. Ludwig, A. Bode, and K. H. Schleifer. 2004. ARB: a software environment for sequence data. Nucleic Acids Res. 32:1363-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manz, W., R. Amann, W. Ludwig, M. Vancanneyt, and K. H. Schleifer. 1996. Application of a suite of 16S rRNA-specific oligonucleotide probes designed to investigate bacteria of the phylum Cytophaga-Flavobacter-Bacteroides in the natural environment. Microbiology 142:1097-1106. [DOI] [PubMed] [Google Scholar]
- 32.McOrist, A. L., M. Jackson, and A. R. Bird. 2002. A comparison of five methods for extraction of bacterial DNA from human faecal samples. J. Microbiol. Methods 50:131-139. [DOI] [PubMed] [Google Scholar]
- 33.Reischer, G. H., D. C. Kasper, R. Steinborn, R. L. Mach, and A. H. Farnleitner. 2006. Quantitative PCR method for sensitive detection of ruminant fecal pollution in freshwater and evaluation of this method in alpine karstic regions. Appl. Environ. Microbiol. 72:5610-5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rigottier-Gois, L., V. Rochet, N. Garrec, A. Suau, and J. Dore. 2003. Enumeration of Bacteroides species in human faeces by fluorescent in situ hybridisation combined with flow cytometry using 16S rRNA probes. Syst. Appl. Microbiol. 26:110-118. [DOI] [PubMed] [Google Scholar]
- 35.Salyers, A. A., S. E. West, J. R. Vercellotti, and T. D. Wilkins. 1977. Fermentation of mucins and plant polysaccharides by anaerobic bacteria from the human colon. Appl. Environ. Microbiol. 34:529-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidt, T. M., E. F. DeLong, and N. R. Pace. 1991. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J. Bacteriol. 173:4371-4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sipos, R., A. J. Szekely, M. Palatinszky, S. Revesz, K. Marialigeti, and M. Nikolausz. 2007. Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targetting bacterial community analysis. FEMS Microbiol. Ecol. 60:341-350. [DOI] [PubMed] [Google Scholar]
- 38.Song, Y. L., C. X. Liu, M. McTeague, and S. M. Finegold. 2004. “Bacteroides nordii” sp. nov. and “Bacteroides salyersae” sp. nov. isolated from clinical specimens of human intestinal origin. J. Clin. Microbiol. 42:5565-5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suau, A., R. Bonnet, M. Sutren, J. J. Godon, G. R. Gibson, M. D. Collins, and J. Dore. 1999. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl. Environ. Microbiol. 65:4799-4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wagner, M., R. Amann, H. Lemmer, and K. H. Schleifer. 1993. Probing activated sludge with oligonucleotides specific for proteobacteria: inadequacy of culture-dependent methods for describing microbial community structure. Appl. Environ. Microbiol. 59:1520-1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu, J. H., and W. T. Liu. 2007. Quantitative multiplexing analysis of PCR-amplified ribosomal RNA genes by hierarchical oligonucleotide primer extension reaction. Nucleic Acids Res. 35:e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu, J., M. K. Bjursell, J. Himrod, S. Deng, L. K. Carmichael, H. C. Chiang, L. V. Hooper, and J. I. Gordon. 2003. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science 299:2074-2076. [DOI] [PubMed] [Google Scholar]
- 43.Zheng, D., E. W. Alm, D. A. Stahl, and L. Raskin. 1996. Characterization of universal small-subunit rRNA hybridization probes for quantitative molecular microbial ecology studies. Appl. Environ. Microbiol. 62:4504-4513. [DOI] [PMC free article] [PubMed] [Google Scholar]