

Abstract
Deletion of the A56R or K2L gene of vaccinia virus (VACV) results in the spontaneous fusion of infected cells to form large multinucleated syncytia. A56 and K2 polypeptides bind to one another (A56/K2) and together are required for interaction with the VACV entry fusion complex (EFC); this association has been proposed to prevent the fusion of infected cells. At least eight viral polypeptides comprise the EFC, but no information has been available regarding their interactions either with each other or with A56/K2. Utilizing a panel of recombinant VACVs designed to repress expression of individual EFC subunits, we demonstrated that A56/K2 interacted with two polypeptides: A16 and G9. Both A16 and G9 were required for the efficient binding of each to A56/K2, suggesting that the two polypeptides interact with each other within the EFC. Such an interaction was established by the copurification of A16 and G9 from infected cells under conditions in which a stable EFC complex failed to assemble and from detergent-treated lysates of uninfected cells that coexpressed A16 and G9. A recombinant VACV that expressed G9 modified with an N-terminal epitope tag induced the formation of syncytia, suggesting partial interference with the functional interaction of A56/K2 with the EFC during infection. These data suggest that A16 and G9 are physically associated within the EFC and that their interaction with A56/K2 suppresses spontaneous syncytium formation and possibly “fuse-back” superinfection of cells.
The simplest infectious form of vaccinia virus (VACV), called the mature virion (MV), consists of an outer membrane with at least 20 associated proteins surrounding a core containing the double-stranded DNA genome, enzymes and factors required for the transcription of early genes, and numerous structural proteins (9, 19). During virus assembly, some MVs are wrapped with additional membranes that facilitate intracellular movement, exocytosis, and cell-to-cell spread (29). Extracellular enveloped virions (EVs) are essentially MVs with an additional membrane, which is opened or removed prior to the fusion of the MV and cell membrane during virus entry (18). Thus, the viral fusion proteins are components of the MV membrane rather than the EV-specific membrane (20). Mutagenesis and affinity purification studies have shown that at least eight MV membrane polypeptides are required for entry and have no other known function (4, 16, 21, 22, 25, 26, 28, 30, 31). The components of this entry/fusion complex (EFC) are conserved in all poxviruses, indicating a common mechanism of infection. Studies with VACV indicate that fusion may occur in a pH-independent manner at the plasma membrane (1, 7, 8) or in a pH-dependent manner within endosomes (32, 33).
The EFC is also required for cell-cell fusion (28, 37), which can be induced by briefly lowering the pH (10, 13) or preventing expression of either the A56 (15, 24) or K2 (17, 34, 38) polypeptide. The glycosylated A56 polypeptide, referred to as the hemagglutinin (HA), is found on the plasma membrane of infected cells as well as in the outer EV membrane (3, 23). The glycosylated K2 polypeptide belongs to the serine protease inhibitor family, although the active site is not required for its fusion inhibitory function (35). Because K2 does not have a transmembrane segment, localization to the EV and plasma membrane is dependent on an association with A56 to form an A56/K2 multimer (5, 36). The physical interaction of A56/K2 with the EFC complex suggests a basis for the fusion regulatory role of A56/K2 (37). Prior to the present study, the interactions of individual EFC polypeptides with one another or with A56/K2 had not been elucidated. Here we describe the physical association of two EFC polypeptides, A16 and G9, and their specific interactions with A56/K2.
MATERIALS AND METHODS
Cells and viruses.
BS-C-1 (ATCC CCL-26) cells were maintained in minimum essential medium with Earle's balanced salts (Quality Biological, Gaithersburg, MD) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, penicillin, and streptomycin. HeLa S3 (ATCC CCL-2.2) suspension cells were grown in minimum essential medium with the spinner modification (Quality Biological) with 5% equine serum. 293TT cells that stably express the large T antigen, a gift from Chris Buck (6), were grown in Dulbecco minimum essential medium (DMEM; Quality Biological) supplemented with 10% FBS, 2 mM l-glutamine, and 400 μg/ml hygromycin (Invitrogen, Carlsbad, CA). The Western Reserve (WR) strain of VACV was used in the construction of all recombinant viruses unless otherwise noted. The general procedures used for preparing and titrating the stocks have been described previously (11).
Plasmid and recombinant VACV construction.
The recombinant viruses constructed for this study (Table 1) were vA28iA56TAP, vA21iA56TAP, vA28iA56TAPJ5Flag, vA28iA56TAPG93XFlag, vA28iA163XFlag, vA28iG93XFlag, vA16iA56TAPG93XFlag, vG9iA56TAP, vG9-HA(N), vG9-HA(C), and vG9-AU1(N) [where “v” refers to virus, “i” indicates an inducible gene, “TAP” refers to a tandem affinity purification tag, “3XFlag” indicates three copies of the Flag epitope, “HA” and “AU1” refer to the influenza virus HA epitope tag and the AU1 epitope tag, respectively, and (N) and (C) refer to the amino and carboxy termini, respectively, of the protein]. Recombinant viruses were screened by PCR to confirm the absence of parental virus, and sequencing was used to verify the inserted DNA. vA28iA56TAP, vA21iA56TAP, vA16iA56TAP, and vG9iA56TAP were constructed from vA28i (27), vA21i (31), vA16i (22), and vG9i (21), respectively, by appending the codons for a C-terminal TAP tag to the A56 gene. The DNA used to construct the C-terminal TAP tag has been described previously (37). vA28iA56TAP was the parental virus for the construction of vA28iA56TAPJ5Flag. An overlapping PCR (AccuPrime Pfx; Invitrogen) was used to assemble the DNA for the subsequent virus recombination. The layout of the DNA sequence for the J5Flag construct from the 5′ to 3′ end was as follows: (i) 500 bp of DNA sequence upstream of the J5R gene, (ii) enhanced green fluorescent protein (GFP) expressed from the I1L promoter, (iii) 70 nucleotides containing the J5R promoter, and (iv) the initial methionine of J5R, followed by the DNA sequence for the Flag epitope (DYKDDDK) and then the remaining DNA sequence of the J5R gene. The recombinant PCR product was cloned into pCR-BluntII-TOPO (Invitrogen) and verified by DNA sequencing. The J5Flag plasmid was linearized by cleavage with a unique restriction endonuclease. BS-C-1 cells were infected with vA28iA56TAP for 1 h and then transfected with the linearized plasmid, using Lipofectamine 2000 (Invitrogen). Isopropyl-β-d-thiogalactopyranoside (IPTG; 100 μM) was added to the medium to allow expression of the inducible A28 gene. The parental and recombinant viruses were distinguished by the GFP fluorescence of the latter. Recombinant virus plaques were clonally isolated through three rounds of plaque purification.
TABLE 1.
Recombinant VACV
| Recombinant virus | Parent | Description | Reference |
|---|---|---|---|
| vA16i | vT7lacOI | Inducible A16 | 22 |
| vA16iA56TAP | vA16i | Inducible A16; A56-TAPa | |
| vA16iA56TAPG93XFlag | vA16iA56TAP | Inducible A16; A56-TAPa; G9-Flagb | |
| vA21i | vT7lacOI | Inducible A21 | 31 |
| vA21iA56TAP | vA21i | Inducible A21; A56-TAPa | |
| vA28i | vT7lacOI | Inducible A28-HAc | 27 |
| vA28iA163XFlag | vA28i | Inducible A28-HAc; A16-Flagb | |
| vA28iA56TAP | vA28i | Inducible A28-HAc; A56-TAPa | |
| vA28iA56TAPG93XFlag | vA28iA56TAP | Inducible A28-HAc; A56-TAPa; G9-Flagb | |
| vA28iA56TAPJ5Flag | vA28iA56TAP | Inducible A28-HAc; A56-TAPa; J5-Flagd | |
| vA28iG93XFlag | vA28i | Inducible A28-HAc; G9-Flagb | |
| vA56TAP | vΔA56 | A56-TAPa | 37 |
| vG9i | vT7lacOI | Inducible G9-HAe | 21 |
| vG9iA56TAP | vG9i | Inducible G9-HAe; A56-TAPa | |
| vL5i | vT7lacOI | Inducible L5 | 30 |
| vG3i | vT7lacOI | Inducible G3 | A. Townsley |
| vH2i | vT7lacOI | Inducible H2 | 25 |
| vJ5i | vT7lacOI | Inducible J5-Flagd | |
| vG9-HA(N) | vG9i | G9-HAe | |
| vG9-HA(C) | VACV WR | G9-HAc | |
| vG9-AU1(N) | VACV WR | G9-AU1f |
TAP tag at C terminus.
3XFlag tag at C terminus.
C-terminal HA tag.
Flag tag (1X) at N terminus.
N-terminal HA tag.
AU1 tag at C terminus.
vA28iA56TAPG93XFlag and vA28iG93XFlag were derived by modification of vA28iA56TAP and vA28i, respectively. vA16iA56TAPG93XFlag was constructed sequentially by first generating vA16iA56TAP from vA16i. The 3XFlag epitope was added to the G9 protein to form vA16iA56TAPG93XFlag, vA28iA56TAPG93XFlag, and vA28iG93XFlag as follows. The G9R gene was PCR amplified from genomic DNA of the WR strain (ATCC VR-1354; accession number, AY243312) of VACV. The 3XFlag epitope was appended to the C terminus of G9 preceding the stop codon. The coding sequence of the Discosoma sp. red fluorescent protein (DsRed) expressed from the I1L intermediate promoter for screening of recombinant viruses was inserted following the tagged G9R gene. We next PCR amplified the L1R gene along with 90 bp upstream of the gene to include the L1R promoter. We then used recombinant PCR to add L1R with the promoter after the DsRed coding sequence. The promoter for the LIR gene is located within the C-terminal codons of G9. To conserve expression of the L1R gene, we had to duplicate the C terminus of G9, causing a direct repeat of the DNA sequence before and after the DsRed gene. Direct repeats are unstable in the virus genome (12). Therefore, to prevent the eventual loss of the DsRed gene, the codons of the final 33 amino acids from G9 were altered, while the expressed amino acid sequence was conserved. The recombinant G93XFlag PCR was cloned into pCR-BluntII-TOPO and verified by DNA sequencing. Recombinant viruses were generated by infecting BS-C-1 cells with the appropriate parental virus and then transfecting a linearized G93XFlag plasmid. Recombinant viruses were distinguished from parental viruses by DsRed fluorescence and were clonally isolated through three rounds of plaque purification.
vG9-HA(N) was constructed from DNA containing the promoter and coding sequence of G9 with an N-terminal HA tag and ∼500 bp of the upstream and downstream flanking sequences. The DNA was transfected into cells infected with vG9i (21), which contains a GFP expression cassette upstream of the G9R gene. vG9-HA(N) was clonally purified by picking nonfluorescent plaques. Recombinant vG9-HA(C) was constructed from DNA containing the promoter and coding sequence of G9 with a C-terminal HA tag and a GFP expression cassette and ∼500 bp of the upstream and downstream flanking sequences. vG9-HA(C) was clonally purified from the parental WR virus by picking fluorescent plaques. vG9-AU1(N) was made with DNA containing the promoter and open reading frame of G9 with an AU1 epitope tag at the N terminus and a cyan fluorescent protein expression cassette and flanking sequence.
The DNA sequences for the VACV WR A16L and G9R genes were optimized (Geneart, Regensburg, Germany) to alter codon usage and G-C content to improve RNA processing and translation. The optimized A16L and G9R genes were PCR amplified with oligonucleotides that contained the sequence of the influenza virus HA or 3XFlag epitope appended to the C terminus of the respective open reading frames. The PCR products of A16HA and G93XFlag were cloned into the directional TOPO vector pcDNA3.1 (Invitrogen) and sequenced to confirm proper insertion and sequence.
Affinity purification.
BS-C-1 cells (6 ×106) were infected at a multiplicity of 3 to 5 PFU per cell in Earle's balanced salts with 2% FBS. After 24 h, the cells were scraped into the medium and subjected to low-speed centrifugation. The cell pellet was washed once by resuspension in 150 mM NaCl with 50 mM Tris-HCl (pH 7.5). The cells were then disrupted with ice-cold lysis buffer (1% Triton X-100 [Sigma, St. Louis, MO], 150 mM NaCl, 50 mM Tris-HCl [pH 7.5]) supplemented with complete protease inhibitor (Roche, Indianapolis, IN) and rotated at 4°C for 30 min. The lysate was centrifuged at 4°C at 20,000 × g for 10 min, the postnuclear supernatant was collected, and 50 μl of the supernatant was saved for analysis. TAP was carried out as previously described (37). Single-step purifications were as follows. Packed beads (20 to 30 μl) of either streptavidin Sepharose (Millipore, Billerica, MA) or anti-Flag conjugated agarose (Sigma) were washed once with 1 ml of lysis buffer, and the postnuclear supernatant was added to the affinity resin and rotated overnight at 4°C. The affinity resin was washed five times with 1 ml of lysis buffer prior to elution. The bound material was eluted from the anti-Flag agarose by 50 μl of 1× lithium dodecyl-sulfate sample buffer (Invitrogen) supplemented with reducing agent (Invitrogen) and incubated at 100°C for 5 min. The beads were pelleted by centrifugation, and the supernatant was collected. Bound material was eluted from the streptavidin Sepharose by incubation with 300 μl of lysis buffer supplemented with 2 mM d-biotin (US Biological, Swampscott, MA). Elution was repeated two times, and the eluates were combined prior to being concentrated by precipitation with trichloroacetic acid. The precipitated material was resuspended in 40 μl of 1× lithium dodecyl sulfate sample buffer with reducing agent.
Transfection and coimmunoprecipitation.
293TT cells were plated at a density of 2 ×106 per 9.2 cm2 the day before being transfected in 10% DMEM plus glutamine, but without hygromycin. Cells were transfected with 2 μg of total DNA, using Lipofectamine 2000 (Invitrogen). After 24 h, fresh 10% DMEM was added and the incubation was continued for an additional 24 h, at which time cell extracts were prepared and subjected to immunoprecipitation.
Western blotting and antibodies.
Samples were loaded onto a 4% to 12% Novex NuPAGE acrylamide gel (Invitrogen) and separated by electrophoresis, using 2-(N-morpholino)ethanesulfonic acid buffer (Invitrogen). The protein samples were transferred to a nitrocellulose membrane and then blocked with 5% nonfat milk in Tris-buffered saline with 0.05% Tween 20 (TBS-t). A primary antibody was incubated a minimum of 1 h prior to extensive washing with TBS-t. Secondary antibodies (Pierce, Rockford, IL) were diluted in 5% nonfat milk TBS-t and incubated for at least 1 h. The nitrocellulose membrane was washed and then developed with Dura or Femto chemiluminescent substrate (Pierce). The antibodies were stripped from the nitrocellulose by a 20-min incubation at 55°C with Restore (Pierce). Antibodies to A56 (37), K2 (37), A21 (31), L5 (31), A16 (22), and p4a/p4b (R. Doms and B. Moss, unpublished data) were used in Western blot analysis. A monoclonal antibody (conjugated to horseradish peroxidase) against the influenza virus HA epitope was acquired from Bethyl Laboratories (Montgomery, TX). The anti-Flag M2 monoclonal antibody was obtained from Sigma (Kansas City, MO), and a monoclonal antibody to cellular glyceraldehyde-3-phosphate dehydrogenase was obtained from Covance Research (Princeton, NJ). Rabbit antiserum to H2 and A28 was generated by immunizing rabbits with purified recombinant H2 or A28 proteins, respectively, and was provided by Gretchen Nelson, NIAID. Peptide antibodies to the G3 protein were generated by immunizing rabbits with a synthetic peptide (SLNGKKHTFNLYDDNDIRT) coupled to keyhole limpet hemocyanin through an N-terminal cysteine (Covance).
RESULTS
A56/K2 interacts with a subset of the polypeptide components of the EFC.
We recently identified a novel interaction between the EFC and the fusion regulatory protein A56/K2 in detergent-treated extracts of infected cells (37). Neither A56 nor K2 polypeptides alone interact with the EFC; instead, a multimer of the two proteins is required. We sought to identify individual polypeptides within the EFC that interact with A56/K2. Our approach was based on previous observations that (i) the viral membrane is required for assembly of the EFC and (ii) that the EFC fails to assemble when expression of either A28 or A21 is repressed even though the remaining EFC proteins are incorporated into the viral membrane (26). By repressing A28 and A21 synthesis, we hoped to identify either individual polypeptides or previously uncharacterized subcomplexes of the EFC capable of interacting with A56/K2. To implement this strategy, we constructed two recombinant viruses, vA28iA56TAP and vA21iA56TAP, in which A28 and A21, respectively, were conditionally expressed. Conditional expression of A28 and A21 was regulated by components of the Escherichia coli lac operon in combination with the T7 phage DNA-dependent RNA polymerase such that viral gene expression depended on the addition of IPTG. Both recombinant viruses were constructed with a TAP tag appended to the C terminus of the A56R gene to allow purification of the A56/K2 heterodimer and associated polypeptides. In addition, A28 had a C-terminal HA epitope tag.
HeLa cells were infected with VACV strain WR, vA56TAP (37), vA28iA56TAP (with and without IPTG), vA21iA56TAP (with and without IPTG), or mock infected. VACV WR with untagged A56 served as a negative control for affinity purification, while vA56TAP with a constitutively expressed A28 was used as a positive control. The stocks of vA28iA56TAP and vA21iA56TAP were prepared in the presence of IPTG so that the virus particles contained A28 and A21, respectively, and were infectious, but synthesis of A28 or A21 during the next cycle depended on the addition of IPTG. At 24 h postinfection, the cells were lysed with Triton X-100 detergent, and the postnuclear supernatant was tandem affinity purified on streptavidin and calmodulin Sepharose columns. The proteins in the final eluate were concentrated, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and detected by Western blotting with specific antibodies.
Inspection of the Western blots prepared with the lysates prior to affinity purification confirmed that the synthesis of A28 and A21, respectively, was regulated by IPTG in cells infected with vA28iA56TAP and vA21iA56TAP (Fig. 1). The electrophoretic migration of A28 from cells infected with vA28iA56TAP in the presence of IPTG is slightly slower than that of A28 made by other recombinant viruses, due to the HA epitope tag. Note that repression of either A28 or A21 had no effect on the synthesis or stability of the other proteins examined. Due to alternative initiation codons for A56 as well as glycosylation, A56 and K2 were resolved as multiple bands.
FIG. 1.

A56/K2 physically associates with A16. HeLa cells were mock infected (M) or infected with VACV WR, vA56TAP, vA28iA56TAP (with [+] or without [−] IPTG), or vA21iA56TAP (with or without IPTG). After 24 h, the cells were lysed with Triton X-100 and the A56 protein was tandem affinity purified. The starting material (PreTAP) and purified samples (TAP) were separated by SDS-PAGE and analyzed by Western blotting, using antibodies to the entry proteins A16, A28, A21, and L5, as well as antibodies to the A56 and K2 proteins. Secondary antibodies conjugated to horseradish peroxidase were used for detection by chemiluminescence. The recombinant viruses are listed at the top, and the targets of the specific antibodies are listed on the side.
As anticipated, affinity-purified A56 from vA56TAP, vA28iA56TAP (with IPTG), and vA21iA56TAP (with IPTG) associated with K2 and with the EFC, as represented by four component proteins, A16, A28, A21, and L5 (Fig. 1). However, when A28 was repressed by omission of IPTG, K2 and A16 still copurified with A56, but only trace amounts of A21 and L5 were detected (Fig. 1). Similarly, when A21 was repressed, K2 and A16 were detected after affinity purification of A56, but there were only trace amounts of A28 and L5. We surmised that the fully assembled EFC is not required for interaction with A56/K2 and that the latter can interact with A16 itself or a subcomplex of polypeptides that includes A16. However, the association of A56/K2 with the four other entry proteins (G3, G9, H2, and J5) was not examined because we lacked the appropriate antibodies for detection at the time of the experiment.
A16 and G9 selectively copurify with A56/K2.
To determine if G3, G9, H2, and J5 were important for the interaction between the EFC and A56/K2, we constructed additional recombinant viruses with epitope tags on G9 or J5 and acquired antibodies to G3 and H2. The recombinant virus vA28iA56TAPJ5Flag encoded an inducible A28, TAP-tagged A56, and J5 with the Flag epitope fused to the N terminus. Cells were infected separately with VACV WR, vA56TAP, and vA28iA56TAPJ5Flag (with and without IPTG). After 24 h, the cells were lysed and analyzed directly or after TAP. Western blots of the lysate showed that A28 was stringently repressed and that both J5 and G3 could be detected using a Flag tag and a G3 peptide antibody, respectively (Fig. 2A). The true J5 band could be distinguished from the upper and lower background bands by its absence from cells infected with VACV WR and vA56TAP. We also probed for the core protein A3 (also called p4b), which served as a negative specificity control for the affinity purification. Bands corresponding to A56, K2, A16, A28, A21, L5, and G3 were detected after affinity purification of A56 from cells infected with vA56TAP (Fig. 2A). The same proteins, as well as J5 Flag, were detected after affinity purification of cells infected with vA28iA56TAPJ5Flag in the presence of IPTG (Fig. 2A). However, when cells were infected with vA28iA56TAPJ5Flag in the absence of IPTG, the A28 protein was repressed and only K2 and A16 copurified with A56. Thus, neither J5 nor G3 interacted with A56/K2 under these conditions.
FIG. 2.

A16 and G9 selectively copurify with A56/K2. (A) HeLa cells were mock infected (M) or infected with VACV WR, vA56TAP, or vA28iA56TAPJ5Flag with (+) or without (−) IPTG. After 24 h, the A56 protein was isolated from the infected cell lysates by successive bindings to streptavidin and calmodulin beads. Western blotting was performed on the starting material (PreTAP) and affinity purified proteins (TAP) as described in the legend to Fig. 1. (B) BS-C-1 cells were mock infected or infected with VACV WR or vA28iA56TAPG93XFlag (with or without IPTG). After 24 h, the A56 proteins were isolated by binding to streptavidin Sepharose. Western blotting was performed on the starting material (Start) and affinity purified proteins (Streptavidin) as described in the legend to Fig. 1.
Another virus we constructed was called vA28iA56TAPG93XFlag. As its name implies, expression of A28 was inducible, A56 was TAP-tagged, and G9 had three repeats of the Flag tag (at the C terminus). Cells were infected with VACV WR or vA28iA56TAPG93XFlag (with and without IPTG). At 24 h after infection, the cells were lysed and analyzed directly or after streptavidin affinity purification, as the calmodulin step was found not to be essential. Western blots of the lysate from cells infected with vA28iA56TAPG93XFlag showed that A28 was stringently repressed and that the proteins, including G9 with 3XFlag and H2, were detected (Fig. 2B). The use of three copies of the Flag epitope enhanced the detection of G9 over background bands. A56, K2, A16, G9, A28, and H2 were detected after affinity purification of proteins from cells infected with vA28iA56TAPG93XFlag in the presence of IPTG, but only K2, A16, and G9 copurified with A56 in the absence of IPTG (Fig. 2B). The A3 core protein was not detected in the presence or absence of IPTG. In a similar experiment (not shown), we also demonstrated by Western blotting that neither the F9 nor the L1 MV membrane protein associated with A56 in the absence of IPTG. Collectively, the affinity purification experiments indicated that of the eight EFC proteins, A16 and G9 interacted most directly with A56/K2.
A56/K2 copurifies with A16 and G9.
A reciprocal experiment was carried out to confirm the interaction of A56/K2 with A16 and G9. To implement the experiment, we constructed two additional epitope-tagged viruses. Both recombinant VACVs inducibly expressed A28, while G9 and A16 had a 3XFlag epitope attached to the C terminus in vA28iG93XFlag and vA28iA163XFlag, respectively. Cells were infected with VACV WR, vA28iG93XFlag (with or without IPTG), or vA28iA163XFlag (with or without IPTG). After 24 h, the cells were collected, and the postnuclear supernatant was incubated overnight with anti-Flag antibody covalently linked to agarose beads, which were then washed extensively prior to elution of the bound material. The eluted proteins were resolved by SDS-PAGE and detected by Western blotting. Analysis of the starting material indicated that A28 was stringently regulated and that each of the constructs expressed A56, K2, A16, and A21 (Fig. 3). The slower migration of A16 from samples infected with vA28iA163XFlag compared to that from cells infected with the wild type was due to the 3XFlag epitope. A21 was analyzed as a representative EFC protein to confirm that the complex was not assembled in the absence of A28. As anticipated, A16 or G9 interacted with A21 only when A28 was synthesized (with IPTG) (Fig. 3). Nevertheless, A16 and G9 interacted with A56 and K2 even when A28 was repressed (Fig. 3). Thus, we could show interaction of A56/K2 with A16 and G9 regardless of the affinity tag and whether it resided in A56, A16, or G9.
FIG. 3.

A56/K2 binds epitope-tagged A16 and G9. BS-C-1 cells were mock infected (M) or infected with VACV WR, vA28iA163XFlag (with [+] or without [−] IPTG), or vA28iG93XFlag (with or without IPTG). Infected cells were harvested after 24 h and lysed with Triton X-100. The lysate was cleared by centrifugation, and the A16 and G9 polypeptides were isolated by binding to agarose beads conjugated with Flag antibody. The eluate (Flag IP) and starting material (Start) were separated by SDS-PAGE and analyzed by Western blotting with antibodies to the viral proteins A56, K2, A28, A16, and A21 as indicated on the side. The viruses are indicated at the top of the figure.
Mutual dependence of A16 and G9 for their association with A56/K2.
In the above experiments, A16 and G9 always copurified with A56/K2. Additional recombinant viruses were constructed to determine whether expression of both A16 and G9 was required for a stable interaction with A56/K2. The recombinant vA16iA56TAPG93XFlag expressed an inducible form of A16, TAP-tagged A56, and G9 with a 3XFlag tag. Cells were infected with either VACV WR or vA16iA56TAPG93XFlag (with or without IPTG) for 24 h, and the postnuclear supernatant was analyzed directly or after streptavidin affinity purification. Analysis of the starting material prior to affinity purification demonstrated the stringent repression of A16 in the absence of IPTG (Fig. 4A). Importantly, the other EFC proteins examined, namely G9, A21, and L5, were stable even in the absence of A16. Curiously, the faster migrating A56 band predominated in the lysates of cells infected with vA16iA56TAPG93XFlag, suggesting initiation predominantly at the second start codon, but this was independent of IPTG. In the presence of IPTG, A56 interacted with K2, A16, G9, A21, and L5, as shown by their copurification (Fig. 4A). In contrast, only K2 interacted with A56 when synthesis of A16 was repressed. Therefore, G9 cannot interact independently with A56/K2.
FIG. 4.

Both A16 and G9 are required for binding A56/K2. (A) BS-C-1 cells were mock infected (M) or infected with VACV WR or vA16iA56TAPG93XFlag (with [+] or without [−] IPTG). The cells were lysed with Triton X-100 after 24 h, and the A56 protein was isolated by binding to streptavidin beads. The starting material (Start) and the affinity-purified proteins (Streptavidin) were resolved by SDS-PAGE and analyzed by Western blotting, using antibodies to the viral proteins indicated on the side. (B) HeLa cells were infected with vG9iA56TAP (with or without IPTG), and the A56 proteins were tandem affinity purified. The starting material (PreTAP) and the purified proteins (TAP) were analyzed as for panel A.
Recombinant vG9iA56TAP, which expressed an inducible form of G9 with an HA epitope tag at the N terminus and TAP-tagged A56, was used to test whether A16 can interact independently with A56/K2. Western blotting showed that K2, A16, A28, A21, and L5 copurified with A56 when cells were infected in the presence of IPTG (Fig. 4B). However, only K2 copurified with A56 when cells were infected in the absence of IPTG. Because of the relatively weak signal produced by the antibody to A16, this analysis was repeated in two independent experiments. With a more intense A16 signal in the lane with IPTG, we could detect a low amount of the protein copurifying with A56 when G9 was repressed (data not shown). Therefore, both A16 and G9 are needed for efficient interaction of either with A56/K2.
Association of A56/K2 with G9 requires A16.
Next, we carried out a reciprocal experiment in which we sought to determine whether A56/K2 copurified with Flag-tagged G9 in the absence of A16. Cells were mock infected or infected with VACV WR or vA16iA56TAPG93XFlag (with or without IPTG). After 24 h, the G9 protein was purified from the postnuclear supernatant by incubation with the Flag antibody conjugated to agarose beads. The beads were washed, and the bound proteins were eluted, separated by SDS-PAGE, and analyzed by Western blotting. Glyceraldehyde phosphate dehydrogenase served as a control for loading and nonspecific binding. Analysis of the lysate prior to immunopurification confirmed the stringent control of A16 expression (Fig. 5). When A16 (with IPTG) was expressed, the EFC represented by A28 and G9, as well as A56 and K2, copurified with G9 (Fig. 5). When A16 synthesis was repressed, however, neither A28, A56, nor K2 copurified with G9. Therefore, A56/K2 did not stably bind to G9 in the absence of A16.
FIG. 5.
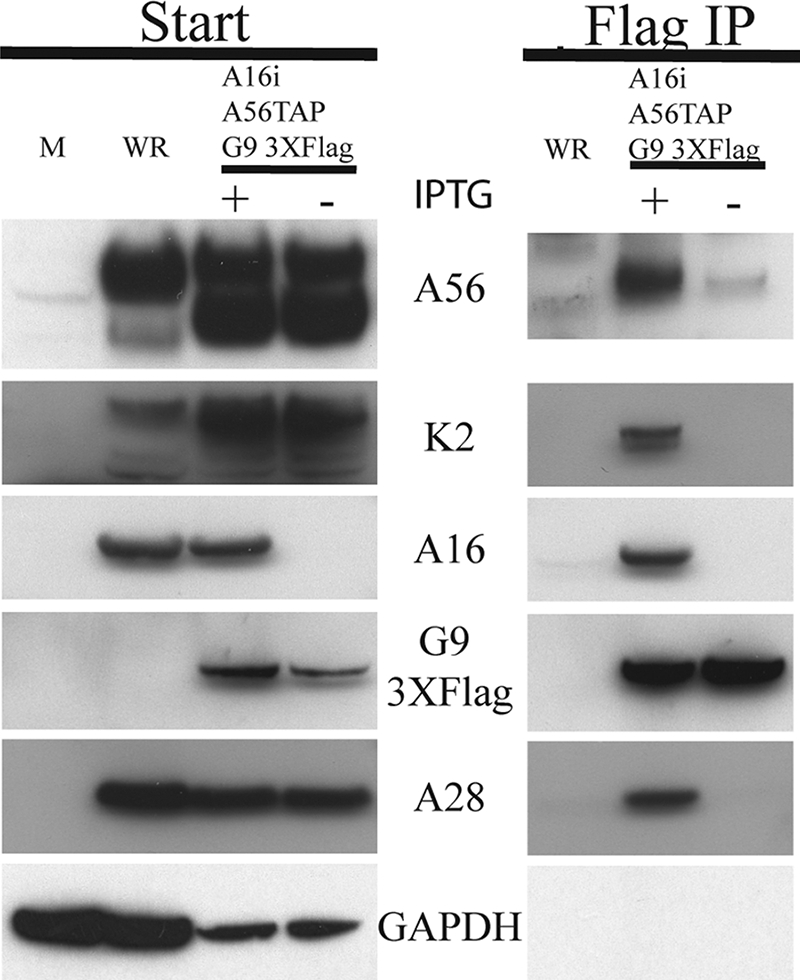
A16 is required for G9 to bind A56/K2. Cells were mock infected (M) or infected with VACV WR or vA16iA56TAPG93XFlag (with [+] or without[−] IPTG). Infected BS-C-1 cells were harvested at 24 h and lysed with Triton X-100. Flag antibody conjugated to agarose beads was used to purify the G9 protein. The bound material (Flag IP) was eluted from the agarose beads and separated along with the starting material (Start) by SDS-PAGE and then transferred to nitrocellulose. Western blotting was performed, using antibodies to the proteins A56, K2, A16, or A28, the Flag epitope, or glyceraldehyde phosphate dehydrogenase (GAPDH), as indicated.
A16 and G9 stably associate with each other in uninfected cells.
The inability of G9 or A16 to independently associate with A56/K2 suggested to us that these two EFC polypeptides exist as a heterodimer or higher-order multimer. To test this hypothesis, A16 and G9 were codon optimized for expression in human cells and tagged with a C-terminal influenza virus HA epitope or a 3XFlag epitope, respectively. A16HA and G93XFlag were cloned separately into pcDNA3.1 under the control of the human cytomegalovirus major immediate-early promoter. Human 293TT cells were transfected with the individual plasmids or cotransfected with both. After 48 h, the cells were lysed with Triton X-100, and synthesis of the recombinant proteins was demonstrated by SDS-PAGE and Western blotting of portions of the postnuclear supernatants (Fig. 6A and B). Additional portions of the postnuclear supernatants were incubated with agarose conjugated to Flag or HA antibody. The beads were washed extensively, and the eluted proteins were analyzed by SDS-PAGE and Western blotting. The A16HA protein was detected after Flag immunoprecipitation only when coexpressed with G93XFlag (Fig. 6A). Likewise, the G93XFlag was detected after HA immunoprecipitation only when coexpressed with A16HA (Fig. 6B). Thus, A16 and G9 can associate with each other in the absence of other viral proteins.
FIG. 6.
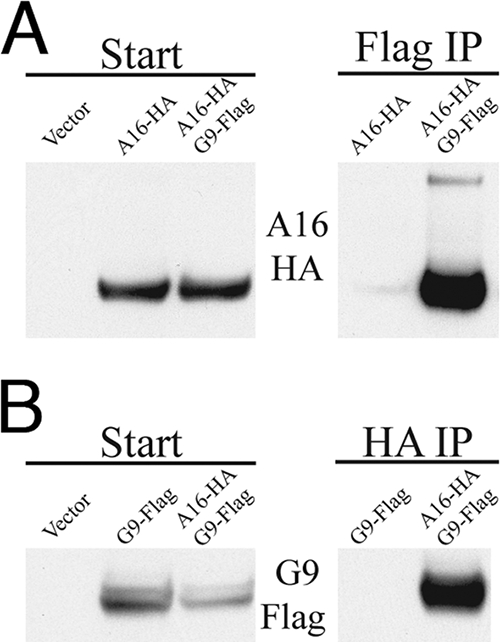
A16 and G9 interact in uninfected cells. (A) 293TT cells were transfected with empty vector or plasmid DNA expressing A16 with a C-terminal influenza virus HA epitope (A16-HA) or cotransfected with plasmids expressing A16-HA and G9 with a C-terminal 3XFlag tag (G9-Flag). After 48 h, the cells were lysed with Triton X-100, the postnuclear supernatant was incubated with the Flag antibody bound to beads. The starting material (Start) and the captured proteins (Flag IP) were analyzed by Western blotting with the anti-HA antibody. (B) Cells were transfected with empty vector or G9-Flag or cotransfected with A16-HA and G9-3XFlag. The lysates were incubated with anti-HA antibody bound to beads. The starting material (Start) and the captured proteins (HA IP) were analyzed by Western blotting with the anti-Flag antibody.
Modification of the G9 protein can result in a syncytial phenotype.
If A56/K2 prevents syncytia by interacting with A16 and G9, it seemed possible that some A16 or G9 mutants might also induce syncytia. In this respect, we noticed the presence of syncytia in cell monolayers that were infected with vG9i. Initially we thought that cell-cell fusion might be related to the overexpression of an EFC protein, since G9 was regulated by a bacteriophage T7 promoter and lac operon in place of the natural promoter. However, when we analyzed recombinant VACVs that inducibly express other EFC proteins, none of them induced more syncytia than the parent VACV WR (Fig. 7A). Syncytium formation was due to G9 expression, since cell fusion did not form in the absence of IPTG (data not shown). The G9 protein synthesized by vG9i had an N-terminal HA tag, which did not prevent the interaction with A56/K2, as shown in Fig. 4B. Nevertheless, to determine if the HA tag was related to the syncytium phenotype, two new recombinant VACVs were made. One had the HA tag at the N terminus of G9, and the other had the tag at the C terminus of G9. Only the virus expressing G9 with an N-terminal HA induced syncytia (Fig. 7C). The N-terminal modification was not sequence specific, as another VACV with an AU1 epitope tag at the N terminus of G9 also induced syncytia (Fig. 7B). We suggest that N-terminal modifications impair the functional interaction of G9 with A56/K2 without abrogating the binding per se.
FIG. 7.

Syncytium formation induced by modified G9. (A) HeLa cell monolayers were infected with 2 PFU per cell of VACVs expressing inducible A16, A21, L5, H2, A28, J5, G3, G9, or VACV WR for 1 h at 37°C and then washed and incubated for 21 h in the presence of IPTG. The cells were stained with Hoechst dye to visualize DNA and Alexa Fluor 594-phalloidin to visualize the actin cytoskeleton. The percentage of nuclei in syncytia was determined by counting the number of nuclei in fused cells that contained three or more nuclei and dividing by the total number of nuclei. Standard error bars are shown. (B and C) HeLa cell monolayers were infected with 2 PFU per cell of VACV expressing G9 with an HA epitope tag at the N or C terminus or an AU1 tag at the N terminus for 21 h and then stained as for panel A. Fluorescent and phase-contrast microscopic images are shown.
DISCUSSION
An unusual feature of VACV reproduction is that the infectious virus particles are assembled in the cytoplasm rather than at the plasma membrane and are subsequently transported to the periphery and exocytosed. Large numbers of progeny virus particles remain adherent to the cell surface, and these are chiefly responsible for virus spread to neighboring cells (2). As argued elsewhere (20), we believe that syncytia form when large numbers of virus particles “fuse-back,” i.e., deposit their fusion proteins into the plasma membrane of the parent cell. The fact that only small numbers of syncytia form normally implies a negative regulation of fuse-back. The EV membrane surrounding the MV may form one barrier to fuse-back, although MVs with broken EV membranes are detected on the surfaces of cells (14). The A56 and K2 polypeptides, which form a higher-order multimer on the cell surface and EV membrane, provide another barrier, since a syncytium phenotype occurs with null mutants of either (15, 17, 24, 34, 36, 38). In theory, A56/K2 could impede fuse-back either by preventing the disruption of the EV membrane and exposure of the MV or by the subsequent interaction of the MV and plasma membranes. The recent finding that A56/K2 interacts with the EFC suggests the latter mechanism is important (37). However, the EFC is still poorly characterized both structurally and functionally. Indeed, we do not know whether the EFC is a positive regulator or a mediator of fusion.
The EFC has at least eight component proteins, and multiple protein-protein interactions are needed to form a stable complex (26). In our current study, we exploited this instability by repressing synthesis of individual EFC polypeptides and determining the binding partners of A56/K2. In the presence of each other, the A16 and G9 EFC polypeptides were found to bind A56/K2 in detergent-treated extracts. Alone, however, these polypeptides did not bind or bound weakly to A56/K2, suggesting that a complex of A16 and G9 binds A56/K2. The idea that A16 and G9 bind to each other was substantiated by coimmunoprecipitation of the two polypeptides following transfection of expression vectors into uninfected cells. Thus, if A16 and G9 bind independently albeit weakly to A56/K2, the stability of the interaction could be enhanced by the A16/G9 complex. Alternatively, a unique A56/K2 binding site may be formed by the interaction of A16 and G9. The model in Fig. 8 depicts A56/K2 in the plasma membrane of an infected cell interacting with the A16 and G9 subunits of the EFC in the viral membrane.
FIG. 8.

Model of G9/A16 in MV membrane binding to A56/K2 in plasma membrane. G9 and A16 are anchored in MV membrane in association with the EFC. A56/K2 is anchored in plasma membrane through the transmembrane domain of A56. The interaction of A56/K2 with A16 and G9 is postulated to prevent fusion of the MV particle with the plasma membrane.
A16 and G9 appear to have two roles: each is required for membrane fusion and virus entry as well as for interaction with A56/K2. Since viruses with mutations in A56 or K2 form syncytia, modifications of A16 or G9 that perturb the interaction with A56/K2 could result in a similar phenotype. Indeed, while characterizing a recombinant virus with an inducible G9 (21), we noticed the formation of syncytia. Syncytium formation depended on the presence of an epitope tag at the N terminus of G9. The modification of G9 did not abrogate the interaction of the EFC with A56/K2 per se, although we did not investigate the possibility that the interaction was weakened or altered.
Under our experimental conditions, the interaction of the EFC and A56/K2 likely occurred after treatment of the infected cells with detergent. Naturally, however, we suggest that the interaction would occur between the EFC in the MV membrane and A56/K2 in the plasma membrane. Thus, the presence of A56/K2 in the plasma membrane may provide a way of differentiating infected from uninfected cells, presumably ensuring that VACVs preferentially fuse with the latter and initiate a new infection. We are currently carrying out experiments to test this prediction.
Acknowledgments
We thank Norman Cooper for preparing cells, Gretchen Nelson for antibodies, and Chris Buck for supplying the 293TT cells.
This work was supported by the Division of Intramural Research, NIAID, NIH.
Footnotes
Published ahead of print on 19 March 2008.
REFERENCES
- 1.Armstrong, J. A., D. H. Metz, and M. R. Young. 1973. The mode of entry of vaccinia virus into L cells. J. Gen. Virol. 21533-537. [DOI] [PubMed] [Google Scholar]
- 2.Blasco, R., and B. Moss. 1992. Role of cell-associated enveloped vaccinia virus in cell-to-cell spread. J. Virol. 664170-4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown, C. K., P. C. Turner, and R. W. Moyer. 1991. Molecular characterization of the vaccinia virus hemagglutinin gene. J. Virol. 653598-3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown, E., T. G. Senkevich, and B. Moss. 2006. Vaccinia virus F9 virion membrane protein is required for entry but not virus assembly, in contrast to the related L1 protein. J. Virol. 809455-9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brum, L. M., P. C. Turner, H. Devick, M. T. Baquero, and R. W. Moyer. 2003. Plasma membrane localization and fusion inhibitory activity of the cowpox virus serpin SPI-3 require a functional signal sequence and the virus encoded hemagglutinin. Virology 306289-302. [DOI] [PubMed] [Google Scholar]
- 6.Buck, C. B., D. V. Pastrana, D. R. Lowy, and J. T. Schiller. 2004. Efficient intracellular assembly of papillomaviral vectors. J. Virol. 78751-757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carter, G. C., M. Law, M. Hollinshead, and G. L. Smith. 2005. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J. Gen. Virol. 861279-1290. [DOI] [PubMed] [Google Scholar]
- 8.Chang, A., and D. H. Metz. 1976. Further investigations on the mode of entry of vaccinia virus into cells. J. Gen. Virol. 32275-282. [DOI] [PubMed] [Google Scholar]
- 9.Condit, R. C., N. Moussatche, and P. Traktman. 2006. In a nutshell: structure and assembly of the vaccinia virion. Adv. Virus Res. 6631-124. [DOI] [PubMed] [Google Scholar]
- 10.Doms, R. W., R. Blumenthal, and B. Moss. 1990. Fusion of intra- and extracellular forms of vaccinia virus with the cell membrane. J. Virol. 644884-4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Earl, P. L., N. Cooper, S. Wyatt, B. Moss, and M. W. Carroll. 1998. Preparation of cell cultures and vaccinia virus stocks, p. 16.16.1-16.16.3. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 2. John Wiley and Sons, New York, NY. [Google Scholar]
- 12.Falkner, F. G., and B. Moss. 1990. Transient dominant selection of recombinant vaccinia viruses. J. Virol. 643108-3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong, S. C., C. F. Lai, and M. Esteban. 1990. Vaccinia virus induces cell fusion at acid pH and this activity is mediated by the N-terminus of the 14-kDa virus envelope protein. Virology 17881-91. [DOI] [PubMed] [Google Scholar]
- 14.Husain, M., A. S. Weisberg, and B. Moss. 2007. Resistance of a vaccinia virus A34R deletion mutant to spontaneous rupture of the outer membrane of progeny virions on the surface of infected cells. Virology 366424-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ichihashi, Y., and S. Dales. 1971. Biogenesis of poxviruses: interrelationship between hemagglutinin production and polykaryocytosis. Virology 46533-543. [DOI] [PubMed] [Google Scholar]
- 16.Izmailyan, R. A., C. Y. Huang, S. Mohammad, S. N. Isaacs, and W. Chang. 2006. The envelope G3L protein is essential for entry of vaccinia virus into host cells. J. Virol. 808402-8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Law, K. M., and G. L. Smith. 1992. A vaccinia serine protease inhibitor which prevents virus-induced cell fusion. J. Gen. Virol. 73549-557. [DOI] [PubMed] [Google Scholar]
- 18.Law, M., G. C. Carter, K. L. Roberts, M. Hollinshead, and G. L. Smith. 2006. Ligand-induced and nonfusogenic dissolution of a viral membrane. Proc. Natl. Acad. Sci. USA 1035989-5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moss, B. 2007. Poxviridae: the viruses and their replication, p. 2905-2946. In D. M. Knipe (ed.), Fields virology, vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 20.Moss, B. 2006. Poxvirus entry and membrane fusion. Virology 34448-54. [DOI] [PubMed] [Google Scholar]
- 21.Ojeda, S., A. Domi, and B. Moss. 2006. Vaccinia virus G9 protein is an essential component of the poxvirus entry-fusion complex. J. Virol. 809822-9830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ojeda, S., T. G. Senkevich, and B. Moss. 2006. Entry of vaccinia virus and cell-cell fusion require a highly conserved cysteine-rich membrane protein encoded by the A16L gene. J. Virol. 8051-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Payne, L. G., and E. Norrby. 1976. Presence of hemagglutinin in the envelope of extracellular vaccinia virus particles. J. Gen. Virol. 3263-72. [DOI] [PubMed] [Google Scholar]
- 24.Seki, M., M. Oie, Y. Ichihashi, and H. Shida. 1990. Hemadsorption and fusion inhibition activities of hemagglutinin analyzed by vaccinia virus mutants. Virology 175372-384. [DOI] [PubMed] [Google Scholar]
- 25.Senkevich, T. G., and B. Moss. 2005. Vaccinia virus H2 protein is an essential component of a complex involved in virus entry and cell-cell fusion. J. Virol. 794744-4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Senkevich, T. G., S. Ojeda, A. Townsley, G. E. Nelson, and B. Moss. 2005. Poxvirus multiprotein entry-fusion complex. Proc. Natl. Acad. Sci. USA 10218572-18577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Senkevich, T. G., B. M. Ward, and B. Moss. 2004. Vaccinia virus A28L gene encodes an essential protein component of the virion membrane with intramolecular disulfide bonds formed by the viral cytoplasmic redox pathway. J. Virol. 782348-2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Senkevich, T. G., B. M. Ward, and B. Moss. 2004. Vaccinia virus entry into cells is dependent on a virion surface protein encoded by the A28L gene. J. Virol. 782357-2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith, G. L., and M. Law. 2004. The exit of vaccinia virus from infected cells. Virus Res. 106189-197. [DOI] [PubMed] [Google Scholar]
- 30.Townsley, A., T. G. Senkevich, and B. Moss. 2005. The product of the vaccinia virus L5R gene is a fourth membrane protein encoded by all poxviruses that is required for cell entry and cell-cell fusion. J. Virol. 7910988-10998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Townsley, A., T. G. Senkevich, and B. Moss. 2005. Vaccinia virus A21 virion membrane protein is required for cell entry and fusion. J. Virol. 799458-9469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Townsley, A. C., and B. Moss. 2007. Two distinct low-pH steps promote entry of vaccinia virus. J. Virol. 818613-8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Townsley, A. C., A. S. Weisberg, T. R. Wagenaar, and B. Moss. 2006. Vaccinia virus entry into cells via a low-pH-dependent endosomal pathway. J. Virol. 808899-8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turner, P. C., and R. W. Moyer. 1992. An orthopoxvirus serpin-like gene controls the ability of infected cells to fuse. J. Virol. 662076-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turner, P. C., and R. W. Moyer. 1995. Orthopoxvirus fusion inhibitor glycoprotein SPI-3 (open reading frame K2L) contains motifs characteristic of serine protease inhibitors that are not required for control of cell fusion. J. Virol. 695978-5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turner, P. C., and R. W. Moyer. 2006. The cowpox virus fusion regulator proteins SPI-3 and hemagglutinin interact in infected and uninfected cells. Virology 34788-99. [DOI] [PubMed] [Google Scholar]
- 37.Wagenaar, T. R., and B. Moss. 2007. Association of vaccinia virus fusion regulatory proteins with the multicomponent entry/fusion complex. J. Virol. 816286-6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou, J., X. Y. Sun, G. J. P. Fernando, and I. H. Frazer. 1992. The vaccinia virus K2L gene encodes a serine protease inhibitor which inhibits cell-cell fusion. Virology 189678-686. [DOI] [PubMed] [Google Scholar]