

Abstract
Previously, we designed an internally controlled quantitative nested real-time (QNRT) PCR assay for Mycobacterium tuberculosis DNA in order to rapidly diagnose tuberculous meningitis. This technique combined the high sensitivity of nested PCR with the accurate quantification of real-time PCR. In this study, we attempted to improve the original QNRT-PCR assay and newly developed the wide-range QNRT-PCR (WR-QNRT-PCR) assay, which is more accurate and has a wider detection range. For use as an internal-control “calibrator” to measure the copy number of M. tuberculosis DNA, an original new-mutation plasmid (NM-plasmid) was developed. It had artificial random nucleotides in five regions annealing specific primers and probes. The NM-plasmid demonstrated statistically uniform amplifications (F = 1.086, P = 0.774) against a range (1 to 105) of copy numbers of mimic M. tuberculosis DNA and was regarded as appropriate for use as a new internal control in the WR-QNRT-PSR assay. In addition, by the optimization of assay conditions in WR-QNRT-PCR, two-step amplification of target DNA was completely consistent with the standard curve of this assay. Due to the development of the NM-plasmid as the new internal control, significantly improved quantitative accuracy and a wider detection range were realized with the WR-QNRT-PCR assay. In the next study, we will try to use this novel assay method with actual clinical samples and examine its clinical usefulness.
Tuberculous meningitis (TBM) is the severest form of infection of Mycobacterium tuberculosis, causing death or severe neurological defects in more than half of those affected in spite of antituberculosis treatment (25). The diagnosis of TBM remains a complex issue because the most widely used conventional bacteriological detection methods, such as direct smear for acid-fast bacilli and culture for M. tuberculosis, are unable to rapidly detect M. tuberculosis with sufficient sensitivity in the acute phase of TBM (7, 8, 11, 12, 18, 19, 21, 22, 23, 25). At present, the detection of M. tuberculosis DNA in cerebrospinal fluid (CSF) by use of PCR is widely used as a more rapid, sensitive, and specific diagnostic method (1, 7, 8, 10, 11, 12, 15, 17, 18, 19, 21, 22, 23, 26). Recently, we designed a novel internally controlled quantitative nested real-time PCR (QNRT-PCR) assay based on TaqMan PCR (Applied Biosystems) (22). This novel assay technique combines the high sensitivity of nested PCR with the accurate quantification of real-time PCR (22, 23). However, this original QNRT-PCR (OR-QNRT-PCR) assay is still unstable and has many points that should be improved (22, 23).
In this study, to reliably detect M. tuberculosis DNA in CSF samples with a wider detection range, we attempted to improve on the OR-QNRT-PCR technique; therefore, a new internal control for use as a “calibrator” was prepared. We named this improved assay technique wide-range QNRT-PCR (WR-QNRT-PCR) and examined its ability to quantitatively detect M. tuberculosis DNA in samples. In this paper, the development and methodology of the WR-QNRT-PCR assay are stated.
MATERIALS AND METHODS
This study was approved by the Nihon University Institutional Review Board.
Preparation of the new internal control (plasmid) for use as a calibrator.
For the WR-QNRT-PCR assay, two types of the original plasmid, wild plasmids (W-plasmids) and new-mutation plasmids (NM-plasmids), were prepared for a quantitative detection of M. tuberculosis DNA, and this was done as well for the OR-QNRT-PCR assay (22, 23).
W-plasmid, which was inserted into a 239-bp DNA fragment of the gene sequence encoding the MPB64 protein of M. tuberculosis (MPT64; GenBank accession no. NC_000962) (22, 23) into pCR 2.1 vector (Invitrogen Corp., San Diego, CA) was constructed for use as the standard template by the previously reported procedure (22, 23).
NM-plasmid was developed based on the previously reported M-plasmid (22, 23) for use as a new internal-control “calibrator” in the WR-QNRT-PCR assay. In NM-plasmid, a total of four regions, where two pairs of (outer and inner) forward and reverse primers annealed, were replaced with the artificial random nucleotides added to the TaqMan probe annealing region in the M-plasmid (Fig. 1). The sequences of the artificial random nucleotides were set to have the same nucleotide composition as MPT64 of wild M. tuberculosis. Replacing procedures were gradually performed by two steps (forward and reverse primer blocks) using the following four pairs of primers: Avr2-F1 and Avr2-R1, Avr2-F2 and Avr2-R2, Mlu1-F and Sal1-R, and Sal1-F and Mlu1-R (Fig. 1). The sequences and positions of these primers are shown in Table 1 and Fig. 1. These four pairs of primers were also specific for MPT64 and contained additional artificial random nucleotides including one restriction enzyme site at the 5′ end. Since each restriction enzyme site (Avr2, Mlu1, and Sal1) in these primers was not contained within pCR2.1 vector, it was possible to accurately digest both ends of each PCR product. In respective replacing steps, each PCR product was digested by restriction enzymes and then ligated (Fig. 1). The final ligation product, i.e., NM-plasmid, was cloned using a TA cloning kit (Invitrogen Corp.) (Fig. 1). The 103 copies of NM-plasmid were adopted as a new internal-control “calibrator.” This copy number was determined by the preliminary experiments (described below).
FIG. 1.

Position of primers and probes in the MPB64 protein encoding gene (MPT64), and the procedure for the development of the NM-plasmid for use as a new internal control. TAMRA, 6-carboxytetramethylrhodamine.
TABLE 1.
Sequences of primers and TaqMan probes for PCR assays
| Objective | First- or second-step PCR | Target | PCR product size (bp) | Type | Sequencea |
|---|---|---|---|---|---|
| Developing NM-plasmid | Forward primer block (NM-plasmid) | 4146 | Avr2-F1: Avr2 random forward primer-1 | 5′-GGATAGCCAGCACGGAGACAACAGGATGGGACGGC-3′ | |
| Avr2-R1: Avr2 random reverse primer-1 | 5′-GGTGGGACAGAATCGAAAGCCGAATTCCAGCACACT-3′ | ||||
| 4178 | Avr2-F2: Avr2 random forward primer-2 | 5′-ATCCTAGGAGAGATCGGATAGCCAGCACGGAGACA-3′ | |||
| Avr2-R2: Avr2 random reverse primer-2 | 5′-CTCCTAGGATAGACGGCGGTGGGACAGAATCGAAAGC-3′ | ||||
| Reverse primer block (NM-plasmid) | 4048 | Mlu1-F: Mlu1 random forward primer: | 5′-CTACGCGTCGAGTCTAAGCCGAATTCTGCAGATAT-3′ | ||
| Sal1-R: Sal1 random reverse primer | 5′-GCGTCGACATATTCTAAAGGACGGATTGCTAGCCGT-3′ | ||||
| 136 | Sal1-F: Sal1 random forward primer | 5′-ATGTCGACGCAGCGCATTCGCAGTCACGAACGACGG-3′ | |||
| Mlu1-R: Mlu1 random reverse primer | 5′-CGACGCGTAGTCCTCGCGAGTCGATCGCGGAACGTG-3′ | ||||
| WR-QNRT-PCR assay | First-step PCR | Wild M. tuberculosis DNA (MPT64) or W-plasmid | 239 | WF1: outer wild forward primer | 5′-ATCCGCTGCCAGTCGTCTTCC-3′; total of 21 nucleotides, A:2, T:6, G:4, C:9 (G+C, 62%) |
| WR1: outer wild reverse primer | 5′-CTCGCGAGTCTAGGCCAGCAT-3′; total of 21 nucleotides, A:4, T:4, G:6, C:7 (G+C, 62%) | ||||
| New internal control (NM-plasmid) | MF1: outer mutation forward primer | 5′-TCGATTCTGTCCCACCGCCGT-3′; total of 21 nucleotides, A:2, T:6, G:4, C:9 (G+C, 62%) | |||
| MR1: outer mutation reverse primer | 5′-AGACTCGACGCGTAGTCCTCG-3′; total of 21 nucleotides, A:4, T:4, G:6, C:7 (G+C, 62%) | ||||
| Second-step PCR | Wild M. tuberculosis DNA (MPT64) or W-plasmid | 77 | TqMn-WF2: TaqMan inner wild forward primer | 5′-GTGAACTGAGCAAGCAGACCG-3′; total of 21 nucleotides, A:7, T:2, G:7, C:5 (G+C, 57%) | |
| TqMn-WR2: TaqMan inner wild reverse primer | 5′-GTTCTGATAATTCACCGGGTCC-3′; total of 22 nucleotides, A:4, T:7, G:5, C:6 (G+C, 50%) | ||||
| New internal control (NM-plasmid) | TqMn-MF2: TaqMan inner mutation forward primer | 5′-AGATCGGATAGCCAGCACGGA-3′; total of 21 nucleotides, A:7, T:2, G:7, C:5 (G+C, 57%) | |||
| TqMn-MR2: TaqMan inner mutation reverse primer | 5′-TGCGCTGCGTCGACATATTCTA-3′; total of 22 nucleotides, A:4, T:7, G:5, C:6 (G+C, 50%) | ||||
| Wild M. tuberculosis DNA (MPT64) or W-plasmid | TqMn-W-VIC: TaqMan probe-wild-VIC | 5′-VIC-TATCGATAGCGCCGAATGCCGG-TAMRA-3′; total of 22 nucleotides, A:5, T:4, G:7, C:6 (G+C, 59%) | |||
| New internal control (NM-plasmid) | TqMn-M-FAM: TaqMan probe-mutation-FAM | 5′-FAM-ATGGGACGGCTAGCAATCCGTC-TAMRA-3′; total of 22 nucleotides, A:5, T:4, G:7, C:6 (G+C, 59%) | |||
| OR-QNRT-PCR assay | First-step PCR | Wild M. tuberculosis DNA (MPT64) and old internal control (M-plasmid) | 239 | WF1 | 5′-ATCCGCTGCCAGTCGTCTTCC-3′ |
| WR1 | 5′-CTCGCGAGTCTAGGCCAGCAT-3′ | ||||
| Second-step PCR | Wild M. tuberculosis DNA (MPT64) and old internal control (M-plasmid) | 77 | TqMn-WF2 | 5′-GTGAACTGAGCAAGCAGACCG-3′ | |
| TqMn-WR2 | 5′-GTTCTGATAATTCACCGGGTCC-3′ | ||||
| Wild M. tuberculosis DNA (MPT64) | TqMn-W-VIC | 5′-VIC-TATCGATAGCGCCGAATGCCGG-TAMRA-3′ | |||
| Old internal control (M-plasmid) | TqMn-M-FAM | 5′-FAM-ATGGGACGGCTAGCAATCCGTC-TAMRA-3′ |
Underlining indicates artificial sequence; double underlining indicates restriction site. TAMRA, 6-carboxytetramethylrhodamine.
(The NM-plasmid is available from us through the laboratory at the High-Tech Research Center, Nihon University School of Medicine, Tokyo, Japan: please e-mail corresponding author Teruyuki Takahashi to request.)
Primers and probes for WR-QNRT-PCR.
For use in the WR-QNRT-PCR assay, four pairs of new specific primers and two types of specific (TaqMan) probes were prepared. The sequences and positions of these new primers and probes are shown in Table 1 and Fig. 1. In the first step of WR-QNRT-PCR assay, two pairs of outer forward and reverse primers, WF1 and WR1, as well as MF1 and MR1, were used. WF1 and WR1 were specific for MRT64 of wild M. tuberculosis or W-plasmid, whereas MF1 and MR1 were specific for the artificial random nucleotides in the NM-plasmid for use as a new internal-control “calibrator.” In the second step, two pairs of inner forward and reverse primers, TqMn-WF2 and TqMn-WR2, as well as TqMn-MF2 and TqMn-MR2, were used. TqMn-WF2 and TqMn-WR2 were specific for wild MPT64. TqMn-MF2 and TqMn-MR2 were specific for the artificial random nucleotides in the NM-plasmid. In addition, two types of probes, TqMn-W-VIC and TqMn-M-FAM, were used. TqMn-W-VIC was labeled with fluorescent reporter dye VIC and specifically annealed to wild MPT64. While TqMn-M-FAM was labeled with fluorescent reporter dye 6-carboxyfluorescein (FAM) and specifically annealed to the artificial random nucleotides in the NM-plasmid. These primers and probes were set to have the same nucleotide composition but a different and random sequence (Table 1). Therefore, the annealing efficiencies of these primers and probes to wild MPT64 or NM-plasmid as a template can be regarded as the same.
In the OR-QNRT-PCR assay, two consecutive PCR amplification steps were performed by using the common two pairs of primers WF1 and WR1 at the first step and TqMn-WF2 and TqMn-WR2 at the second step for both M. tuberculosis DNA and M-plasmid as the old internal control (Table 1 and Fig. 1). Two types of probes, TqMn-W-VIC and TqMn-M-FAM, were also used to specifically detect each M. tuberculosis DNA or M-plasmid (Table 1 and Fig. 1).
Extraction and purification of DNA from CSF samples.
A 500-μl aliquot of original lysis buffer containing 20 mM Tris-HCl (pH 8.0), 300 mM NaCl, 0.8% (vol/vol) sodium dodecyl sulfate, and 0.5 mg of proteinase K was prepared. This lysis buffer was added to 500 μl of the CSF sample, followed by incubation in a water bath at 65°C overnight. After incubation, the total 1,000-μl suspension was divided into two 500-μl aliquots for use in the WR-QNRT-PCR assay.
In advance, the 103 copies of NM-plasmid as a new internal-control “calibrator” were added to one of the 500-μl aliquot containing 250 μl each of CSF and lysis buffer. The DNA specimens including M. tuberculosis DNA and NM-plasmid were extracted and purified from these 500-μl aliquots by a previously reported conventional phenol-chloroform method and ethanol precipitation (7, 8, 22). To efficiently extract a small amount of DNA, a high-molecular-weight carrier, Ethachinmate (Nippon Gene, Tokyo, Japan), was used as a coprecipitating agent for the nucleotides in the ethanol precipitation. After complete vacuum desiccation, the extracted DNA specimen was resuspended in 20 μl of pure water and then stored at −20°C until use.
Assay condition of WR-QNRT-PCR.
Both WR and OR-QNRT-PCR assays consist of two consecutive PCR amplification steps, which were conventional PCR at the first step and real-time (TaqMan) PCR at the second step. M. tuberculosis DNA and NM-plasmid were amplified and detected in separate tubes and wells. However, the entire procedure was performed simultaneously under the same assay conditions.
In the first-step PCR, 18-μl conventional PCR solution mixtures containing 10 mM Tris-HCl (pH 8.0), 50 mM KCl, 1.5 mM MgCl2, 400 μM of each deoxynucleoside triphosphate mix, 20 pM each of outer primers WF1 and WR1 or MF1 and MR2, and 2.5 U of Taq DNA polymerase were prepared. As a template, 2 μl of the extracted DNA specimen, including M. tuberculosis DNA and the new internal control (NM-plasmid), was added to the PCR solution mixture (each total reaction volume was 20 μl). This preparation was subjected to the protocol shown in Table 2 at 25 amplification cycles, using the GeneAmp PCR system 9700 (Perkin Elmer, Norwalk, CT). The assay protocol of OR-QNRT-PCR is additionally described in Table 2. The first-step PCR in the OR-QNRT-PCR assay was set at 35 amplification cycles (22).
TABLE 2.
PCR assay conditions
| Step | Parameter at indicated step for:
|
|
|---|---|---|
| WR-QNRT-PCR | OR-QNRT-PCR | |
| First-step PCR | ||
| Initial denaturing | 96.0°C; 3 min | 96.0°C; 3 min |
| Amplification | 25 cyclesa | 35 cycles |
| Denaturing | 95.0°C; 30 s | 95.0°C; 30 s |
| Annealing | 60.0°C; 30 s | 60.0°C; 30 s |
| Extension | 72.0°C; 1 min | 72.0°C; 1 min |
| Final extension | 72.0°C; 10 min | 72.0°C; 10 min |
| Second-step PCR | ||
| Incubation | 50.0°C; 2 min | 50.0°C; 2 min |
| Initial denaturing | 95.0°C; 10 min | 95.0°C; 10 min |
| Amplification | 40 cycles | 40 cycles |
| Denaturing | 95.0°C; 15 s | 95.0°C; 15 s |
| Annealing-extension | 60.0°C; 1 min | 60.0°C; 1 min |
Improved amplification cycle number.
In the second-step PCR, 23-μl PCR solution mixtures containing 12.5 μl of TaqMan universal PCR master mix, 0.9 μM each of inner primers TqMn-WF2 and TqMn-WR2 or TqMn-MF2 and TqMn-MR2, and 0.2 μM TaqMan probe TqMn-W-VIC or TqMn-M-FAM were prepared. As a template, 2 μl of the first-step PCR product was added to this PCR solution mixture (each total reaction volume was 25 μl). This preparation was subjected to the protocol shown in Table 2, using the ABI Prism 7700 sequence detector (Applied Biosystems, Foster City, CA).
Quantitative detection of M. tuberculosis DNA.
In the WR-QNRT-PCR assay, the procedures of extraction, amplification, and detection for both M. tuberculosis DNA and the new internal control were performed simultaneously by using two pairs of primers and two probes that had annealing efficiencies equivalent to those of the templates. Therefore, the initial copy number of M. tuberculosis DNA in CSF samples was able to be calculated based on the amplification ratio against the new internal control (103 copies of NM-plasmid) as a “calibrator.” Therefore, we adopted equation 1 as well as the OR-QNRT-PCR assay (22, 23).
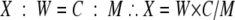 |
(1) |
where X is the initial copy number of M. tuberculosis DNA per 1 ml of CSF sample, C is the initial copy number of the new internal control (i.e., “calibrator” [103 copies of NM-plasmid]), and W and M are the copy numbers of M. tuberculosis DNA and NM-plasmid, respectively, after passing through extraction and PCR amplification procedures. In M. tuberculosis, it was universally acceptable that a single copy of the MPT64 gene represented one bacterial cell (7, 8). Therefore, we considered that the copy numbers calculated by the WR-QNRT-PCR assay corresponded to the bacterial cell numbers of M. tuberculosis in CSF samples.
Statistical analysis.
The statistical analysis was calculated using data analysis software program SPSS 13.0 for Windows. A P value of <0.05 was considered statistically significant.
RESULTS
Precision of the standard curves.
For the WR-QNRT-PCR assay, two specific standard curves for the quantitative detection of M. tuberculosis DNA and the new internal control are needed, and this is also the case for OR-QNRT-PCR assay (22). Therefore, the precision of these two specific standard curves was statistically evaluated using the standard templates in the previously reported preliminary experimental protocols (22). The two specific standard curves are shown in Fig. 2A and B. In simple regression analysis, both of these two standard curves demonstrated a significant linear relationship (R2 = >0.99) between the threshold cycle numbers (CT values) (y axis) and log of the starting copy numbers for each standard template (x axis). In both standard curves, no significant differences were found among the plots in each preliminary experiment (F = 1.007, P = 0.65 and F = 1.015, P = 0.53) by two-way analysis of variance (ANOVA). The PCR efficiency (PCR-Eff) of real-time PCR can be calculated by the slope of the standard curve by the following equation: PCR-Eff = 10(−1/slope) − 1 (14). In the WR-QNRT-PCR assay, the PCR-Eff values calculated by this equation based on the slopes (−3.33 and −3.28) of two standard curves were 99.7 and 101.8%, respectively.
FIG. 2.


Statistical evaluation of CT value data in preliminary experiments. (A) Specific standard curve for use in the quantitative detection of M. tuberculosis DNA or W-plasmid. VIC (TqMn-W-VIC) was used for analysis. (B) Specific standard curve for use in the quantitative detection of the NM-plasmid as a new internal control. FAM (TqMn-M-FAM) was used for analysis. (C) Amplification curves for W-plasmids after first-step PCR at 25 cycles. (D) Result of simple regression analysis between CT values (y axis) and the log of the starting copy numbers of W-plasmids (x axis) in setting first-step PCR at 25 cycles. (E) Result of simple regression analysis between CT values (y axis) and the log of the starting copy numbers of W-plasmids (x axis) in setting first-step PCR at 35 cycles (OR-QNRT-PCR assay). (F) Amplification curves for 103 copies of NM-plasmids as the new internal control. (G) Comparative results of one-way ANOVA against CT values for 103 copies of NM-plasmid (WR-QNRT-PCR assay) or M-plasmid (OR-QNRT-PCR assay) as an internal control.
Optimization of WR-QNRT-PCR assay conditions.
For the WR-QNRT-PCR assay, two important parameters may affect assay conditions: the amplification cycle number for the first-step PCR and the copy number of the new internal control. These two parameters were determined by previously reported preliminary experimental protocols (22) using five serial sets of 10-fold-diluted W-plasmids (1 to 105 copies) as the templates instead of actual M. tuberculosis DNA. The CT value data collected under various assay conditions were statistically analyzed.
(i) Optimal amplification cycle number for first-step PCR.
To determine the optimal amplification cycle number for the first-step PCR, cycle numbers were set at 5-cycle intervals in the range from 20 to 35 cycles. When the first-step PCR was set at 25 cycles, the most constantly isolated amplification curves were demonstrated in all starting copy numbers of W-plasmids (Fig. 2C). The CT value data (means ± standard deviations) at 25 cycles in first-step PCR are shown in Table 3. Reflecting Fig. 2C, a significant linear relationship (R2 = 0.996) was demonstrated between CT values (y axis) and the log of the starting copy numbers of W-plasmids (x axis) by simple regression analysis (Fig. 2D). The slope of this linear regression curve (−3.33) was completely consistent with that of the standard curve shown in Fig. 2A. Therefore, 25 cycles was adopted as the optimal cycle number in the first-step PCR. Whereas, when the first-step PCR was set at the previously reported 35 cycles in the OR-QNRT-PCR assay (22), a significant linear relationship (R2 = 0.991) was also demonstrated by simple regression analysis for the CT value data (Table 3 and Fig. 2E). However, the slope of this linear regression curve (−1.46) indicated overamplification (PCR-Eff = 384.1%).
TABLE 3.
CT value data collected under different assay conditions in the WR- and OR-QNRT-PCR assays
| W-plasmid starting copy no. |
CT value fora:
|
|||||
|---|---|---|---|---|---|---|
| WR-QNRT-PCR assay (internal control, NM-plasmid [103 copies]); 25 cycles
|
OR-QNRT-PCR assay (internal control, M-plasmid [103 copies])
|
|||||
| 35 Cycles
|
25 Cycles
|
|||||
| VIC | FAM | VIC | FAM | VIC | FAM | |
| 105 | 7.64 ± 0.17 | 11.62 ± 0.20 | 5.79 ± 0.01 | 25.27 ± 0.95 | 8.80 ± 0.03 | 40.00 (NA)b |
| 104 | 10.16 ± 0.18 | 11.66 ± 0.18 | 6.99 ± 0.05 | 16.78 ± 0.37 | 12.85 ± 0.3 | 28.67 ± 0.05 |
| 103 | 13.20 ± 0.26 | 11.62 ± 0.27 | 10.19 ± 0.05 | 14.03 ± 0.08 | 16.87 ± 0.09 | 22.80 ± 0.13 |
| 102 | 16.37 ± 0.20 | 11.62 ± 0.26 | 11.29 ± 0.07 | 13.39 ± 0.06 | 19.61 ± 0.09 | 19.36 ± 0.21 |
| 10 | 20.48 ± 0.18 | 11.65 ± 0.25 | 12.71 ± 0.09 | 13.37 ± 0.06 | 19.99 ± 0.04 | 16.64 ± 011 |
| 1 | 24.20 ± 0.22 | 11.66 ± 0.20 | 14.25 ± 0.05 | 13.32 ± 0.04 | 20.04 ± 0.24 | 16.45 ± 0.33 |
CT value data represent the means ± standard deviations in duplicate for five independent experiments. The numbers of cycles are for first-step PCR. Reporter dyes VIC and FAM are indicated.
NA, no amplification.
(ii) Optimal copy number of new internal control.
To determine the optimal copy number of the new internal control, 103, 104, and 105 copies of NM-plasmids were examined. When 103 copies of NM-plasmid were set as the new internal control, the amplification curves of NM-plasmids revealed extremely uniform patterns in all starting copy numbers of W-plasmids (Table 3 and Fig. 2F). Reflecting Fig. 2F, the CT values for 103 copies of the NM-plasmid also revealed significantly uniform variance between all starting copy numbers of W-plasmids (F = 1.086, P = 0.774) by one-way ANOVA (Fig. 2G). Therefore, 103 copies of NM-plasmid were adopted as the sufficient and optimal copy number of the new internal control for use as a “calibrator.” However, when 103 copies of M-plasmid were set as the old internal control in the OR-QNRT-PCR assay (22), the CT values for M-plasmid revealed unbalanced patterns (Table 3 and Fig. 2G). In particular, a significant difference was demonstrated for 103 to 105 copies of W-plasmid (Fig. 2G). In addition, when the first-step PCR was set at 25 cycles, as same as the WR-QNRT-PCR assay, the CT values for the old M-plasmid were inconstant, and there was no difference in the CT values for the 1 to 102 copies of W-plasmid (Table 3). These results indicated that M-plasmid as the old internal control was incomplete and needed more improvement.
DISCUSSION
We have developed an improved WR-QNRT-PCR assay technique for the accurate quantitative detection of M. tuberculosis DNA in CSF samples collected from patients with clinically suspected TBM. In the WR-QNRT-PCR assay, the initial copy number of M. tuberculosis DNA in CSF samples was calculated from the amplification ratio of the specific new internal control used as a “calibrator,” as was the case for the OR-QNRT-PCR assay. For use as the specific new internal control, the original NM-plasmid was designed to have equivalent amplification and detection efficiency against actual M. tuberculosis DNA. Based on a similar concept, specific primers and probes were prepared. Consequently, we were able to formulate equation 1, which can be used to determine initial copy number.
For the accurate quantitative detection of a small amount of M. tuberculosis DNA in CSF samples by WR-QNRT-PCR assay, it is extremely important that both M. tuberculosis DNA and the new internal control are amplified with sufficient balance. Therefore, the precision of the two specific standard curves was strictly examined by statistical evaluation in a series of preliminary experiments. Previously, many investigators have reported that the precision of the standard curve is the principal factor for quantitative detection in real-time (TaqMan) PCR assays (1-6, 9, 10, 13, 15-17, 20, 24, 26, 27). In this study, the two specific standard curves demonstrated statistically significant precision (R2 > 0.99, F = 1.007 or 1.015) (Fig. 2A and B). Therefore, we consider that any overall errors relating to the dilution procedure or within each experiment can be disregarded. In addition, the PCR-Eff values of two standard curves calculated by the slopes (−3.33 and −3.28) were 99.7 and 101.8%, respectively. These results indicated that the efficiency of amplification and detection for both M. tuberculosis DNA and the new internal control was almost equivalent in the WR-QNRT-PCR assay. Therefore, our hypothesis was proved experimentally by these results.
The optimal assay conditions were examined in detail by statistical analysis for CT value data collected from the preliminary experiments. In setting 25 cycles as the optimal cycle number for the first-step PCR, the primary concentration gradient for all starting copy numbers of W-plasmid was completely preserved (Fig. 2C and D). Moreover, in setting 103 copies of NM-plasmid as the optimal copy number of the new internal control, extremely uniform amplifications were demonstrated for all starting copy numbers of W-plasmids (Fig. 2F and G). These results indicate that there is no interference between M. tuberculosis DNA and the new internal control in the entire PCR amplification procedure. Therefore, the new internal control could be regarded as appropriate for use as a “calibrator” in the WR-QNRT-PSR assay.
In the OR-QNRT-PCR assay, both M. tuberculosis DNA and the old internal control (M-plasmid) were simultaneously amplified using two pairs of common primers (22). This system is the most serious weak point of the OR-QNRT-PCR assay because the amplification for a small copy number of template (M. tuberculosis DNA or M-plasmid) was poor owing to the interference by a high copy number of template. In order to obtain sufficient amplification for a small amount of M. tuberculosis DNA (<100 copies), the first-step PCR cycle number need to be set at a large number (35 cycles) in the OR-QNRT-PCR assay (22). This led to the problems of overamplification (Fig. 2E) and the instability of the M-plasmid for a large copy number (>1,000) of M. tuberculosis DNA (Fig. 2G). Therefore, the OR-QNRT-PCR assay was limited necessarily to being within a narrow detection range (22). Due to the development of NM-plasmid as the new internal control, the stable and accurate quantitative detection of M. tuberculosis DNA was possible in a detection range wider than that for the OR-QNRT-PCR assay.
In this study, we attempted to improve the OR-QNRT-PCR assay and developed NM-plasmid for use as a new internal control. Due to the development of NM-plasmid, significantly improved quantitative accuracy and a wider detection range were realized with the WR-QNRT-PCR assay. In the clinical application of the WR-QNRT-PCR assay, the advantages of this method would be powerful tool for rapid and accurate diagnosis in the difficult cases in which it is impossible to detect M. tuberculosis by conventional assay methods. In our next study, we plan to examine and evaluate the clinical usefulness of the WR-QNRT-PCR assay for the rapid and accurate diagnosis of TBM and for assessing the clinical course of TBM.
Acknowledgments
We thank Hiroki Nagase and many doctors who collected CSF samples in the following institutions: Department of Neurology and Pediatrics, Kasugai Hospital; Department of Neurology, Nagoya University School of Medicine; Department of Neurology, Kariya Hospital; Department of Neurology, Tokai University School of Medicine; Fourth Department of Internal Medicine, Saitama Medical Center; Department of Neurology, Metropolitan Bokutoh Hospital; Kohnodai Hospital; National Center of Neurology and Psychiatry, Japan.
This work was supported by “Academic Frontier” Project for Private Universities: matching fund subsidy from MEXT (Ministry of Education, Culture, Sports, Science and Technology), 2006-2010.
Footnotes
Published ahead of print on 12 March 2008.
REFERENCES
- 1.Aldous, W. K., J. I. Pounder, J. L. Cloud, and G. L. Woods. 2005. Comparison of six methods of extracting Mycobacterium tuberculosis DNA from processed sputum for testing by quantitative real-time PCR. J. Clin. Microbiol. 432471-2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heid, C. A., J. Stevens, K. J. Livak, and P. M. Williams. 1996. Real time quantitative PCR. Genome Res. 6986-994. [DOI] [PubMed] [Google Scholar]
- 3.Kawada, J., H. Kimura, Y. Ito, Y. Hoshino, N. Tanaka-Kitajima, Y. Ando, M. Futamura, and T. Morishima. 2004. Comparison of real-time and nested PCR assays for detection of herpes simplex virus DNA. Microbiol. Immunol. 48411-415. [DOI] [PubMed] [Google Scholar]
- 4.Kimura, H., Y. Ito, M. Futamura, Y. Ando, Y. Yabuta, Y. Hoshino, Y. Nishiyama, and T. Morishima. 2002. Quantitation of viral load in neonatal herpes simplex virus infection and comparison between type 1 and type 2. J. Med. Virol. 67349-353. [DOI] [PubMed] [Google Scholar]
- 5.Kohmoto, M., M. Enomoto, Y. Yano, S. Otani, S. Minamitani, A. Tamori, D. Habu, T. Takeda, S. Shiomi, S. Seki, T. Arakawa, and S. Nishiguchi. 2003. Detection of serum hepatitis B virus DNA by real-time quantitative polymerase chain reaction (TaqMan PCR) during lamivudine treatment: comparison with three other assays. Hepatol. Res. 26125-133. [DOI] [PubMed] [Google Scholar]
- 6.Larsen, H. H., H. Masur, J. A. Kovacs, V. J. Gill, V. A. Silcott, P. Kogulan, J. Maenza, M. Smith, D. R. Lucey, and S. H. Fischer. 2002. Development and evaluation of a quantitative, touch-down, real-time PCR assay for diagnosing Pneumocystis carinii pneumonia. J. Clin. Microbiol. 40490-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee, B. W., J. A. Tan, S. C. Wong, C. B. Tan, H. K. Yap, P. S. Low, J. N. Chia, and J. S. Tay. 1994. DNA amplification by the polymerase chain reaction for the rapid diagnosis of tuberculous meningitis. Comparison of protocols involving three mycobacterial DNA sequences, IS6110, 65 kDa antigen, and MPB64. J. Neurol. Sci. 123173-179. [DOI] [PubMed] [Google Scholar]
- 8.Liu, P. Y., Z. Y. Shi, Y. J. Lau, and B. S. Hu. 1994. Rapid diagnosis of tuberculous meningitis by a simplified nested amplification protocol. Neurology 441161-1164. [DOI] [PubMed] [Google Scholar]
- 9.Locatelli, G., F. Santoro, F. Veglia, A. Gobbi, P. Lusso, and M. S. Malnati. 2000. Real-time quantitative PCR for human herpesvirus 6 DNA. J. Clin. Microbiol. 384042-4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marin, M., D. Garcia de Viedma, M. J. Ruiz-Serrano, and E. Bouza. 2004. Rapid direct detection of multiple rifampin and isoniazid resistance mutations in Mycobacterium tuberculosis in respiratory samples by real-time PCR. Antimicrob. Agents Chemother. 484293-4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Medical Research Council. 1948. Streptomycin treatment of tuberculous meningitis. Lancet i582-596. [PubMed] [Google Scholar]
- 12.Nakajima, H., K. Ashida, H. Yamasaki, K. Shinoda, and N. Ohsawa. 1995. Intracranial tuberculoma with spontaneous recovery. Rinsho Shinkeigaku 35521-525. (In Japanese.) [PubMed] [Google Scholar]
- 13.O'Neill, H. J., D. E. Wyatt, P. V. Coyle, C. McCaughey, and F. Mitchell. 2003. Real-time nested multiplex PCR for the detection of herpes simplex virus types 1 and 2 and varicella zoster virus. J. Med. Virol. 71557-560. [DOI] [PubMed] [Google Scholar]
- 14.Pfaffl, M. W., G. W. Horgan, and L. Dempfle. 2002. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 30e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rindi, L., N. Lari, D. Bonanni, and C. Garzelli. 2004. Detection of Mycobacterium tuberculosis genotypic groups by a duplex real-time PCR targeting the katG and gyrA genes. J. Microbiol. Methods 59283-287. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez-Lazaro, D., M. Hernandez, and M. Pla. 2004. Simultaneous quantitative detection of Listeria spp. and Listeria monocytogenes using a duplex real-time PCR-based assay. FEMS Microbiol. Lett. 233257-267. [DOI] [PubMed] [Google Scholar]
- 17.Ruiz, M., M. J. Torres, A. C. Llanos, A. Arroyo, J. C. Palomares, and J. Aznar. 2004. Direct detection of rifampin- and isoniazid-resistant Mycobacterium tuberculosis in auramine-rhodamine-positive sputum specimens by real-time PCR. J. Clin. Microbiol. 421585-1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scarpellini, P., S. Racca, P. Cinque, F. Delfanti, N. Gianotti, M. R. Terreni, L. Vago, and A. Lazzarin. 1995. Nested polymerase chain reaction for diagnosis and monitoring treatment response in AIDS patients with tuberculous meningitis. AIDS 9895-900. [DOI] [PubMed] [Google Scholar]
- 19.Shankar, P., N. Manjunath, K. K. Mohan, K. Prasad, M. Behari, Shriniwas, and G. K. Ahuja. 1991. Rapid diagnosis of tuberculous meningitis by polymerase chain reaction. Lancet 3375-7. [DOI] [PubMed] [Google Scholar]
- 20.Stranska, R., R. Schuurman, M. de Vos, and A. M. van Loon. 2004. Routine use of a highly automated and internally controlled real-time PCR assay for the diagnosis of herpes simplex and varicella-zoster virus infections. J. Clin. Virol. 3039-44. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi, T., T. Nakayama, M. Tamura, K. Ogawa, H. Tsuda, A. Morita, M. Hara, M. Togo, H. Shiota, Y. Suzuki, M. Minami, H. Ishikawa, K. Miki, E. Shikata, S. Takahashi, T. Kuragano, K. Matsumoto, S. Sawada, and T. Mizutani. 2005. Nested polymerase chain reaction for assessing the clinical course of tuberculous meningitis. Neurology 641789-1793. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi, T., and T. Nakayama. 2006. Novel technique of quantitative nested real-time PCR assay for Mycobacterium tuberculosis DNA. J. Clin. Microbiol. 441029-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi, T., M. Tamura, S. N. Takahashi, K. Matsumoto, S. Sawada, E. Yokoyama, T. Nakayama, T. Mizutani, T. Takasu, and H. Nagase. 2007. Quantitative nested real-time PCR assay for assessing the clinical course of tuberculous meningitis. J. Neurol. Sci. 25569-76. [DOI] [PubMed] [Google Scholar]
- 24.Templeton, K. E., S. A. Scheltinga, A. W. Graffelman, J. M. van Schie, J. W. Crielaard, P. Sillekens, P. J. Van Den Broek, H. Goossens, M. F. Beersma, and E. C. Claas. 2003. Comparison and evaluation of real-time PCR, real-time nucleic acid sequence-based amplification, conventional PCR, and serology for diagnosis of Mycoplasma pneumoniae. J. Clin. Microbiol. 414366-4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thwaites, G. E., D. B. Nguyen, H. D. Nguyen, T. Q. Hoang, T. T. Do, T. C. Nguyen, Q. H. Nguyen, T. T. Nguyen, N. H. Nguyen, T. N. Nguyen, N. L. Nguyen, H. D. Nguyen, N. T. Vu, H. H. Cao, T. H. Tran, P. M. Pham, T. D. Nguyen, K. Stepniewska, N. J. White, T. H. Tran, and J. J. Farrar. 2004. Dexamethasone for the treatment of tuberculous meningitis in adolescents and adults. N. Engl. J. Med. 3511741-1751. [DOI] [PubMed] [Google Scholar]
- 26.Wada, T., S. Maeda, A. Tamaru, S. Imai, A. Hase, and K. Kobayashi. 2004. Dual-probe assay for rapid detection of drug-resistant Mycobacterium tuberculosis by real-time PCR. J. Clin. Microbiol. 425277-5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whiley, D. M., I. M. Mackay, M. W. Syrmis, M. J. Witt, and T. P. Sloots. 2004. Detection and differentiation of herpes simplex virus types 1 and 2 by a duplex LightCycler PCR that incorporates an internal control PCR reaction. J. Clin. Virol. 3032-38. [DOI] [PubMed] [Google Scholar]