

Abstract
The high-resolution structural data for simian virus 40 large-T-antigen helicase revealed a set of nine residues bound to ATP/ADP directly or indirectly. The functional role of each of these residues in ATP hydrolysis and also the helicase function of this AAA+ (ATPases associated with various cellular activities) molecular motor are unclear. Here, we report our mutational analysis of each of these residues to examine their functionality in oligomerization, DNA binding, ATP hydrolysis, and double-stranded DNA (dsDNA) unwinding. All mutants were capable of oligomerization in the presence of ATP and could bind single-stranded DNA and dsDNA. ATP hydrolysis was substantially reduced for proteins with mutations of residues making direct contact with the γ-phosphate of ATP or the apical water molecule. A potentially noncanonical “arginine finger” residue, K418, is critical for ATP hydrolysis and helicase function, suggesting a new type of arginine finger role by a lysine in the stabilization of the transition state during ATP hydrolysis. Interestingly, our mutational data suggest that the positive- and negative-charge interactions in the uniquely observed residue pairs, R498/D499 and R540/D502, in large-T-antigen helicase are critically involved in the transfer of energy of ATP binding/hydrolysis to DNA unwinding.
Simian virus 40 encodes large tumor antigen (LTAg), an oncogenic protein required for viral DNA replication (1). LTAg has been used as a model system for eukaryotic and archaeal replication by minichromosome maintenance (MCM) proteins (6, 17), as it requires many of the same cellular components for viral genome replication (20). During viral DNA replication, LTAg assembles as a double hexamer at the origin of replication, melts the origin DNA, and initiates double-stranded DNA (dsDNA) unwinding (9, 21).
LTAg is a superfamily 3 helicase and belongs to the AAA+ (ATPases associated with various cellular activities) family (2, 24). LTAg contains three domains. The first domain (amino acids [aa] 1 to 82) has homology with DnaJ and is involved in Hsc70 binding (23). The second domain (aa 131 to 259), the origin binding domain, recognizes the viral origin of replication (14, 16). The C terminus (aa 251 to 627) is the hexameric-helicase domain that unwinds dsDNA (13).
For the high-resolution structures of LTAg helicase bound to ATP and ADP, Gai et al. (8) noted several distinct interactions between LTAg residues and the ATP at the binding pocket. An “arginine finger” has been proposed to facilitate ATP hydrolysis by stabilizing the transition state (15). The arginine finger in LTAg appears to be unique in two ways. First, there are two, instead of one, potential finger residues. Second, one of the fingers is a lysine residue. Both potential finger residues are supplied in trans (tR540 and tK418, where “t” denotes trans) from a neighboring monomer interacting with ATP (Fig. 1A). tR540 binds γ-phosphate from the end, consistent with an “associative” ATP hydrolysis mechanism in which the developing charge on the γ-phosphate of ATP is neutralized by the arginine finger. tK418 interacts with the γ-β oxygen of ATP from the side, appropriately positioned for neutralization of the developing charge at the γ-β oxygen during a “dissociative” ATP hydrolysis mechanism. It is unclear if LTAg uses a combination of associative and dissociative mechanisms for ATP hydrolysis or if one mechanism is favored over the other (8). Other trans residues include the following: tK419, which interacts with the ribose; tR498, which binds the apical water molecule to the γ-phosphate; tD499, which binds tR498 and ADP in the ADP-LTAg form (Fig. 1B) and was hypothesized to be involved in the translation of the energy of ATP binding/hydrolysis into mechanical motion; and tD502, which interacts with tR540, possibly to stabilize and orient the arginine finger residue (8).
FIG. 1.

Nine residues around the nucleotide binding pocket of LTAg which interact with ATP or ADP (Protein Data Bank accession no. 1svl and 1svm). (A) Residues that bind ATP at the nucleotide binding cleft between two monomers of LTAg. The backbone of the cis subunit, defined by the location of the ATP binding pocket, is highlighted in gray. The backbone of the trans subunit is highlighted in red. (B) Residues that bind ADP.
Two cis residues, cD474, of the Walker B motif, and cN529 (where “c” denotes cis), bind to the γ-phosphate of ATP as well as to the apical water molecule. Residue cT527 establishes a hydrogen bond with a water molecule that coordinates Mg2+ in the ADP-bound form. All of these residues are potentially critical for sensing and modulating ATP binding and hydrolysis, as well as for coupling the energy of ATP binding and hydrolysis to the mechanical motion in DNA unwinding. For other AAA+ proteins, the critical roles of the canonical arginine finger (equivalent to the tR540 LTAg), Sensor I (equivalent to the cN529 LTAg), and Walker B (equivalent to the cD474 LTAg) for ATP hydrolysis and motor function have been previously confirmed (reviewed in reference 5). However, the functional role of the other residues contacting ATP or the apical water molecule seen in the set of high-resolution structures of LTAg in different nucleotide-bound states is unclear. These residues are conserved in many AAA+ family proteins (10).
The goal of this work was to test two distinct hypotheses. First, we hypothesized that the observed movement of residue pairs tR498/tD499 and tR540/tD502 in response to ATP binding and hydrolysis is indicative of their involvement in coupling dsDNA unwinding to nucleotide binding and hydrolysis (8). Second, we hypothesized that the hydrolysis of ATP may be dependent upon two finger residues, tK418 and tR540. We also wanted to confirm the roles of the Sensor I (cN529) and Walker B (cD474) residues. Therefore, to obtain the mechanistic details of LTAg helicase, we systematically mutated these structurally defined residues near the nucleotide binding site and examined their physical and biochemical properties. Our results have revealed the novel roles of two pairs of positively and negatively charged residues during ATP binding/hydrolysis and in the coupling of ATP binding/hydrolysis to the conformational changes for DNA unwinding. Moreover, we have identified residues critical for the coupling of DNA interaction and ATP binding/hydrolysis by LTAg helicase. Because many of these mutated residues (tK418, tK419, cD474, tR498, tD499, tD502, cT527, cN529, tR540) are conserved across AAA+ ATPases, our results should provide insights for understanding the general mechanism of ATP hydrolysis and motor function for AAA+ helicases and other motor proteins.
MATERIALS AND METHODS
Mutagenesis, protein expression, purification, and oligomeric properties.
The DNA encoding residues 131 to 627 of LTAg was subcloned into the pGEX-KG vector using BamHI and EcoRI. The resulting construct was used for site-directed mutagenesis. We generated alanine mutations at positions tK418, tK419, cD474, tR498, tD499, tD502, cT527, cN529, and tR540 (Fig. 1A and B). All mutations were confirmed by sequencing the entire LTAg coding sequence. Wild-type (WT) and mutated LTAg proteins were expressed in Escherichia coli and purified to homogeneity as previously described (13). All proteins were quantified by the Bradford method (Bio-Rad Laboratories) and confirmed/calibrated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis with Coomassie blue staining. The oligomerization properties of LTAg were measured by gel filtration chromatography. Equal amounts of LTAg (1 mg per 500 μl) were loaded onto a Superdex 200 column equilibrated with 25 mM Tris (pH 8.0), 250 mM NaCl, and 1 mM dithiothreitol (DTT), with or without 1 mM ATP. No Mg2+ was included in the buffers. The integration of absorbance data at 280 nm was used to determine the total ratio of hexameric to monomeric LTAg.
DNA binding assay.
Rotational anisotropy was used to measure single-stranded DNA (ssDNA) and dsDNA binding by WT and mutant LTAg proteins. Measurements were taken on a QuantaMaster QM-1 fluorometer (Photon Technology International). Reaction mixtures (70 μl) contained 20 mM Tris (pH 7.5), 10 mM MgCl2, 1 mM DTT, 0.1 mg/ml bovine serum albumin, 50 nM DNA, LTAg from 0 to 1,200 nM hexamer, and 1 mM adenosine 5′-(gamma-thio)triphosphate (ATP-γ-S), if indicated, at 25°C. An 80-nucleotide ssDNA substrate labeled with 5-carboxyfluorescein was used for all ssDNA anisotropy experiments. No substantial secondary structure was predicted to form, nor was this ssDNA self complementary. dsDNA binding experiments used a 64-base-pair substrate labeled with 6-carboxyfluorescein at the 5′ end of one strand; this substrate did not contain the viral origin. Nonlinear regression was used to fit anisotropy data by the use of Enzfitter (Biosoft) to the following equation (18):
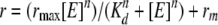 |
(1) |
where r, rmax, and rmin are the observed anisotropy, maximum anisotropy, and initial anisotropy, respectively. [E] is the concentration of hexameric LTAg, Kd is the concentration of LTAg that produces 0.5rmax, and n is the Hill coefficient.
ATPase and helicase assays.
The ATPase assay, in the absence of ssDNA, and the helicase assay were performed as described previously (19). The degree of stimulation of ATP hydrolysis by ssDNA was quantified in the presence of 1 μM poly(deoxyribosyladenine)50 and a saturating amount of cold ATP, as estimated from Km measurements (see Table 2) for the WT and each mutant. The degree of stimulation was estimated as a ratio of the percentage of ATP hydrolyzed in the presence of ssDNA to the percentage of ATP hydrolyzed in the absence of ssDNA, both at the same concentration of ATP over the same time scale.
TABLE 2.
Kinetic constants for ATP hydrolysis for WT and mutated LTAgsa
| Protein | kcat (min−1) | Km (μM) | ssDNA stimulation (fold)b |
|---|---|---|---|
| WT | 20 ± 1 | 270 ± 40 | 7.8 ± 1.4 |
| tK418A mutant | 2.8 ± 0.3 | 110 ± 30 | 1.4 ± 0.2 |
| tK419A mutant | 14 ± 1 | 280 ± 50 | 3.2 ± 0.5 |
| tR498A mutant | 6.6 ± 1.7 | 400 ± 200 | 5.1 ± 0.3 |
| tD499A mutant | 3.3 ± 0.5 | 80 ± 40 | 12.2 ± 0.6 |
| tD502A mutant | 0.60 ± 0.04 | 30 ± 7 | 1.5 ± 0.3 |
| tR540A mutant | 0.70 ± 0.10 | 170 ± 60 | 2.2 ± 0.3 |
| cD474A mutant | 0.70 ± 0.10 | 80 ± 30 | 1.4 ± 0.2 |
| cT527A mutant | 12 ± 0.8 | 50 ± 10 | 7.1 ± 1.2 |
| cN529A mutant | 1.8 ± 0.3 | 120 ± 50 | 1.6 ± 0.4 |
Michaelis-Menten kinetic constants for WT and mutated LTAgs in the absence of ssDNA. The data were generated as described in reference 17 and were an average of at least four independent experiments.
ssDNA stimulation is defined as the ratio of the amount of ATP hydrolyzed by a given protein in the absence of ssDNA versus that hydrolyzed by the same protein in the presence of a saturating concentration of ssDNA.
RESULTS
Oligomerization properties of the mutants.
We replaced all nine residues involved in ATP and/or ADP binding with alanine (Fig. 1A and B) in order to understand ATP hydrolysis and helicase function. To rule out possible structural disruption due to mutations, mutant LTAgs were tested for their ability to form hexamers by using gel filtration chromatography. Proteins (2 mg/ml, 1 mg total protein) were injected onto a Superdex 200 column both in the presence and in the absence of 1 mM ATP. The tK419A, tD499A, and tD502A LTAg mutants formed no detectable oligomers in the absence of ATP. All mutants supported oligomer formation in the presence of 1 mM ATP (Table 1), suggesting that the mutations caused no gross folding defects. Further, the presence of 1 mM ATP increased the hexamer-to-monomer ratio for all proteins. However, compared to the case with the WT, the mutation of residues around the ATP binding pocket widely reduced the extent of ATP-induced oligomerization, except in the cD474A and cT527A mutants.
TABLE 1.
Oligomerization of WT and mutated LTAgs
| Protein | Ratio of hexamer to monomera with:
|
|
|---|---|---|
| 1 mM ATP | No ATPb | |
| WT | 4.2 | 1.2 |
| tK418A mutant | 1.4 | 1.3 |
| tK419A mutant | 0.3 | 0 |
| tR498A mutant | 1.8 | 0.3 |
| tD499A mutant | 0.3 | 0 |
| tD502A mutant | 0.2 | 0 |
| tR540A mutant | 2.6 | 1.1 |
| cD474A mutant | 20.4 | 0.4 |
| cT527A mutant | 4.7 | 0.2 |
| cN529A mutant | 1.3 | 1.2 |
Ratio of LTAg hexamer to monomer measured by Superdex 200 gel filtration chromatography. Approximately 1 mg of each protein was injected onto a size exclusion column; the areas under each peak as measured by UV spectrophotometry were calculated and compared for the WT and the mutants.
Only a monomer peak was detected for the tK419A, tD499A, and tD502A mutants.
ATPase activity.
Measurements of ATPase activity in the absence of ssDNA revealed that the mutation of all of the nine residues interacting with nucleotides uniformly decreased kcat (see Table 2). However, the tK419A and cT527A variant proteins and, to a lesser extent, the tK418A, tR498A, and tD499A mutants were still competent as ATPases. Mutations at position tD502 and at the classically conserved residues cD474, cN529, and tR540 reduced kcat to a level that is ≤10% of the kcat of the WT LTAg. In some cases, Km was also increased for mutant proteins, but no uniform trend was detected.
Results for ATP hydrolysis by the WT and mutated LTAgs in the presence of 1 μM ssDNA, which is the saturating concentration for the WT and for most variant LTAgs as determined by rotational fluorescence anisotropy experiments (see below), were compared with results for ATP hydrolysis in the absence of ssDNA (Table 2; Fig. 2). Compared with the WT LTAg, only four mutants, the tK419A, cT527A, tR498A, and tD499A mutants, had appreciable levels of stimulation of hydrolysis (Fig. 2). All other mutants, due to their poor ability to hydrolyze ATP and a weaker stimulation of ATPase activity by ssDNA, had less than 3% of the WT activity in the presence of 1 μM ssDNA.
FIG. 2.

Relative levels of ATP hydrolysis by WT and mutated LTAgs in the presence of 1 μM poly(deoxyribosyladenine)50 and 1 mM ATP. The presence of helicase activity is indicated below the designation for each protein (+, present; −, absent). The data represent an average of three or more experiments.
Helicase activity.
We tested the WT and mutant LTAgs for their ability to displace a 32P-labeled ssDNA annealed to the M13 DNA substrate. A typical result, chosen from three independent experiments, is shown in Fig. 3. Two of the mutant proteins, the tK419A and cT527A mutants, displayed significantly reduced helicase activity compared to the WT and displayed little dependence of activity on protein concentration. Neither tK419 nor cT527 directly interacts with the γ-phosphate of ATP or the apical water molecule. All other mutants showed no detectable helicase functionality. In general, we noted a good correlation between ssDNA-dependent and ssDNA-independent ATPase activities and the helicase activities of the mutants. Mutants having significantly reduced ssDNA-stimulated ATPase activity compared to the WT (the tK418A, cD474A, tD502A, cN529A, and tR540A mutants) did not show any detectable dsDNA unwinding.
FIG. 3.

Results of helicase assay of LTAg mutations. Each lane in all panels contains 15 fmol dsDNA substrate. Three protein concentrations (50, 100, 200 ng protein or 0.85, 1.7, 3.4 pmol monomer) for each protein sample (labeled above the gels) were used in the assay. Lane 1, boiled dsDNA substrate; lane 2, dsDNA substrate alone. The circle labeled with an asterisk indicates radiolabeled ssDNA annealed to nonradiolabeled M13 ssDNA. The horizontal line labeled with an asterisk indicates unwound radiolabeled ssDNA.
DNA binding.
In order to quantify the ssDNA and dsDNA binding activities of mutant LTAgs, we used rotational fluorescence anisotropy to examine DNA binding using a fluorescently labeled ss- or dsDNA probe both in the absence and in the presence of ATP or ATP-γ-S. Fluorescence anisotropy titrations were fit to a hyperbolic binding curve modified with the Hill coefficient to account for the possible cooperative binding of DNA (see equation 1). Results for ssDNA binding of the WT LTAg in the presence and absence of 1 mM ATP are shown in Fig. 4. Binding curves for ssDNA and dsDNA were similar for most variant proteins, with the exception of the tD502A mutant, which bound ssDNA but could not be fit to equation 1. Results for the WT and mutants are summarized in Table 3. Binding of ssDNA was essentially unaltered by mutations around the ATP binding pocket (except for tD502A), either in the presence or in the absence of ATP-γ-S. However, the binding of dsDNA in the absence of ATP-γ-S was weakened for the tK418A, tK419A, cD474A, tD499A, and especially tR540A mutant proteins, compared with that of the WT. Notably, the dsDNA binding affinity of the tR540A mutant was 10-fold less than that of the WT in the absence of ATP-γ-S.
FIG. 4.

ssDNA binding curves for WT LTAg in the presence (•) and absence (▪) of 1 mM ATP. ssDNA binding experiments contained 50 nM 80-nucleotide, 5-carboxyfluorescein-labeled ssDNA, 20 mM Tris (pH 7.5), 10 mM MgCl2, 1 mM DTT, 0.1 mg/ml bovine serum albumin, and the indicated concentration of hexameric LTAg. The ssDNA binding curve with 1 mM ATP fits equation 1, where rmax is 0.24 ± 0.01, apparent Kd (Kd,app) is (84 ± 3) nM, n is 2.1 ± 0.1, and c is 0.144 ± 0.001. The ssDNA binding curve without ATP fits equation 1, where rmax is 0.22 ± 0.01, Kd,app is (180 ± 10) nM, n is 1.8 ± 0.1, and c is 0.131 ± 0.001.
TABLE 3.
Binding of ss- and dsDNA by WT and mutated LTAgsa
| Protein | Binding of ssDNAb
|
Binding of dsDNAc
|
||||||
|---|---|---|---|---|---|---|---|---|
| −ATP-γ-S
|
+ATP-γ-S
|
−ATP-γ-S
|
+ATP-γ-S
|
|||||
| Kd,app (nM) | n | Kd,app (nM) | n | Kd,app (nM) | n | Kd,app (nM) | n | |
| WT | 180 ± 10 | 1.8 | 64 ± 1 | 1.2 | 110 ± 10 | 1.8 | 95 ± 5 | 1.8 |
| tK418A mutant | 160 ± 20 | 1.5 | 100 ± 5 | 1.5 | 230 ± 20 | 1.5 | 100 ± 10 | 1.8 |
| tK419A mutant | 140 ± 20 | 1.2 | 48 ± 5 | 1.8 | 200 ± 30 | 2.0 | 53 ± 3 | 2.0 |
| tR498A mutant | 80 ± 30 | 1.6 | 64 ± 3 | 1.4 | 70 ± 7 | 1.9 | 60 ± 6 | 1.8 |
| tD499A mutant | 110 ± 20 | 1.2 | 100 ± 20 | 1.4 | 160 ± 10 | 1.3 | 180 ± 40 | 1.2 |
| tD502A mutant | + | + | − | − | ||||
| tR540A mutant | 67 ± 3 | 1.8 | 46 ± 2 | 2.5 | 1200 ± 300 | 1.2 | 290 ± 20 | 1.4 |
| cD474A mutant | 75 ± 4 | 1.5 | 50 ± 1 | 1.9 | 290 ± 150 | 1.2 | 100 ± 10 | 1.5 |
| cT527A mutant | 100 ± 10 | 1.0 | 40 ± 2 | 1.9 | 100 ± 10 | 1.7 | 50 ± 3 | 2.3 |
| cN529A mutant | 55 ± 3 | 1.3 | 57 ± 4 | 1.4 | 55 ± 5 | 1.8 | 52 ± 4 | 1.8 |
For the WT LTAg and each mutant, increasing amounts of LTAg were titrated into a solution containing 50 nM fluorescently labeled ssDNA or dsDNA substrate. The apparent values of Kd (Kd,app) for ss- and dsDNA binding by WT and mutated LTAgs in the presence (+ATP-γ-S) and absence (−ATP-γ-S) of 1 mM ATP-γ-S, measured by rotational fluorescence anisotropy, were calculated by fitting the resultant binding curve to equation 1. Errors in Hill coefficients (n) were typically less than 10%.
+, binding detected but data could not be fit to equation 1.
−, weak or no binding detected.
DISCUSSION
Despite efforts to characterize ATP binding/hydrolysis for hexameric helicases, no systematic characterization of the roles of all the residues around the ATP binding pocket has been carried out, mainly due to the lack of a set of high-resolution structures of different nucleotide-bound states for a particular hexameric-helicase machine. Taking advantage of the high-resolution structures of LTAg hexamers in different nucleotide-bound states (8), we studied a series of ATP-binding-pocket mutations. Our mutational analysis has revealed the functional role of these residues from cis and trans in ATP hydrolysis and other functions related to the helicase motor function.
Oligomeric state and DNA binding of LTAg mutants.
Even though each mutated LTAg was capable of forming hexamers to a certain degree in the presence of nucleotides (Table 1), we observed a uniform decrease in the degree of hexamer formation for all trans residue mutants compared to the WT LTAg. This result suggests that these trans residues contribute to oligomerization by interacting with the ATP bound to the adjacent cis monomer. This may explain why hexamerization is favored when ATP is bound by LTAg.
DNA binding studies showed that ssDNA binding properties for all mutants other than the tD502A mutant were essentially unchanged from that of the WT (Table 3). Functional unwinding defects of these mutants are therefore not due to the disruption of ssDNA binding. In most cases, the addition of 1 mM ATP-γ-S increased the apparent affinity of LTAg for ssDNA. Even though the WT LTAg binds ds- and ssDNA with similar affinities, mutants appear to have a weaker binding affinity for dsDNA than for ssDNA (7). Binding curves for ssDNA were best fit with a Hill coefficient of greater than unity for the WT and most mutant proteins, indicative of cooperativity during DNA binding. One likely explanation for this cooperativity is that the binding of ssDNA is enhanced by hexamerization and that the assembly of the hexameric form is a cooperative process. This interpretation is consistent with the increase in affinity seen upon the addition of ATP-γ-S, which enhances hexamerization (Table 1).
We note that the DNA binding curves may reflect only the binding of DNA by hexameric LTAg, and thus, the apparent Kd for DNA binding would actually be an apparent Kd for oligomeric assembly. If this were the case, we would expect variant proteins that hexamerize poorly to also bind DNA with a lower apparent affinity. Since this is not the case (Tables 1 and 3), we conclude that the observed Kd mainly reflects DNA binding.
ATP hydrolysis by the mutants.
Our systematic alanine scanning of all nine residues involved in nucleotide binding revealed a wide variation of effects on DNA-independent ATPase behavior (Fig. 1; Table 2). All nine residues were required for maximal ATP hydrolysis.
For other reported helicases and ATPases, the apical water molecule of the γ-phosphate of ATP is normally shown to be coordinated by one Walker B residue (equivalent to cD474 in LTAg) (3-5, 12, 22). Thus, it is unusual in LTAg that the apical water molecule is coordinated by four different residues (cD474, cN529, tR498, and tR540). A single point mutation of each of these four residues resulted in decreased ATP hydrolysis, both in the presence and in the absence of ssDNA. To the best of our knowledge, these are the first data supporting the structural observation that the apical water orientation and/or activation may be regulated by a group of four residues that are from two adjacent monomers. One of the four residues, tR540, may have an additional role for ATP hydrolysis, as discussed below.
Another unique feature in the LTAg ATP-bound structure is the possibility that dual finger residues, tR540 and tK418, are potentially required to stabilize the transition state during ATP hydrolysis (8). In addition to the mutation of the usual arginine finger residue (tR540A), we found that the unconventional lysine finger mutation (tK418A) caused significant disruption in ATPase activity. This provides evidence for the first time that supports the presence of a second finger residue in LTAg to work together with the normal arginine finger for ATP hydrolysis. Although the lysine finger is not universally conserved among all AAA+ ATPases, several family members, including TorsinA, the dynein heavy chain, the Chl12 protein, and the HPV18 E1 protein, show sequence conservation at this position (data not shown), suggesting that this additional lysine finger may be a feature in some AAA+ family members.
Helicase activity of the mutants.
Our mutational studies revealed significant disruption of DNA strand separation in all variant proteins except one (the tK419A mutant) (Fig. 3). This result is consistent with the binding and hydrolysis of ATP being tightly coupled to dsDNA strand separation. We showed that lack of helicase activity was not due to lack of DNA binding, as each mutant other than tD502A bound both ssDNA and dsDNA (Table 3). The absence of helicase activity was also not due to the inability to hexamerize (Table 1).
Two mutant proteins, the tK419A (Fig. 3, lanes 6 to 8) and cT527A (Fig. 3, lanes 9 to 11) mutants, showed reduced helicase activity compared to the WT. Based on our biochemical evidence, the reduction of helicase activity for the tK419A and cT527A mutants may be due to decreased ssDNA-dependent ATPase stimulation (Fig. 2). For the cT527A mutant, the hydrogen bond established between cT527 and a water molecule in the ADP-bound form of LTAg may also be necessary for full helicase activity.
A very surprising result was the lack of detectable helicase activity for the tR498A and tD499A mutants (Fig. 3, lanes 21 to 26), which were competent as ATPases. To determine the cause of this behavior, we found it useful to compare the individual biochemical properties of these mutants with those of the helicase-competent tK419A mutant. Both the tR498A and tD499A mutants oligomerized to the same extent or better than the tK419A mutant (Table 1). The binding of ss- and dsDNA was minimally affected by all three mutations (Table 3). ssDNA-stimulated ATP hydrolysis rates are nearly identical for all three mutants (Fig. 2). The only difference between the tR498A and tD499A mutants and the tK419A mutant is a decrease in DNA-independent ATP hydrolysis rates, approximately twofold for the tR498A mutant and fourfold for the tD499A mutant (Table 2). These results suggest that defects in oligomerization, DNA binding, and DNA-dependent ATP hydrolysis cannot be used to explain the lack of helicase activity of the tR498A and tD499A mutants. Thus, one likely explanation for the loss of helicase activity is that tR498 and tD499 may be critical for coupling the energy of ATP binding/hydrolysis to the conformational changes that are necessary for DNA unwinding. This coupling function of tR498 and tD499 is consistent with the structural evidence that tR498 and tD499 interact with each other and move together as a lever arm in response to ATP binding as well as to ATP hydrolysis (8). An alternative explanation is that robust DNA-independent ATP hydrolysis is required for helicase activity. Previous work by Jiao and Simmons (11) suggested that tD499 may be involved in positioning residues K512 and H513, residues which have been shown to be necessary for DNA unwinding (19). In this respect, tD499 may ultimately couple ATP hydrolysis to the motion of K512 and/or H513.
The structural evidence also shows that another pair of positively charged/negatively charged residues, tR540/tD502, interact with each other and move together in response to ATP binding and hydrolysis (8). While tR540 contacts ATP directly as an arginine finger, tD502 has no direct contact with ATP. Therefore, it is unexpected that the tD502A mutant exhibited drastically reduced ATPase activity and no detectable helicase activity. One suggestion is that tD502 is required to interact with and stabilize the positive charge of tR540 in the ADP-bound form of LTAg. In the absence of this stabilization, the positive charge of tR540 may create structural instability, resulting in the poor hexamerization of the tD502A protein (Table 1). The drastic loss of ATPase activity of the tD502A mutant in the presence and absence of ssDNA contrasts that of the tD499A mutant, which retained appreciable ATPase activity, even though both mutants have a complete loss of helicase activity.
In summary, our mutational studies identified the residues required to support efficient ATP hydrolysis and helicase activity. All of these residues, except tR498 and tD499, were required for ssDNA-dependent stimulation of ATP hydrolysis. We have also demonstrated for the first time that two distinct pairs of positively charged/negatively charged residues were critical for LTAg helicase function. The pair tD502/tR540 was required for ATP hydrolysis and thus helicase activity. However, mutation of the other pair, tR498/tD499, had only moderate effects on ATP hydrolysis and DNA binding. Nonetheless, the helicase function was abolished if either residue of the tR498/tD499 pair was mutated to alanine, suggesting that these residues are involved in coupling ATP binding/hydrolysis to dsDNA unwinding. The functional roles of these residues in linking ATP hydrolysis, DNA binding, and DNA unwinding may be similar within the subclass of AAA+ helicases and for other closely related AAA+ hexameric machines.
Acknowledgments
This work was supported by NIH grant R01AI055926, awarded to X.S.C., and NIH grant 5T32CA009320-22, awarded to W.B.G.
Footnotes
Published ahead of print on 9 April 2008.
REFERENCES
- 1.Ahuja, D., M. T. Saenz-Robles, and J. M. Pipas. 2005. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 247729-7745. [DOI] [PubMed] [Google Scholar]
- 2.Borowiec, J. A., and J. Hurwitz. 1988. Localized melting and structural changes in the SV40 origin of replication induced by T-antigen. EMBO J. 73149-3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cho, H.-S., N.-C. Ha, L.-W. Kang, K. M. Chung, S. H. Back, S. K. Jang, and B.-H. Oh. 1998. Crystal structure of RNA helicase from genotype 1b hepatitis C virus. A feasible mechanism of unwinding duplex RNA. J. Biol. Chem. 27315045-15052. [DOI] [PubMed] [Google Scholar]
- 4.Crampton, D. J., S. Mukherjee, and C. C. Richardson. 2006. DNA-induced switch from independent to sequential dTTP hydrolysis in the bacteriophage T7 DNA helicase. Mol. Cell 21165-174. [DOI] [PubMed] [Google Scholar]
- 5.Erzberger, J. P., and J. M. Berger. 2006. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 3593-114. [DOI] [PubMed] [Google Scholar]
- 6.Fletcher, R. J., B. E. Bishop, R. P. Leon, R. A. Sclafani, C. M. Ogata, and X. S. Chen. 2003. The structure and function of MCM from archaeal M. Thermoautotrophicum. Nat. Struct. Biol. 10160-167. [DOI] [PubMed] [Google Scholar]
- 7.Fradet-Turcotte, A., C. Vincent, S. Joubert, P. A. Bullock, and J. Archambault. 2007. Quantitative analysis of the binding of simian virus 40 large T antigen to DNA. J. Virol. 819162-9174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gai, D., R. Zhao, D. Li, C. V. Finkielstein, and X. S. Chen. 2004. Mechanisms of conformational change for a replicative hexameric helicase of SV40 large tumor antigen. Cell 11947-60. [DOI] [PubMed] [Google Scholar]
- 9.Gomez-Lorenzo, M. G., M. Valle, J. Frank, C. Gruss, C. O. S. Soranzo, X. S. Chen, L. E. Donate, and J. M. Carazo. 2003. Large T antigen on the simian virus 40 origin of replication: a 3D snapshot prior to DNA replication. EMBO J. 226205-6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iyer, L. M., D. D. Leipe, E. V. Koonin, and L. Aravind. 2004. Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 14611-31. [DOI] [PubMed] [Google Scholar]
- 11.Jiao, J., and D. T. Simmons. 2003. Nonspecific double-stranded DNA binding activity of simian virus 40 large T antigen is involved in melting and unwinding of the origin. J. Virol. 7712720-12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korolev, S., J. Hsieh, G. H. Gauss, T. M. Lohman, and G. Waksman. 1997. Major domain swiveling revealed by the crystal structures of complexes of E. coli Rep helicase bound to single-stranded DNA and ADP. Cell 90635-647. [DOI] [PubMed] [Google Scholar]
- 13.Li, D., R. Zhao, W. Lilyestrom, D. Gai, R. Zhang, J. A. DiCaprio, E. Fanning, A. Jochimiak, G. Szakonyi, and X. S. Chen. 2003. Structure of the replicative helicase of the oncoprotein SV40 large tumor antigen. Nature 423512-518. [DOI] [PubMed] [Google Scholar]
- 14.Luo, X., D. G. Sanford, P. A. Bullock, and W. W. Bachovchin. 1996. Solution structure of the origin DNA-binding domain of SV40 T-antigen. Nat. Struct. Biol. 31034-1039. [DOI] [PubMed] [Google Scholar]
- 15.Maegley, K. A., S. J. Admiraal, and D. Herschlag. 1996. Ras-catalyzed hydrolysis of GTP: a new perspective from model studies. Proc. Natl. Acad. Sci. USA 938160-8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meinke, G., P. A. Bullock, and A. Bohm. 2006. Crystal structure of the simian virus 40 large T-antigen origin-binding domain. J. Virol. 804304-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mendez, J., and B. Stillman. 2003. Perpetuating the double helix: molecular machines at eukaryotic DNA replication origins. Bioessays 251158-1167. [DOI] [PubMed] [Google Scholar]
- 18.Nakazawa, K., H. Ojima, R. Ishii-Nozawa, K. Takeuchi, and Y. Ohno. 2004. Amino acid substitutions from and indispensable disulfide bond affect P2X2 receptor activation. Eur. J. Pharmacol. 48329-35. [DOI] [PubMed] [Google Scholar]
- 19.Shen, J., D. Gai, A. Patrick, W. B. Greenleaf, and X. S. Chen. 2005. The roles of the residues on the channel beta-hairpin and loop structures of simian virus 40 hexameric helicase. Proc. Natl. Acad. Sci. USA 10211248-11253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simmons, D. T., D. Gai, R. Parsons, A. Debes, and R. Roy. 2004. Assembly of the replication initiation complex on SV40 origin DNA. Nucleic Acids Res. 321103-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smelkova, N. V., and J. A. Borowiec. 1997. Dimerization of simian virus 40 T-antigen hexamers activates T-antigen DNA helicase activity. J. Virol. 718766-8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soultanas, P., M. S. Dillingham, S. S. Velankar, and D. B. Wigley. 1999. DNA binding mediates conformational changes and metal ion coordination in the active site of PcrA helicase. J. Mol. Biol. 290137-148. [DOI] [PubMed] [Google Scholar]
- 23.Sullivan, C. S., and J. M. Pipas. 2002. T antigens of simian virus 40: molecular chaperones for viral replication and tumorigenesis. Microbiol. Mol. Biol. Rev. 66179-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wold, M. S., J. J. Li, and T. J. Kelly. 1987. Initiation of simian virus 40 DNA replication in vitro: large-tumor-antigen- and origin-dependent unwinding of the template. Proc. Natl. Acad. Sci. USA 843643-3647. [DOI] [PMC free article] [PubMed] [Google Scholar]