

Abstract
Signal-to-noise-ratio (SNR) thresholds for microarray data analysis were experimentally determined with an oligonucleotide array that contained perfect-match (PM) and mismatch (MM) probes based upon four genes from Shewanella oneidensis MR-1. A new SNR calculation, called the signal-to-both-standard-deviations ratio (SSDR), was developed and evaluated, along with other two methods, the signal-to-standard-deviation ratio (SSR) and the signal-to-background ratio (SBR). At a low stringency, the thresholds of the SSR, SBR, and SSDR were 2.5, 1.60, and 0.80 with an oligonucleotide and a PCR amplicon as target templates and 2.0, 1.60, and 0.70 with genomic DNAs as target templates. Slightly higher thresholds were obtained under high-stringency conditions. The thresholds of the SSR and SSDR decreased with an increase in the complexity of targets (e.g., target types) and the presence of background DNA and a decrease in the compositions of targets, while the SBR remained unchanged in all situations. The lowest percentage of false positives and false negatives was observed with the SSDR calculation method, suggesting that it may be a better SNR calculation for more accurate determination of SNR thresholds. Positive spots identified by SNR thresholds were verified by the Student t test, and consistent results were observed. This study provides general guidance for users to select appropriate SNR thresholds for different samples under different hybridization conditions.
Microarrays have become a routine tool for studying gene functions, regulations, and networks in a variety of biological systems. The technology has been also applied to drug discovery and validation (7), microbial diagnostics (4, 10, 16, 20, 22, 31), mutation and single-nucleotide polymorphism detection (9), strain comparison and genotyping (1, 8, 21), species identification (32), array sequencing (35), environmental detection and monitoring (5, 6, 13, 24, 27, 28, 33), and evolutionary processes (14). However, due to small spot sizes, different degrees of uniformity of printing pins, and uneven hybridization, microarray spots inherently have relatively high noise, which presents a variety of challenges for quantitative analysis of microarray data. For example, how to distinguish a real signal from its background is still an unsolved problem, and a subset of this question is what parameters and thresholds should be used to differentiate a signal from noise.
The signal-to-noise ratio (SNR) has been used to define a positive spot, and two general methods are currently used to calculate SNR values. One is to use the ratio of the differences between the signal mean and background noise divided by the background standard deviation (2). This calculation method has been commonly used in many signal-processing disciplines, such as radio, electronics, and imaging (2, 30), and the threshold is usually set to 3.0 (30). The other method is to use the ratio of the signal median divided by the background median with the threshold set to 1.50 (26), and it was modified to calculate the SNR for a probe with replicate spots and to set a threshold of 2.0 (18, 19). However, the determination of these thresholds is arbitrary and has not been experimentally validated. Although the background standard deviation of pixel intensities for each spot is included in the first calculation method, the signal standard deviation is not considered in either of the two SNR calculation methods. In addition, an SNR threshold may vary with different types of targets, target compositions, and hybridization conditions, and hence, it could be difficult to set a universal SNR threshold. Therefore, new SNR calculation methods that include both signal and background standard deviations and experimental evaluations of SNR thresholds are needed.
The objectives of this study were to (i) evaluate a new method for SNR calculation, (ii) determine appropriate SNR thresholds for differentiating signals from noise based on different SNR calculation methods, and (iii) examine the effects of target types, background DNA, and target compositions on the threshold determination. Our results demonstrated that our new calculation performed better than two other existing calculations and that SNR thresholds were affected by the hybridization stringency, types of target templates, background DNAs, and compositions of the target templates. The results provide general guidance for users to select appropriate SNR thresholds under different conditions.
MATERIALS AND METHODS
Oligonucleotide probe design and microarray construction.
Fifty-mer and 70-mer perfect-match (PM) and mismatch (MM) oligonucleotide probes were prepared as previously described (12). Briefly, four genes (SO1679, SO1744, SO2680, and SO0848) were selected from the Shewanella oneidensis MR-1 genome. For each gene, 1 50- or 70-mer PM probe and 45 MM probes (with 1 to 37 mismatches) were generated with 3 random MM probes at each level. All 368 designed oligonucleotides were commercially synthesized without modification by MWG Biotech Inc. (High Point, NC). The concentrations of oligonucleotide probes were adjusted to 100 pmol/μl. Oligonucleotide probes prepared in 50% dimethyl sulfoxide (Sigma Chemical Co., Missouri) were spotted onto UltraGAPS glass slides (Corning Life Science, New York) using a PixSys 5500 robotic printer (Cartesian Technologies Inc., California). Each probe had four replicates on a single slide. In total, there were 1,472 (368 × 4) spots on the array. After being printed, the oligonucleotide probes were fixed onto the slides by UV cross-linking (600 mJ of energy) according to the protocol of the manufacturer (Corning Life Science, New York).
Target template preparations.
Four 70-mer artificial targets (T1-SO1679, T2-SO1744, T3-SO2680, and T4-SO0848) that were complementary to the 70-mer PM probes were synthesized by the Molecular Structure Facility at Michigan State University (East Lansing, MI). The artificial oligonucleotide targets were labeled at the 5′ ends with Cy5 (T1-SO1679, T2-SO1744, and T3-SO2680) or Cy3 (T4-SO0848) fluorescent dye during synthesis. The 70-mer oligonucleotide targets also contained the sequences of the 50-mer oligonucleotide targets.
Gene-specific primers were chosen for the four selected genes (see Table S1 in the supplemental material), with each PCR product about 500 bp, covering both 50-mer and 70-mer probe sequences. Each gene was amplified with S. oneidensis MR-1 genomic DNA (gDNA) as a template using the standard PCR amplification protocol. The amplified PCR products were purified using the Qiaquick PCR purification kit (Qiagen Inc., California) according to the protocol of the manufacturer. The purified PCR fragments were visualized, and the sizes via were checked by agarose gel electrophoresis, and then the fragments were quantified using the PicoGreen dsDNA Assay Kit (Invitrogen, California).
Genomic DNAs from four bacteria were also used as target DNAs. S. oneidensis MR-1, Escherichia coli S17, and Pseudomonas sp. strain G179 were grown in LB medium to stationary phase, and Desulfovibrio vulgaris Hildenborough was grown in the standard lactate and sulfate (LS) medium (20a). The cells were collected by centrifugation at 4,000 × g at room temperature for 10 min. Their gDNAs were isolated and purified as described previously (34). Methanococcus maripludis gDNA was provided by Sergey Stolyar at the University of Washington (Seattle). The yeast Saccharomyces cerevisiae was grown in yeast-peptone-dextrose medium to saturation, and its gDNA was extracted using the glass bead method as described by Hoffman and Winston (15).
To test how bacterial ratios affect the determination of SNRs, S. oneidensis MR-1 gDNA was mixed with four other bacterial gDNAs (D. vulgaris Hildenborough, E. coli S17, Pseudomonas sp. strain G179, and M. maripludis) at three different ratios: A (10 [S. oneidensis MR-1]:1:1:1:1), B (1 [S. oneidensis MR-1]:1:1:1:1), and C (1 [S. oneidensis MR-1]:10:10:10:10). Each sample had the same amount of total gDNA (2.5 μg).
Probe labeling, microarray hybridization, and image quantification.
PCR amplicons, the purified gDNAs from pure cultures (500 ng), and mixed gDNAs (2.5 μg) were fluorescently labeled by random priming using the Klenow fragment of DNA polymerase (12). Mixture I (35 μl), containing certain amounts (as indicated for different experiments) of gDNA and 20 μl of random primers (Invitrogen, California), was heated at 98°C for 3 to 5 min, cooled on ice, and then centrifuged. Mixture II (15 μl), containing 1 μl of 5 mM dATP, dGTP, and dTTP and 2.5 mM dCTP, 2 μl (80 U) of Klenow (Invitrogen, CA), and 0.5 μl of Cy3 dye (Amersham BioSciences, United Kingdom), was added to mixture I. A total of 50 μl labeling-reaction solution was incubated for 3 h at 42°C. The labeling reaction was terminated by heating the solution at 98°C for 3 min. The tubes were removed and placed on ice. The labeled cDNA targets were purified immediately using a QIAquick PCR purification column and concentrated in a Savant Speedvac centrifuge (Savant Instruments Inc., Holbrook, NY).
The labeled PCR amplicons or gDNAs were resuspended in 25 μl of hybridization solution containing 50% formamide, 5× saline-sodium citrate (SSC) (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), 0.1% sodium dodecyl sulfate (SDS), and 0.1 mg/ml of herring sperm DNA (Invitrogen, California). The hybridization solution was incubated at 95 to 98°C for 5 min, centrifuged to collect condensation, and kept at 50°C. The solution was immediately applied to the microarray slide, and hybridization was carried out in a waterproof Corning hybridization chamber (Corning Life Science, New York) submerged in a 45°C water bath in the dark for 16 h (12). Washing was performed immediately in the following steps: (i) in a solution containing 2× SSC and 0.1% SDS at 40°C for 5 min, repeated once; (ii) in a solution containing 0.1× SSC and 0.1% SDS at room temperature for 10 min, repeated once; and (iii) in 0.1× SSC at room temperature for 2 min, repeated once. The slides were dried with compressed air prior to being scanned. The same batch slides and the same settings were used for all experiments. The laser power was set to 95%, and photomultiplier tube efficiency was set to 70%. Five slides (with four replicated spots on each slide) were used for each condition, and hence, each spot had up to 20 data points. The hybridized microarray slides were scanned using a ScanArray Express microarray analysis system (Perkin Elmer, Massachusetts). The spot signals, spot quality, and background fluorescence intensities of scanned images were quantified with ImaGene version 6.0 (Biodiscovery Inc., Los Angeles, CA).
Data analysis.
Data analysis included four major steps.
(i) Defining positive and negative spot pools.
Microarray detection mainly depends on probe specificity and hybridization stringency (e.g., temperature), and two levels of stringency were used in this study. High-level stringency is expected to eliminate cross-hybridization for the probes with a higher probe-target similarity, a longer continuous stretch length, and a lower free energy. At both stringencies, positive and negative pools were defined (see Tables S2 and S3 in the supplemental material). At high stringency, a positive 50-mer probe had a sequence identity of >90%, a stretch length of >20, and free energy of <−35 kcal/mol with its nontargets, and a negative probe had a sequence identity of ≤90%, a stretch length of ≤20, and free energy of ≥−35 kcal/mol with its nontargets. Our previous experimental results showed that such high-stringency hybridization could be achieved at 50°C with 50% formamide (17). Similarly, a positive 70-mer probe had a sequence identity of >90%, a stretch length of >25, and free energy of <−50 kcal/mol with its nontargets, and a negative probe had a sequence identity of ≤90%, a stretch length of ≤25, and free energy of ≥−50 kcal/mol with its nontargets. At low stringency, a positive 50-mer probe had a sequence identity of >85%, a stretch length of >15, and free energy of <−30 kcal/mol with its nontargets, and a negative probe had a sequence identity of ≤85%, a stretch length of ≤15, and free energy of ≥−30 kcal/mol with its nontargets (12). The low stringency generally corresponded to hybridization at 42°C with 50% formamide. Similarly, a positive 70-mer probe had a sequence identity of >85%, a stretch length of >20, and free energy of <−40 kcal/mol with its nontargets, and a negative probe had a sequence identity of ≤85%, a stretch length of ≤20, and free energy of ≥−40 kcal/mol with its nontargets (12). In addition, the probes that did not qualify for either the positive pool or the negative pool were ignored for further analysis.
(ii) Microarray spot analysis.
Spot intensity data were extracted from ImaGene output files. The values for gene ID, flag, signal mean (S̄), background mean (B̄), signal standard deviation (σs), and background standard deviation (σb) were extracted from ImaGene output files. After the removal of bad spots, the rest of the spots (including potential empty spots and good spots) were kept for further analysis. All processes were conducted with Microsoft Excel software.
(iii) Calculation of SNR values.
For each spot, three methods were used to calculate SNR values:
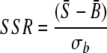 |
(1) |
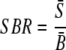 |
(2) |
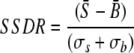 |
(3) |
where S̄ and B̄ are the signal mean and the background mean of pixel intensities, respectively, and σs and σb are the standard deviations of signal and background, respectively. Based on false-positive (FP) and false-negative (FN) spots at different values of the signal-to-standard-deviation ratio (SSR), the signal-to-background ratio (SBR), and the signal-to-both-standard-deviations ratio (SSDR) (in comparison with the defined positive and negative spot pools), their thresholds were determined by (i) minimizing FPs, (ii) minimizing FNs, and (iii) optimizing the overall percentage of FPs and FNs.
(iv) Student t test analysis of threshold-identified positive spots.
The values of signal (S) and background (B) for a probe with replicate spots were extracted from ImaGene output files, and their means (S̄m and B̄m, respectively) and standard deviations (σs,m and σb,m, respectively) were calculated. Outliers were removed if S − S̄m was greater than or equal to 2.0 × σs,m or B − B̄m was greater than or equal to 2.0 × σb,m, and this process continued until outliers were recursively removed. The final S̄m, B̄m, σs,m, and σb,m were used for the Student t test, and the significance between S̄m and B̄m was statistically evaluated for each probe at a given P value.
Data analysis for D. vulgaris Hildenborough microarrays.
Both wild-type and Δfur mutant D. vulgaris cells were grown in LS4D medium with 60 μM of iron, and microarray data were obtained as previously described (3). The SSDR method was used to detect positive spots with a threshold of 0.80, and details of data analysis were conducted as previously described (3).
Data analysis for GeoChip with a soil sample.
A soil sample was taken from a plot at BioCON (23), and 5 g of soil was used to extract DNA. GeoChip (13) was used to detect functional genes in such a microbial soil community. SSR, SBR, and SSDR were used to detect positive spots with thresholds of 2.0, 1.6, and 0.8, respectively, and details of labeling, hybridization, and scanning were performed as described previously (13).
RESULTS
New SNR calculation method.
To consider the signal intensity and background noise, as well as their standard deviations for each spot, a new calculation method, termed SSDR, was developed. SSDR differs from other two SNR calculation methods (SSR and SBR) in that it takes into account the signal standard deviation as a part of the denominator. The relationship between the SSDR and signal or background intensity (together with their standard deviations) can be simply represented as in Fig. 1, which shows that both signal and background standard deviations are equally important for the determination of SNR thresholds. When the SSDR is ≥1.0, the difference between the signal intensity and the background noise is equal to or larger than the sum of the signal and background standard deviations. In this case, the pixel values of signal intensity are completely separated from those of background noise (Fig. 1). Intuitively, such a spot should represent positive signal. When the SSDR is <1.0, overlaps of the pixel values between signals and background noise exist (Fig. 1). In this case, some spots could be positive while some are not, but the key question is what is the minimum SNR (e.g., the SSDR) threshold for distinguishing the signal from its background noise. Thus, in this study, we experimentally determined the threshold of SSDR for differentiating signals from noise.
FIG. 1.

Schematic presentation of the SSDR calculation method. A, B, and C represent SSDRs of <1.0, 1.0, and >1.0, respectively. All four parameters used in the calculation were extracted from the ImaGene output files (ImaGene manual). The error bars represent standard deviations.
Experimental determination of SNR thresholds.
To determine appropriate thresholds for distinguishing signal from noise for a single spot on the array, four synthesized targets were hybridized with the array at a final concentration of 10 pg per oligonucleotide. Based on the predefined positive and negative pools at low stringency, 60 (27 for 50-mer; 33 for 70-mer) probes were expected to be positive, 249 negative, and 59 ignored (see Table S2 in the supplemental material). The ignored probes failed to satisfy the definition of positive or negative spots. Based on the predicted pools of the positive and negative spots, the numbers of FP and FN spots were calculated for different scenarios. First, FP spots were minimized. To have no FPs, the thresholds of the SSR, SBR, and SSDR should be 5.0, 5.0, and 1.0, respectively (Table 1 and Fig. 2). If 1% FP spots were allowed, the thresholds were 4.0 for the SSR, 3.5 for the SBR, and 0.90 for the SSDR (Table 1 and Fig. 2). The thresholds would be 2.0, 1.8, and 0.70 for the SSR, SBR, and SSDR, respectively, when 5% FP spots could be tolerated (Table 1 and Fig. 2). Second, FNs were minimized. The thresholds of the SSR, SBR, and SSDR should be 0.5, 0.5, and 0.3, respectively, if there were no FN spots (Table 1 and Fig. 2). If 1% FN spots were allowed, the thresholds were 1.5 for the SSR, 1.2 for the SBR, and 0.70 for the SSDR (Table 1 and Fig. 2). The thresholds would be 2.5, 1.6, and 0.85 for the SSR, SBR, and SSDR, respectively, when 5% FN spots were allowed (Table 1 and Fig. 2). In addition, the thresholds of the SSR, SBR, and SSDR were determined by optimizing the total percentage of FP and FN spots. Generally speaking, higher percentages of FPs were observed at a lower threshold of the SSR, SBR, or SSDR. For example, the percentages of FPs were 11.8%, 12.2%, and 7.9% at an SSR of 1.5, an SBR of 1.4, and an SSDR of 0.5, respectively, which led to 13.0%, 14.9%, and 8.3% total percentages of FP and FN spots, respectively (Fig. 2). On the other hand, higher percentages of FNs were observed at a higher threshold of the SSR, SBR, or SSDR. For example, the percentages of FNs were 17.1%, 19.0%, and 12.8% at an SSR of 4.0, an SBR of 4.0, and an SSDR of 1.2, respectively, resulting in 18.2%, 19.9%, and 13.1% total percentages of FP and FN spots, respectively (Fig. 2). However, relatively low and stable percentages of FP and FN spots were observed when the values of the SSR, SBR, or SSDR were in a certain range. For example, when SSRs were between 2.0 and 3.0, the percentages of FP and FN spots were 8.0 to 9.7%; those percentages were 10.0 to 14.9% when SBRs were 1.4 to 3.0; and SSDRs were 0.6 to 1.0 when those percentages were 5.0 to 8.0% (Fig. 2). Therefore, the above-mentioned results indicate that the thresholds of the SSR, SBR, and SSDR can be in a certain range with a relatively low percentage of FP and FN spots, although optimal thresholds were determined to be an SSR of 2.5, an SBR of 1.6, and an SSDR of 0.80.
TABLE 1.
Thresholds of SSR, SBR, and SSDR determined by minimizing the percentage of FP or FN spots on the array using synthesized oligonucleotide targets under low and high stringencies
| Spot | Threshold
|
||
|---|---|---|---|
| SSR | SBR | SSDR | |
| Low stringency | |||
| No FP | 5.0 | 5.0 | 1.00 |
| 1% FP | 4.0 | 3.5 | 0.90 |
| 5% FP | 2.0 | 1.8 | 0.70 |
| 5% FN | 2.5 | 1.6 | 0.85 |
| 1% FN | 1.5 | 1.2 | 0.70 |
| No FN | 0.5 | 0.5 | 0.30 |
| High stringency | |||
| No FP | 5.0 | 5.0 | 1.10 |
| 1% FP | 4.5 | 4.0 | 1.00 |
| 5% FP | 2.5 | 2.0 | 0.70 |
| 5% FN | 3.0 | 1.8 | 0.95 |
| 1% FN | 2.0 | 1.4 | 0.75 |
| No FN | 1.0 | 1.0 | 0.50 |
FIG. 2.

Determination of thresholds of the SSR (A), SBR (B), and SSDR (C) at low stringency by minimizing the percentages of FP and FN spots. Ten picograms of each synthesized oligonucleotide was used to hybridize with the array, and five replicate slides were used. The SSR, SBR, and SSDR were determined to be 2.5, 1.6, and 0.80, respectively. The error bars represent standard deviations.
Under high stringency, 33 (13 for 50-mer and 20 for 70-mer) probes were positive, 280 (147 for 50-mer and 133 for 70-mer) were negative, and 55 were ignored (see Table S3 in the supplemental material). The thresholds of the SSR, SBR, and SSDR were determined using the same strategies described above. First, through the minimization of FPs, the thresholds of the SSR, SBR, and SSDR were determined to be 5.0, 5.0, and 1.1, respectively, when no FP spots were allowed; those thresholds were 4.5 for the SSR, 4.0 for the SBR, and 1.0 for the SSDR if 1% FP spots were allowed; if 5% FP spots were tolerated, those thresholds of the SSR, SBR, and SSDR were 2.5, 2.0, and 0.70, respectively (Table 1 and Fig. 3). Second, through the minimization of FNs, the thresholds of the SSR, SBR, and SSDR were determined to be approximately 1.0, 1.0, and 0.5, respectively, when no FN spots were allowed; if 1% FN spots were allowed, those thresholds were 2.0 for the SSR, 1.4 for the SBR, and 0.75 for the SSDR; they would be 3.0 for the SSR, 1.8 for the SBR, and 0.95 for the SSDR if 5% FN spots were tolerated (Table 1 and Fig. 3). Finally, by optimizing the total percentage of FP and FN spots on the array, the thresholds of the SSR, SBR, and SSDR were determined to be 3.0, 2.0, and 0.90, respectively (Fig. 3). The results demonstrated that the thresholds of the SSR, SBR, and SSDR increased with an increase in the stringencies of defined positive and negative probe pools. In addition, both Fig. 2 and 3 show that the lowest percentages of FP and FN spots were observed with the SSDR calculation and that an optimization of the percentage of FPs and FNs appeared to be the best method for SNR determination. Therefore, for further experiments, the defined positive and negative pools with low stringencies were used, and an optimization of FPs and FNs was considered the best method for SNR determination.
FIG. 3.

Determination of thresholds of the SSR (A), SBR (B), and SSDR (C) at high stringency by minimizing the percentages of FP and FN spots. Ten picograms of each synthesized oligonucleotide was used to hybridize with the array, and five replicate slides were used. The SSR, SBR, and SSDR were determined to be 3.0, 2.0, and 0.90, respectively. The error bars represent standard deviations.
Effects of target types on SNR threshold determination.
To determine the impacts of target types on threshold selection, 100 pg of each PCR amplicon or 500 ng of S. oneidensis MR-1 gDNA was also labeled with Cy3 and hybridized with the array, and the thresholds of the SNR, SBR, and SSDR were determined by optimizing the percentages of FN and FP spots. The same thresholds were obtained for PCR amplicon targets as for the synthesized oligonucleotides, although the PCR amplicon targets caused slightly higher percentages of total FN and FP than synthesized oligonucleotides. For example, the thresholds of the SSR were 2.5 for oligonucleotide and PCR amplicon targets when the percentages of FPs and FNs were 8.0% and 8.7%, respectively (Fig. 4A). However, the SSR threshold of 2.0 (Fig. 4A) and SSDR threshold of 0.70 (Fig. 4C) for gDNA were lower than those for synthesized oligonucleotides or PCR amplicons. The percentages of total FNs and FPs of gDNA were a bit higher than those of synthesized oligonucleotide or PCR amplicon targets (Fig. 4). For example, the percentage of FN and FP was 7.1% for gDNA compared to 5.0% for oligonucleotide targets and 6.51% for PCR targets when SSDR thresholds of 0.8, 0.8, and 0.7 were used for oligonucleotide, PCR amplicon, and gDNA targets, respectively (Fig. 4C). In contrast to the SSR and SSDR, the SBR remained unchanged with different types of targets. The results also confirmed that the lowest percentage of FPs and FNs was observed with the SSDR calculation method.
FIG. 4.

Effects of target types on the thresholds and the percentages of FPs, FNs, and both (FP+FN) for the SSR (A), SBR (B), and SSDR (C). The left y axes present the optimal thresholds, and the right y axes present the percentages of FP, FN, or FP plus FN under the optimal threshold. The targets used were synthesized oligonucleotides (10 pg each), PCR amplicons (100 pg each), and S. oneidensis MR1 gDNA (500 ng). The more significant P value is shown on the top of each column, with the following notations: nd, no difference; one asterisk, P < 0.10; two asterisks, P < 0.05; and three asterisks, P < 0.01 (the Student t test) when one type of target was compared with two others.
Effects of background DNA on threshold determination.
When microarrays are used for community analysis, significant amounts of DNAs from nontarget organisms exist as background, and they could affect SNR threshold determination. To examine the effects of such background DNA on the SSR, SBR, and SSDR thresholds, 500 ng of S. oneidensis gDNA, or 10 pg per oligonucleotide target, was mixed with 1.0 μg of the yeast gDNA, and their thresholds were determined as described in the legend to Fig. 2. With the yeast gDNA as background, the thresholds of the SSR and SSDR for S. oneidensis gDNA were determined to be 1.75 and 0.65, respectively, which were slightly lower than those without the yeast gDNA as background (Fig. 5A). Similarly, the thresholds of the SSR and SSDR changed from 2.5 and 0.80 to 2.0 and 0.70, respectively, when synthesized oligonucleotide targets were spiked into the yeast gDNA (Fig. 5B). However, the thresholds of the SBR did not change with the target type or the background DNA (Fig. 5). These results indicate that the thresholds of the SSR and SSDR decreased with the addition of yeast gDNA as background but that the threshold of the SBR stayed the same.
FIG. 5.

Effects of background DNA on the determination of SSR, SBR, and SSDR thresholds. Five hundred nanograms of S. oneidensis MR-1 gDNA (A) and 10 pg for each synthesized oligonucleotide (oligo) (B) were spiked into 1.0 μg of yeast gDNA. For synthesized oligonucleotide targets, the yeast gDNA was first labeled and then mixed with the spiked oligonucleotides. S. oneidensis MR-1 gDNA was first mixed with the yeast gDNA and then labeled together. The significance is shown on the top of each column, with the following notations: nd, no difference; one asterisk, P < 0.10; two asterisks, P < 0.05; and three asterisks, P < 0.01 (the Student t test) when thresholds with background DNA were compared to those without background DNA.
To further understand why the background DNA caused a decrease in the thresholds of the SSR and SSDR, the changes in signal mean, background mean, and their standard deviations for each spot with the yeast DNA as nontarget DNA were compared with those without the yeast DNA (Fig. 6). When the yeast gDNA was added to the S. oneidensis gDNA, the trends of the signal mean and the background mean did not change, but the average signal and background standard deviations increased to 124% and 134%, respectively, compared to S. oneidensis gDNA only (Fig. 6A). Similarly, when the oligonucleotide targets were used as target templates with the background yeast gDNA, the average signal mean and the average background mean did not change significantly, but the average signal and background standard deviations increased to 129% and 148%, respectively, in comparison with the oligonucleotide targets only (Fig. 6B). These results indicated that an increase in both signal and background standard deviations might result in lower thresholds of the SSR and SSDR when nontarget DNAs are present.
FIG. 6.

Comparison of changes in signal mean, background (Bkgrd.) mean, signal standard deviation (std. dev.), and background standard deviation for each spot on the array when the yeast gDNA was added to the S. oneidensis gDNA (A) or the synthesized oligonucleotide (Oligo) targets (B). The error bars represent standard deviations.
Determination of SNR thresholds for artificial bacterial mixtures.
To examine how DNA mixtures with different compositions affect the SNR threshold determination, S. oneidensis gDNA was mixed with four other bacteria in the ratios A, 10:1:1:1:1; B, 1:1:1:1:1, and C, 1:10:10:10:10, and each mixture had 2.50 μg of gDNA in total. The optimal thresholds of the SSR, SBR, and SSDR were determined to be 2.00, 1.60, and 0.70, respectively, for mixture A and 1.75, 1.60, and 0.60, respectively, for mixture B (Table 2). Only about 23.3% of the defined positive spots were detected on the array for mixture C, so no thresholds of the SSR, SBR, or SSDR could be estimated (Table 2). The results showed that the thresholds of the SSR and SSDR were decreased with a decrease in the percentage of the target (S. oneidensis gDNA) in the sample but that the thresholds of the SBR were not affected, which is also consistent with the results observed with different types of target or with the yeast DNA. It is possible that a decrease in the target concentration in a mixed sample might lead to a higher rate for FNs or/and FNs plus FPs.
TABLE 2.
Thresholds of SSR, SBR, and SSDR and the percentages of FNs, FPs, or both for artificial bacterial mixtures
| Parameter | Valuea
|
||
|---|---|---|---|
| Mixture A (10:1:1:1:1) | Mixture B (1:1:1:1:1) | Mixture C (1:10:10:10:10) | |
| No. of defined positive spots | 300 | 300 | 300 |
| % of detected positive spots | 318 | 311 | 70 |
| SSR | |||
| Threshold | 2.0 | 1.75 | ND |
| % FP | 4.3 | 3.5 | 0 |
| % FN | 3.3 | 3.4 | 76.7 |
| % Total FP and FN | 7.6 | 6.9 | 76.7 |
| SBR | |||
| Threshold | 1.60 | 1.60 | ND |
| % FP | 4.7 | 3.6 | 0 |
| % FN | 3.3 | 4.7 | 76.7 |
| % Total FP and FN | 8.0 | 8.3 | 76.7 |
| SSDR | |||
| Threshold | 0.70 | 0.60 | ND |
| % FP | 2.7 | 2.2 | 0 |
| % FN | 2.8 | 3.7 | 76.7 |
| % Total FP and FN | 5.5 | 5.9 | 76.7 |
Genomic DNAs from mixtures A, B, and C containing S. oneidensis MR-1 (boldface) and four other bacteria at different ratios were used as targets. SSR, SBR, SSDR, and percentages of FPs and FNs were determined as described in the legend to Fig. 2. Five slides were used. ND, not determined.
Verification of identified positive spots.
To further understand if the identified positive spots based on the above-mentioned thresholds had signals significantly higher than their backgrounds, the Student t test was used to determine if a probe with replicate spots was positive at a given P value. Since gDNA is the most commonly used target, this experiment was carried out with S. oneidensis MR-1 gDNA (500 ng). The predefined positives (at a low stringency), the t test-identified positives (at P < 0.01), and SNR threshold-identified (2.0 for SSR, 1.6 for SBR, and 0.70 for SSDR) positives were compared, and relatively consistent results were observed (Table 3). Among 368 probes, 60, 249, and 59 were defined as positive, negative, and ignored, respectively, under low stringency. Based on the t test, a total of 76 probes were identified as positive, with 57 from the defined positives, 4 from the defined negatives, and 15 from the ignored pool at P < 0.01. (A total of 292 probes were identified as negatives, with 3 from the defined positives, 245 from the defined negatives, and 44 from the ignored pool at P < 0.01.) Numbers of positives similar to the t test analysis were identified based on the SNR thresholds determined above. For example, at the SSDR threshold of 0.70, 81, 79, and 75 positives were identified at positive rates of >50%, >70%, and >90%, respectively (Table 3). These results demonstrated that the positive spots or probes identified by SNR thresholds and by the Student t test were very similar, which was also consistent with the predefined positives and negatives.
TABLE 3.
Comparison of positive probes identified by probe design criteria, by the Student t test, and by SNR thresholdsa
| Identifier | Threshold | No. of identified positives (% of t test positives)b
|
||
|---|---|---|---|---|
| PRc > 50% | PR > 70% | PR > 90% | ||
| SSR | 2.0 | 58 + 7 + 21 = 86 (113) | 58 + 5 + 19 = 82 (108) | 57 + 3 + 18 = 78 (103) |
| SBR | 1.6 | 58 + 8 + 25 = 91 (120) | 57 + 6 + 23 = 86 (113) | 56 + 4 + 20 = 80 (105) |
| SSDR | 0.70 | 59 + 4 + 18 = 81 (107) | 59 + 3 + 17 = 79 (104) | 58 + 1 + 16 = 75 (99) |
SNR threshold-identified positive probes at different positive rates; 368 probes were valid for analysis when 500 ng of labeled S. oneidensis MR1 gDNA hybridized with the array. Five slides were used with four replicates in each slide, so each probe had up to 20 spots.
The first three numbers are the size of the defined positive probe pool, the size of the defined negative probe pool, and the number of ignored probes based on defined positive, negative, and ignored probe pools.
PR = (number of positive spots identified by SNR thresholds × 100)/total number of spots for each probe.
Determination of positive spots by SSDR threshold for pure culture and soil samples.
To demonstrate the application of SSDR thresholds for determining positive spots, two sets of data were used. One was pure cultures of wild-type and Δfur mutant (JW707) D. vulgaris Hildenborough with the D. vulgaris Hildenborough oligonucleotide microarray (3), and the other was a BioCON soil sample with GeoChip (13). For the first data set, an SSDR threshold of 0.80 was used. The average SSDR for the fur probe was 0.25 for the Δfur mutant and 2.16 for the wild type, confirming the absence of the gene in the mutant (Table 4). Fur is a transcriptional regulator, and it negatively regulates several genes in the fur regulon when it binds to a promoter. The microarray data did show that genes such as feoA, feoB, fld, and gdp, predicted in the fur regulon (25), were up-regulated in the mutant JW707 (Table 4). The Fur regulator has been shown to be involved in oxidative-stress responses, which are mainly controlled by the PerR regulator (25). Indeed, our results also showed that ahpC, rbr, and perR were overexpressed in the JW707 mutant (Table 4). In addition, it was observed that the expression of genes (cobI, cluster of orthologous groups [COG] fepB, fepC, and COG fepD) involved in iron uptake was repressed and that the expression of genes (bfr and ftn) involved in iron storage was induced (Table 4). This is consistent with the fact that more iron may accumulate in the mutant due to the absence of the Fur protein. It should be noted that different cutoffs for up-regulation and down-regulation were used in this study (twofold) and the previous study (3).
TABLE 4.
Examples of transcriptional changes of genes of known function in Δfur mutant (JW707) and wild-type D. vulgaris Hildenborough
| Category/locus tag | Gene | Annotated function | SSDRa (mean ± SD) (n = 6)
|
Expression ratio (JW707/WT) | |
|---|---|---|---|---|---|
| JW707 | WT | ||||
| Genes in the predicted Fur regulonb | |||||
| DVU0303 | genZ | GenZ, hypothetical protein | 2.16 ± 0.285 | 1.78 ± 0.172 | 2.11 |
| DVU0304 | genY | GenY, hypothetical protein | 2.47 ± 0.277 | 2.15 ± 0.122 | 2.27 |
| DVU0763 | gdp | GGDEF domain protein | 2.28 ± 0.321 | 1.96 ± 0.231 | 4.08 |
| DVU0942 | fur | Fur, transcriptional regulator | 0.25 ± 0.036 | 2.16 ± 0.116 | NDc |
| DVU2571 | feoB | Ferrous iron transport protein B | 1.97 ± 0.166 | 1.95 ± 0.142 | 1.96 |
| DVU2572 | feoA | Ferrous iron transport protein A | 1.72 ± 0.321 | 1.83 ± 0.211 | 1.76 |
| DVU2574 | feoA | Ferrous ion transport protein | 2.17 ± 0.277 | 1.93 ± 0.102 | 2.67 |
| DVU2680 | fld | Flavodoxin | 1.88 ± 0.130 | 2.06 ± 0.133 | 1.50 |
| Genes in the predicted PerR regulonb | |||||
| DVU2247 | ahpC | Antioxidant, AhpC/Tsa family | 2.02 ± 0.220 | 2.03 ± 0.186 | 2.12 |
| DVU2318 | rbr | Rubrerythrin, putative | 2.47 ± 0.277 | 1.76 ± 0.122 | 2.94 |
| DVU3095 | perR | PerR, transcriptional regulator | 1.88 ± 0.213 | 1.90 ± 0.133 | 1.61 |
| Other iron-related genes | |||||
| DVU0646 | cobI | Precorrin-2 C20-methyltransferase | 1.28 ± 0.096 | 2.35 ± 0.182 | 0.30 |
| DVU0647 | COGfepB | Iron compound ABC transporter, iron-binding protein | 0.93 ± 0.071 | 2.11 ± 0.171 | 0.14 |
| DVU0648 | fepC | Iron compound ABC transporter, ATP-binding protein | 1.17 ± 0.277 | 1.84 ± 0.132 | 0.25 |
| DVU0649 | COGfepD | Iron compound ABC transporter, permease protein | 1.20 ± 0.076 | 2.26 ± 0.119 | 0.28 |
| DVU1397 | bfr | Bacterioferritin | 2.27 ± 0.217 | 2.16 ± 0.212 | 1.97 |
| DVU1568 | ftn | Ferritin | 1.89 ± 0.173 | 2.33 ± 0.222 | 1.89 |
The SSDR was calculated from Cy5-labeled cDNA signal, while Cy3-labeled gDNA was used for both JW707 and the wild type (WT).
Predicted by Rodionov et al. (25).
ND, not determined due to lack of a Cy5 signal of the Δfur mutant.
Despite our successful demonstration of the application of the SSDR to pure cultures, a similar demonstration with environmental samples, such as soil, is much more difficult. Thus, in this study, we used one soil sample with three hybridizations to see the number of detected positive spots and their unique and overlap spots among replicates (Table 5). With thresholds of 2.0 for the SSR, 1.6 for the SBR, and 0.80 for the SSDR, the average numbers of detected spots were 3,858, 4,372, and 3,828 for the SSR, SBR, and SSDR, respectively (Table 5). Although the fewest positive spots (3,903) were detected by the SSDR, it had the highest number (3,761) and the highest rate (96.3%) of overlap spots but the lowest number (97) and rate (2.5%) of unique spots, indicating that the SSDR is a more accurate method to discriminate true signals from background noise (Table 5). Therefore, the above-mentioned results demonstrated that the SSDR method, with an appropriate threshold, could be used to determine positive spots for both pure culture and environmental (e.g., soil) samples.
TABLE 5.
Numbers of detected, unique, and overlap spots among replicates A, B, and Ca
| Parameter | Value
|
||
|---|---|---|---|
| SSR | SBR | SSDR | |
| Threshold | 2.0 | 1.6 | 0.80 |
| No. of detected positive spots (mean ± SD) | 3,858 ± 157 | 4,372 ± 322 | 3,828 ± 60 |
| Total no. of positive spots (A ∪ B ∪ C) | 4,132 | 4,743 | 3,903 |
| No. (%) of unique positive spots among three replicates | 232 (5.6) | 566 (12) | 97 (2.5) |
| No. (%) of overlapped positive spots among two replicates [(A ∩ B) U (A ∩ C) U (B ∩ C)] | 263 (6.4) | 521 (11) | 45 (1.2) |
| No. (%) of overlapped positive spots among three replicates (A ∩ B ∩ C) | 3,637 (88) | 3,656 (77) | 3,761 (96.3) |
Three different methods, SSR, SBR, and SSDR, and their predetermined thresholds were used for the detection of positive spots.
DISCUSSION
How to distinguish a real signal from its background remains challenging in microarray data analysis, and this study focused on the experimental determination of SNR thresholds. The determination of SNR thresholds is an important step for the generation of high-quality microarray data, and its accuracy is critical for the subsequent data processing and biological interpretation of microarray results. Thus, this study experimentally determined the thresholds of the SNR in different scenarios. The results of the study should provide guidance for users to select appropriate SNR thresholds for their experiments.
Considering the standard deviations of pixel intensities of both signal and background, a new calculation method was developed. It had two advantages. First, the signal standard deviation was considered as a parameter together with the background standard deviation. Since the pixel intensities of a spot are not uniform, its standard deviation significantly affects the ability to distinguish a true signal from its background. In this case, consideration of the signal standard deviation can more accurately reflect microarray hybridization behaviors and more reliably identify a true spot and its threshold. Second, our experimental data demonstrated that fewer FPs and NPs were observed with this method than with two other methods. The SBR did not change with target types or background DNA, since this calculation does not consider the signal standard deviation or background standard deviation, but it generally had a high percentage of FN and FP spots, and it may not be a good parameter to distinguish a true signal from its background noise. Therefore, this new method may be used for a general SNR calculation, and more accurate thresholds could be obtained with this calculation.
Three possible scenarios, minimizing FPs, minimizing FNs, and optimizing FPs and FNs, were considered to determine the ranges of SNR thresholds for detecting real signals, but the threshold values for optimal FPs and FNs could be used more often. By optimizing the percentage of FP and FN spots, those thresholds of the SSR and SBR determined in this experiment appeared to be lower than other commonly accepted thresholds. For example, the threshold of the SSR was set to 3.0 (30) and that of the SBR to 1.50 (26) or 2.0 (19). Considering all three methods for SNR determination, the ranges of SNR thresholds for gDNA targets are summarized in Table 6. For example, the thresholds of the SSR were in the range of 0.5 (no FN) and 2.0 (optimal) to 4.0 (no FP), and those of the SSDR were in the range of 0.3 (no FN) and 0.7 (optimal) to 0.9 (no FP) under low-stringency conditions. Those ranges provide a general guideline for users to select appropriate SNR thresholds based on their experiments. Two points need to be mentioned. One is that an error rate of 5% (FP plus FN) was used in this study, which is considered reasonable, since microarray data have relatively high variations due to various reasons, such as the small size, degrees of uniformity of printing pins, and uneven hybridization. The other is that the SNR threshold values determined here for DNA microarray studies under different stringencies and different target types or/and concentrations may be applied only to long (50- to 70-mer) oligonucleotide microarrays. The application of such parameters to short (18- to 25-mer) oligonucleotide microarrays remains unclear and needs to be further evaluated.
TABLE 6.
Summary of ranges of experimentally determined SNR thresholds under low- and high-stringency conditions using the S. oneidensis MR1 gDNA target
| Ratio | Threshold
|
|||||
|---|---|---|---|---|---|---|
| Low stringency
|
High stringency
|
|||||
| No FN | Optimal | No FP | No FN | Optimal | No FP | |
| SSR | 0.5 | 2.0 | 4.0 | 1.0 | 2.5 | 4.5 |
| SBR | 0.5 | 1.6 | 4.0 | 1.1 | 1.8 | 4.5 |
| SSDR | 0.3 | 0.7 | 0.9 | 0.4 | 0.8 | 1.0 |
It is known that probe specificity and the stringency of hybridization conditions affect the determination of SNR thresholds. Two stringency conditions were used in this study. As expected, a lower threshold (e.g., SSR = 2.0, SBR = 1.6, and SSDR = 0.80) can be used for detecting specific hybridizations under high-stringency hybridization conditions (e.g., at a high temperature of 50°C), and a higher threshold (e.g., SSR = 3.0, SBR = 2.0, and SSDR = 0.90) may be required for detecting specific hybridizations under low-stringency hybridization conditions (e.g., at a low temperature of 42°C).
Many factors, such as target type, background DNAs, target composition, and target amount in the tested sample, affect the SNR threshold determination. The microarray hybridization signal intensity is determined by the number of probe molecules bound to the microarray surface, the number of labeled targets present in the sample, and their ratios, which are closely related to the target type and their concentrations. In this study, the synthesized oligonucleotides and PCR amplicons were the simplest targets, they are similar, and they had almost the same thresholds. S. oneidensis MR-1 gDNA is more complex, and its threshold was a bit lower. Similarly, the complexity of the target was expected to increase in the presence of background DNA, and hence, a lower threshold was observed. Further analysis revealed that this might be due to an increase in the background standard deviation. This was validated by the fact that the thresholds of the SBR did not change with the target type or with the background DNA. With the mixed templates, mixture A contained >70% real target (S. oneidensis gDNA), and the threshold did not change significantly. However, a slight decrease in threshold was observed in mixture B, with 20% real target, and it became undeterminable for mixture C, containing about 2.5% real target. The decrease in the thresholds with a decrease in the target template composition can be explained by an increase in sample noise when the target concentration decreased. Sample noise is mostly from labeled molecules in a sample. For example, labeled target solutions can react in a nonspecific manner on microarrays, which masks the interactions between a probe and its target and obscures the microarray signal. Therefore, an increase in nontarget concentrations leads to an increase in noise, which may reduce SNR thresholds to compromise microarray detectability. This is also consistent with our observations for different types of target or with background DNAs, since labeled nontargets, such as background DNAs, cause a significant amount of background noise.
As previous studies showed, the detection limits for 50-mer oligonucleotide and 70-mer oligonucleotide arrays were estimated to be 25 to 100 ng of gDNA (11) for a pure culture, although a higher sensitivity (5 to 10 ng gDNA) was also observed (24, 29). In the presence of background DNA, the detection limit for a 50-mer oligonucleotide was estimated to be 50 to 100 ng of gDNA (24, 29). In mixture C, the real target was about 63 ng of gDNA, so it was not surprising that only 23.3% of defined positive probes had true signals. These results suggest that a threshold might change with the target composition, which is closely related to the microarray sensitivity.
It was also noted that the amount of target might affect the threshold determination. For example, a higher threshold might be required when a relatively large amount of target is used. In this study, we used the optimal concentrations of 10 pg for each oligonucleotide, 100 pg for each PCR amplicon, and 500 ng for gDNA, which are considered equivalent amounts of the target in samples. This is a simulation for a pure culture or a mixture of a few known microorganisms. For a sample with many unknown microorganisms, such as microbial communities in soil and the human intestinal tract, a determination of SNR thresholds may be even more challenging. Because of unequal abundances, low-abundance genes/microorganisms may not be detected even at a relatively low threshold.
In summary, three methods were used to calculate SNR values, and the newly developed calculation showed a better performance for distinguishing a true signal from its background than the other two methods. The positives identified based on SNR thresholds were verified by the Student t test across many replicate data, and consistent results were obtained. This study provides guidance for the selection of SNR thresholds for different samples, such as PCR amplicons and gDNAs from pure cultures and simple mixed cultures.
Supplementary Material
Acknowledgments
We thank Meiying Xu for providing GeoChip data on the BioCON soil sample and Yuting Liang for providing D. vulgaris Hildenborough microarray data on both the wild type and the Δfur mutant.
This research was supported by the U.S. Department of Energy under the Genomics:GTL program through the Virtual Institute of Microbial Stress and Survival (VIMSS) (http://vimss.lbl.gov) and the Environmental Remediation Science Program.
Footnotes
Published ahead of print on 14 March 2008.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Aakra, A., O. L. Nyquist, L. Snipen, T. S. Reiersen, and I. F. Nes. 2007. Survey of genomic diversity among Enterococcus faecalis strains by microarray-based comparative genomic hybridization. Appl. Environ. Microbiol. 73:2207-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basarsky, T., D. Verdnik, D. Willis, and J. Zhai. 2000. An overview of a DNA microarray scanner: design essentials for an integrated acquisition and analysis platform. In M. Schena (ed.), Microarray biochip technology. Eaton Publishing, Natick, MA.
- 3.Bender, K. S., B. C. B. Yen, C. L. Hemme, Z. Yang, Z. He, Q. He, J. Zhou, K. H. Huang, E. J. Alm, T. C. Hazen, A. P. Arkin, and J. D. Wall. 2007. Analysis of a ferric uptake regulator (Fur) mutant of Desulfovibrio vulgaris Hildenborough. Appl. Environ. Microbiol. 73:5389-5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bodrossy, L., and A. Sessitsch. 2004. Oligonucleotide microarrays in microbial diagnostics. Curr. Opin. Microbiol. 7:245-254. [DOI] [PubMed] [Google Scholar]
- 5.Bodrossy, L., N. Stralis-Pavese, M. Konrad-Köszler, A. Weilharter, T. G. Reichenauer, D. Schöfer, and A. Sessitsch. 2006. mRNA-based parallel detection of active methanotroph populations by use of a diagnostic microarray. Appl. Environ. Microbiol. 72:1672-1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brodie, E. L., T. Z. DeSantis, J. P. M. Parker, I. X. Zubietta, Y. M. Iceno, and G. L. Andersen. 2007. Urban aerosols harbor diverse and dynamic bacterial populations. Proc. Natl. Acad. Sci. USA 104:299-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Debouck, C., and P. N. Goodfellow. 1999. DNA microarrays in drug discovery and development. Nat. Genet. 21:48-50. [DOI] [PubMed] [Google Scholar]
- 8.Dziejman, M., E. Balon, D. Boyd, C. M. Fraser, J. F. Heidelberg, and J. J. Mekalanos. 2002. Comparative genomic analysis of Vibrio cholerae: genes that correlate with cholera endemic and pandemic disease. Proc. Natl. Acad. Sci. USA 99:1556-1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gresham, D., D. M. Ruderfer, S. C. Pratt, J. Schacherer, M. J. Dunham, D. Botstein, and L. Kruglyak. 2006. Genome-wide detection of polymorphisms at nucleotide resolution with a single DNA microarray. Science 311:1932-1936. [DOI] [PubMed] [Google Scholar]
- 10.Han, W., B. Liu, B. Cao, L. Beutin, U. Krüger, H. Liu, Y. Li, Y. Liu, L. Feng, and L. Wang. 2007. DNA microarray-based identification of serogroups and virulence gene patterns of Escherichia coli isolates associated with porcine postweaning diarrhea and edema disease. Appl. Environ. Microbiol. 73:4082-4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He, Z., L. Wu, M. W. Fields, and J. Zhou. 2005. Comparison of microarrays with different probe sizes for monitoring gene expression. Appl. Environ. Microbiol. 71:5154-5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He, Z., L. Wu, X. Li, M. W. Fields, and J. Zhou. 2005. Empirical establishment of oligonucleotide probe design criteria. Appl. Environ. Microbiol. 71:3753-3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He, Z., T. J. Gentry, C. W. Schadt, L. Wu, J. Liebich, S. C. Chong, Z. Huang, W. Wu, B. Gu, P. Jardine, C. Criddle, and J. Zhou. 2007. GeoChip: a comprehensive microarray for investigating biogeochemical, ecological, and environmental processes. ISME J. 1:67-77. [DOI] [PubMed] [Google Scholar]
- 14.Hinchliffe, S. J., K. E. Isherwood, R. A. Stabler, M. B. Prentice, A. Rakin, R. A. Nichols, P. C. Oyston, J. Hinds, R. W. Titball, and B. W. Wren. 2003. Application of DNA microarrays to study the evolutionary genomics of Yersinia pestis and Yersinia pseudotuberculosis. Genome Res. 13:2018-2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffman, C. S., and S. Winston. 1987. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene 57:267-272. [DOI] [PubMed] [Google Scholar]
- 16.Kim, I. J., H. C. Kang, S. G. Jang, S. A. Ahn, H. J. Yoon, and J. G. Park. 2007. Development and applications of a BRAF oligonucleotide microarray. J. Mol. Diagn. 9:55-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liebich, J., C. W. Schadt, S. C. Chong, Z. He, S. K. Rhee, and J. Zhou. 2006. Improvement of oligonucleotide design criteria for the development of functional gene microarrays for environmental applications. Appl. Environ. Microbiol. 72:1688-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loy, A., A. Lehner, N. Lee, J. Adamczyk, H. Meier, J. Ernst, K.-H. Schleifer, and M. Wagner. 2002. Oligonucleotide microarray for 16S rRNA gene-based detection of all recognized lineages of sulfate-reducing prokaryotes in the environment. Appl. Environ. Microbiol. 68:5064-5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loy, A., C. Schulz, S. Lücker, A. Schöpfer-Wendels, K. Stoecker, C. Baranyi, A. Lehner, and M. Wagner. 2005. 16S rRNA gene-based oligonucleotide microarray for environmental monitoring of the betaproteobacterial order “Rhodocyclales.” Appl. Environ. Microbiol. 71:1373-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maynard, C., F. Berthiaume, K. Lemarchand, J. Harel, P. Payment, P. Bayardelle, L. Masson, and R. Brousseau. 2005. Waterborne pathogen detection by use of oligonucleotide-based microarrays. Appl. Environ. Microbiol. 71:8548-8557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20a.Odom, J. M., and J. D. Wall. 1987. Properties of a hydrogen-inhibited mutant of Desulfovibrio desulfuricans ATCC 27774. J. Bacteriol. 169:1335-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quiñones, B., C. T. Parker, J. M. Janda, Jr., W. G. Miller, and R. E. Mandrell. 2007. Detection and genotyping of Arcobacter and Campylobacter isolates from retail chicken samples by use of DNA oligonucleotide arrays. Appl. Environ. Microbiol. 73:3645-3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ragoussis, J., and G. Elvidge. 2006. Affymetrix GeneChip system: moving from research to the clinic. Exp. Rev. Mol. Diagn. 6:145-152. [DOI] [PubMed] [Google Scholar]
- 23.Reich, P. B., J. Knops, D. Tilman, J. Craine, D. Ellsworth, M. Tjoelker, T. Lee, D. Wedin, S. Naeem, D. Bahauddin, G. Hendrey, S. Jose, K. Wrage, J. Goth, and W. Bengston. 2001. Plant diversity enhances ecosystem responses to elevated CO2 and nitrogen deposition. Nature 410:809-812. [DOI] [PubMed] [Google Scholar]
- 24.Rhee, S. K., X. Liu, L. Wu, S. C. Chong, X. Wan, and J. Zhou. 2004. Detection of biodegradation and biotransformation genes in microbial communities using 50-mer oligonucleotide microarrays. Appl. Environ. Microbiol. 70:4303-4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodionov, D. A., I. Dubchak, A. P. Arkin, E. Alm, and M. S. Gelfand. 2004. Reconstruction of regulatory and metabolic pathways in metal-reducing δ-proteobacteria. Genome Biol. 5:R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schena, M. 2003. Microarray analysis. John Wiley & Sons, Inc., Hoboken, NJ.
- 27.Small, J., D. R. Call, F. J. Brockman, T. M. Straub, and D. P. Chandler. 2001. Direct detection of 16S rRNA in soil extracts by using oligonucleotide microarrays. Appl. Environ. Microbiol. 67:4708-4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taroncher-Oldedburg, G., E. M. Griner, C. A. Francis, and B. B. Ward. 2003. Oligonucleotide microarray for the study of functional gene diversity in the nitrogen cycle in the environment. Appl. Environ. Microbiol. 69:1159-1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tiquia, S. M., L. Wu, S. C. Chong, S. Passovets, D. Xu, Y. Xu, and J. Zhou. 2004. Evaluation of 50-mer oligonucleotide arrays for detecting microbial populations in environmental samples. BioTechniques 36:664-675. [DOI] [PubMed] [Google Scholar]
- 30.Verdick, D., S. Handran, and S. Pickett. 2002. Key considerations for accurate microarray scanning and image analysis, p. 83-98. In G. Kamberova (ed.), DNA array image analysis: nuts and bolts. DNA Press LLC, Salem, MA.
- 31.Vora, G. J., C. E. Meador, M. M. Bird, C. A. Bopp, J. D. Andreadis, and D. A. Stenger. 2005. Microarray-based detection of genetic heterogeneity, antimicrobial resistance, and the viable but nonculturable state in human pathogenic Vibrio spp. Proc. Natl. Acad. Sci. USA 102:19109-19114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu, L., D. K. Thompson, X. D. Liu, M. W. Fields, C. E. Bagwell, J. M. Tiedje, and J. Zhou. 2004. Development and evaluation of microarray-based whole-genome hybridization for detection of microorganisms within the context of environmental applications. Environ. Sci. Technol. 38:6775-6782. [DOI] [PubMed] [Google Scholar]
- 33.Zhou, J. 2003. Microarrays for bacterial detection and microbial community analysis. Curr. Opin. Microbiol. 6:288-294. [DOI] [PubMed] [Google Scholar]
- 34.Zhou, J., M. R. Fries, J. C. Chee-Sanford, and J. M. Tiedje. 1995. Phylogenetic analyses of a new group of denitrifiers capable of anaerobic growth on toluene: description of Azoarcus tolulyticus sp. nov. Int. J. Syst. Bacteriol. 45:500-506. [DOI] [PubMed] [Google Scholar]
- 35.Zhou, S., K. Kassauei, D. J. Cutler, G. C. Kennedy, D. Sidransky, A. Maitra, and J. Califano. 2006. An oligonucleotide microarray for high-throughput sequencing of the mitochondrial genome. J. Mol. Diagn. 8:476-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.