

Abstract
Abelson murine leukemia virus (Ab-MLV) arose from a recombination between gag sequences in Moloney MLV (Mo-MLV) and the c-abl proto-oncogene. The v-Abl oncoprotein encoded by Ab-MLV contains MA, p12, and a portion of CA sequences derived from the gag gene of Mo-MLV. Previous studies indicated that alteration of MA sequences affects the biology of Mo-MLV and Ab-MLV. To understand the role of these sequences in Ab-MLV transformation more fully, alanine substitution mutants that affect Mo-MLV replication were examined in the context of Ab-MLV. Mutations affecting Mo-MLV replication decreased transformation, while alanine mutations in residues dispensable for Mo-MLV replication did not. The altered v-Abl proteins displayed aberrant subcellular localization that correlated to transformation defects. Immunofluorescent analyses suggested that aberrant trafficking of the altered proteins and improper interaction with components of the cytoskeleton were involved in the phenotype. Similar defects in localization were observed when the Gag moiety containing these mutations was expressed in the absence of abl-derived sequences. These results indicate that MA sequences within the Gag moiety of the v-Abl protein contribute to proper localization by playing a dominant role in trafficking of the v-Abl molecule.
Abelson murine leukemia virus (Ab-MLV) arose from a spontaneous recombination event that occurred between the gag gene of Moloney MLV (Mo-MLV) and a cellular proto-oncogene, c-abl. As a result, Ab-MLV encodes the v-Abl protein, a constitutively active tyrosine kinase. The virus transforms pre-B cells and NIH 3T3 cells in vitro and induces pre-B-cell lymphoma in mice (23, 27). The abl-derived region of the protein contains the catalytic domain, the SH2 domain that binds tyrosine-phosphorylated proteins, and the carboxyl terminus, a region that enhances pre-B-cell transformation in a manner not clearly understood (14, 15). Intact tyrosine kinase activity and the SH2 domain are required for transformation by Ab-MLV (9, 11, 23).
Despite the central role played by abl-derived sequences in mediating the biological consequences of Ab-MLV infection, mutational analyses have shown that the Gag region is also important (3, 17, 18, 21, 34). Unlike Mo-MLV Gag, a polyprotein that is processed to yield MA, p12, CA, and NC, the Gag domain retained in v-Abl contains only MA, p12, and a small portion of CA sequences and is not processed to give rise to individual proteins. Mo-MLV Gag proteins play a central role in virion formation and genome packaging and are also critical for early events in infection (reviewed in reference 29). However, the helper virus-dependent nature of Ab-MLV suggests that the gag sequences in this virus contribute to v-Abl function in ways that are distinct from their role in Mo-MLV.
Because the RNA sequence at the site of gag-abl fusion resembles a splice site (4), the Gag residues retained in v-Abl may not reflect biological selection for protein function. Nonetheless, the integrity of the Gag region appears to be important because insertional mutations in gag result in decreased transformation by Ab-MLV (21). In addition, a v-Abl protein containing only the first 34 Gag residues of v-Abl is localized to the nucleus, suggesting that Gag suppresses nuclear localization signals known to be present in v-Abl (34). The myristoylation signal at the amino terminus of MA appears important for directing the v-Abl protein to the plasma membrane. Loss of this signal results in a complete defect in transformation of NIH 3T3 cells but renders the virus capable of transforming BaF3, a factor-dependent hematopoietic cell line (3). Other deletions affecting MA sequences also result in severe defects in Ab-MLV transformation (17, 18, 34).
Because several studies have indicated that MA sequences play an important role in Ab-MLV-mediated transformation and in Mo-MLV replication (8, 17, 18, 28, 34), we examined the way these sequences contribute to transformation in detail. MA sequences important for Mo-MLV replication as well as those that do not affect viral replication (8) and the glycine residue at position 2 that abolishes Mo-MLV replication when mutated to an alanine (28) were targeted in Ab-MLV. Mutants containing alterations known to affect Mo-MLV replication were compromised for transformation, a phenotype that correlated to aberrant localization of the v-Abl proteins. Because expression of these mutations in the context of the Gag sequences present in v-Abl caused similar defects in localization, these data indicate that Gag plays a dominant role in localization of the molecule and suggest that conservation of Gag functions that are important for replicating retroviruses may have played an important role in the origins of Ab-MLV and other transforming retroviruses that encode Gag-v-Onc fusion proteins.
MATERIALS AND METHODS
Cells and viruses.
293T, NIH 3T3, and Ab-MLV-transformed pre-B cells were grown in standard culture media appropriate to these cell types as described elsewhere (34). Viral stocks were prepared by transfection of 293T cells with pMIG vector (9, 31) or pMSCV vector encoding various Ab-MLV gag mutants and the pSV-ψ−-E-MLV retroviral packaging plasmid (12) as previously described (32). To determine the infectious titer of the viral stocks, NIH 3T3 cells were infected with virus containing 8 μg/ml Polybrene for 24 h and analyzed for the frequency of green fluorescent protein (GFP)-positive cells by flow cytometry. To characterize Ab-MLV mutants in a pre-B-cell setting, the temperature-sensitive Ab-MLV(P70/H590)-transformed 7C411 pre-B-cell line (5) was superinfected with various viral stocks in the presence of 8 μg/ml Polybrene by centrifuging the mixture at 1,000 × g for 1.5 h at room temperature. Derivatives of 7C411 cells expressing wild-type or mutant Ab-MLV were collected by sorting for GFP-positive cells by use of a MoFlo instrument at 24 h postinfection. The cells were maintained at 34°C, the permissive temperature for 7C411 cells. To test for the ability of different Ab-MLV mutants to support the growth and viability of pre-B cells, 7C411 derivatives were seeded at 5 × 105 cells in 60-mm-diameter dishes and incubated at 39.5°C, the nonpermissive temperature for 7C411 cells. Proliferation of the cells was monitored by counting cells by use of a hemocytometer; viability was determined by trypan blue exclusion.
Transformation assays.
The transforming efficiency of different virus stocks was tested using an NIH 3T3 cell transformation assay (25) or a bone marrow transformation assay (1). To determine the ability of different virus stocks to transform NIH 3T3 cells, cells were infected with serial dilutions of virus as described above and fed every 5 days with fresh medium. Foci of morphologically distinct, transformed cells were counted 13 to 15 days postinfection. Lymphoid transformation assays were done using bone marrow cells from BALB/cJ mice. The cells were infected with undiluted viral stocks and plated directly onto 35-mm-diameter dishes. The cultures were fed every 5 days and were scored as transformed when the density of transformed lymphoid cells reached 2 × 106 cells/ml (1, 34).
Construction and characterization of viral plasmids.
The Ab-MLV coding region was cloned into TA cloning vector (Stratagene) for use as a template, and mutations were introduced into Ab-MLV coding sequences by use of the QuikChange mutagenesis (Stratagene) method. All mutagenized fragments were sequenced and reinserted into pMIG vector (9, 31). The oligonucleotide sequences used to generate the G2A, 54-56A, 60-62A, 69-71A, 85-87A, and 111-115A mutants are available on request.
Protein analysis.
To confirm the expression of the proteins encoded by the mutants, 293T cells were transfected with the viral plasmids and lysates were analyzed by Western blotting as described elsewhere (34). Erk phosphorylation was examined by transfecting 293T cells with the different viral plasmids and incubating the cells in serum-free medium 24 h after transfection. Protein lysates were prepared after an additional 24 h and analyzed by Western blotting. The antibodies used included anti-Abl (24-21) (26), anti-phospho-Erk (Cell Signaling Technology), anti-Erk (Cell Signaling Technology), anti-phosphotyrosine (Upstate), and alkaline phosphatase-conjugated goat anti-mouse or goat anti-rabbit (Promega) antibodies.
Immunofluorescent staining.
To prepare NIH 3T3 cells for immunofluorescent staining, the cells were incubated in chambered coverglasses (Lab-Tek) at 37°C overnight. The cells were infected the following day and incubated for 2 to 3 h at 37°C. After infection, growth medium was added, and the cells were incubated at 37°C for 30 h. Pre-B-cell transformants were centrifuged onto glass slides by use of a Cytospin instrument (Shandon). All samples were fixed in 3% formaldehyde-phosphate-buffered saline (PBS) for 10 min at room temperature and permeabilized with 0.1% Trition X-100 in PBS for 10 min. After blocking the samples by use of 10% fetal calf serum (FCS)-PBS for 30 min, the cells were treated with various antibodies diluted in 0.5% FCS-PBS for 1 h at room temperature. After washing the cells three times with PBS for 10 min each time, the cells were stained with Alexa 647- or Alexa 594-conjugated goat anti-mouse antibody (Molecular Probes) or Alexa 568-conjugated goat anti-rabbit antibody (Molecular Probes) in 0.5% FCS-PBS for 30 min at room temperature. For this and all subsequent steps, the samples were protected from light. The cells were washed three times with PBS for 10 min each time. In some cases, the samples were stained with 300 nM 4′,6′-diamidino-2-phenylindole (DAPI) in PBS for 5 min and washed three times with PBS for 5 min each time. Samples were visualized using a spinning disc confocal microscope (Zeiss) and UltraVIEW 5.5 software or a fluorescence E400 Nikon microscope and Spot Advanced software. Primary antibodies used included anti-Abl (24-21) (26), anti-EEA1 (Sigma; catalog no. E4156), anti-GM130 (Calbiochem; catalog no. CB1008), anti-γ-tubulin (Sigma; catalog no. T5192), antivimentin (Abcam; catalog no. Ab7783), anticalnexin (Stressgen; catalog no. SPA860), and anti-LAMP3 (Santa Cruz; catalog no. Sc-15363) antibodies.
Analysis of viral sequences in lymphoid transformants.
Approximately 5 × 106 transformed pre-B cells were harvested and washed once with PBS. The cells were lysed using sarcosyl lysis buffer (10 mM Tris [pH 7.5], 10 mM EDTA [pH 8.0], 10 mM NaCl, 0.5% N-lauroylsarcosine) containing 1 mg/ml proteinase K at 55°C overnight. The following day, the samples were treated with Na-EtOH (75 mM NaCl in −20°C ethanol) to precipitate genomic DNA. The DNA was recovered and dissolved in double-distilled water. To clone the gag sequences from transformed pre-B-cell lines, 5 to 10 μg of genomic DNA was digested with EcoRI for 3 h at 37°C to recover the v-abl coding region. Digested DNA was desalted by incubation on a 0.05 μm-pore-size nitrocellulose filter (Millipore) for 15 min. The v-abl coding region, including nucleotides 1 to 758 (which includes all of gag sequences), was amplified using appropriate primers in a PCR mixture containing 50 to 200 ng of template DNA, 1.25 U of native Pfu DNA polymerase, 1× Pfu plus buffer, 0.4 μM primers, and 0.2 mM deoxynucleoside triphosphate. Samples were denatured for 2 min at 94°C and amplified for 30 cycles of 45 s at 94°C, 45 s at 55°C, and 2.5 min at 72°C followed by a 10-min extension at 72°C. PCR products were purified using a PCR purification kit (Qiagen), and 3′A overhangs were added by incubating the purified DNA in 0.2 mM dATP-1× Tαq buffer-0.5 U Taq DNA polymerase (Applied Biosystems) in a 50-μl total volume for 15 min at 72°C. PCR products were cloned into TOPO TA cloning vector (Invitrogen), and the inserts were sequenced.
RESULTS
Alanine substitutions in MA sequences do not affect v-Abl protein expression and tyrosine kinase activity.
Since loss of MA sequences affects the ability of Ab-MLV to induce transformation in a marked way (34), alanine substitution mutants that affect MA were constructed to examine the role these sequences play in transformation in more detail. Sequences that have been shown to affect Mo-MLV replication as well as those that do not influence replication (8, 28) were mutated in the context of Ab-MLV (Fig. 1A). In addition, because others have shown that an Ab-MLV mutant encoding an aspartic acid at position 2 instead of a glycine fails to transform NIH 3T3 cells but renders BaF3 hematopoietic cells factor independent (3), a mutant in which the glycine codon was replaced with an alanine codon was also prepared. Western blot analysis of cell lysates prepared from transfected 293T cells confirmed that all mutants expressed v-Abl proteins (Fig. 1B). In addition, probing the blot with antibodies directed against phosphotyrosine revealed that all of the v-Abl proteins, except the kinase-inactive D484N mutant (20), had intact tyrosine kinase activity (Fig. 1B). Densitometric analysis indicated that all levels of kinase activity differed less than twofold for all of the v-Abl proteins (data not shown). Although samples including cells expressing the G2A mutant have very high levels of kinase activity compared to other samples in the example shown, in most cases, the levels of tyrosine-phosphorylated proteins recovered from cells expressing this mutant were similar to those obtained with wild-type virus.
FIG. 1.

Ab-MLV mutants with alanine substitutions in MA sequences retain intact tyrosine kinase activity. (A) The different regions of the wild-type P120 v-Abl protein are depicted. The MA region of Gag is expanded to show the positions of residues in MA that were changed to alanines. The numbers above the diagram denote amino acid numbers, and the jagged lines indicate the myristoylation site on the protein. (B) 293T cells were transiently transfected with plasmids expressing the different mutants, and cell lysates were prepared for Western blotting using the indicated antibodies. The arrowhead indicates the positions of the v-Abl proteins expressed by the different viruses. D484N encodes a kinase-inactive form of the v-Abl protein (20). α-pTyr, anti-phosphate-tyrosine.
MA sequences important for Mo-MLV replication are important for NIH 3T3 cell transformation.
To examine the effects of alanine substitutions in MA on transformation by Ab-MLV, stocks with matched titer values were prepared and tested for their ability to transform cells in an NIH 3T3 focus assay (25, 34). As is consistent with previous findings (3), the G2A mutant that lacks the myristoylation signal failed to transform NIH 3T3 cells (Table 1). When residues important for Mo-MLV replication were changed to alanines in the 54-56A, 60-62A, and 85-87A mutants, a 30- to 90-fold decrease in the transformation of NIH 3T3 cells was observed (Table 1). In contrast, alanine substitutions in residues dispensable for Mo-MLV replication such as those in the 69-71A mutant did not affect the transformation of NIH 3T3 cells. The titer of the 111-115A mutant was not assessed in these NIH 3T3 cell transformation assays. However, independent analyses revealed that this mutant readily transformed the cells (data not shown). These results suggest that residues important for Mo-MLV replication also play an important role in transformation by Ab-MLV.
TABLE 1.
MA sequences important for Mo-MLV replication are important for transformation of NIH 3T3 cells by Ab-MLVa
| Virus | Transforming titer (FFU/ml)b
|
|
|---|---|---|
| Expt 1 | Expt 2 | |
| P120 | 3.0 × 103 | 4.6 × 103 |
| G2A | <1 | <1 |
| 54-56A | 6.5 × 101 | 6.5 × 101 |
| 60-62A | 8.0 × 101 | 5.0 × 101 |
| 69-71A | 2.5 × 103 | 1.5 × 103 |
| 85-87A | 1.1 × 102 | 5.3 × 101 |
Transformation was assayed by using NIH 3T3 cells (26). Cells were infected with serial dilutions of virus stocks of similar titers. All mutants were tested for transformation in at least two independent experiments.
The numbers of transformed foci formed per milliliter of virus added were determined. Values with a less-than symbol (<) represent the limit of detection. FFU, focus-forming units.
MA sequences important for Mo-MLV replication play a role in pre-B-cell transformation.
Some Ab-MLV mutants such as those affecting the v-Abl carboxyl terminus exhibit differential transformation phenotypes in NIH 3T3 and pre-B cells (14, 15). To assess the ability of the alanine mutants to transform pre-B cells, a liquid transformation assay that allows transformation to be monitored for an extended period was used (1, 32, 34). Bone marrow cells were infected with undiluted virus in a 35-mm-diameter dish and monitored for 4 weeks or until the concentration of primary transformants reached 2 × 106 cells per ml, a density correlated with the ability to derive transformed cell lines from such cultures (1, 32, 34). Consistent with their ability to transform NIH 3T3 cells, the 69-71A mutant and the 111-115A mutant were similar to the wild type in their ability to transform pre-B cells (Table 2). However, the 54-56A, 60-62A, and 85-87A mutants that were defective for NIH 3T3 cell transformation displayed defects in transformation of pre-B cells (Table 2). Only a fraction of cultures infected with these mutants transformed, and transformants appeared following an extended 10- to 32-day period. One of 21 cultures infected with the G2A mutant was scored as transformed 26 days after infection. The cells in this culture initially displayed less than 50% viability and established very slowly over a 3-month period compared to other transformed cultures, which generally establish in 1 month or less (19).
TABLE 2.
Mutations of MA sequences important for Mo-MLV replication show defects in pre-B-cell transformation by Ab-MLVa
| Virus | Expt 1
|
Expt 2
|
||
|---|---|---|---|---|
| Frequency | Day(s) | Frequency | Day(s) | |
| P120 | 5/5 | 8, 9 | 7/7 | 8, 9 |
| G2A | 0/4 | _b | 1/7c | 26 |
| 54-56A | 3/5 | 11, 13 | 1/7 | 32 |
| 60-62A | 3/5 | 11, 14, 16 | 1/7 | 13 |
| 69-71A | 5/5 | 9 | 7/7 | 8 |
| 85-87A | 3/5 | 12, 14 | 2/7 | 16, 30 |
| 111-115A | NTd | NT | 7/7e | 8 |
The frequency of transformation was assayed by infecting bone marrow cells with undiluted virus stocks of similar titers and plating them in liquid medium. Cell cultures were scored as transformed when the density of transformed lymphoblastoid cells reached 2 × 106 cells/ml. Frequency values indicate the numbers of transformed cultures/total number of cultures examined. Day(s), day(s) postinfection when the cultures were scored as transformed. Unless otherwise indicated (see footnote e), data shown are representative of the results of at least three independent experiments.
No evidence of transformation was found during the entire observation period of approximately 40 days.
An additional 10 samples were evaluated with the G2A mutant in an experiment independent from those whose results are represented in the table. None of these G2A-infected cultures gave rise to transformants.
NT, not tested.
A total of 17 dishes were evaluated for this virus in two independent experiments. The second experiment, in which 10 dishes were evaluated, was conducted independently from the experiments shown in the table. All 10 of the dishes were transformed in 7 to 9 days.
Revertants are present in most transformants derived following infection with the defective mutants.
A small number of transformants arose in pre-B-cell cultures infected with the alanine mutants that were compromised for transformation. Analyses of other transformation-defective mutants have documented the presence of Ab-MLV that had acquired additional mutations that compensate for the original defect (18, 32). All of the transformants derived from the transformation-defective alanine mutants expressed a v-Abl protein of the expected size (data not shown), and PCR and sequence analysis of the Ab-MLV provirus revealed that five of the seven transformants tested had lost the original mutation. These transformants arose following infection with mutants 54-56A (one transformant), 85-87A (three transformants), and 60-62A (one transformant). Because the gag sequences present in the backbone of the pMIG vector (9) differ from those in Ab-MLV by a single nucleotide, it was possible to infer that reversion to the wild-type sequence occurred by recombination with gag sequences derived from the pMIG vector. However, one of the 60-62A transformants and the single G2A-derived transformant contained Ab-MLV that retained the original mutation. Consistent with the ability of 69-71A and 111-115A to transform cells at wild-type levels, sequence analyses of one independent transformant from each of the cultures infected with these viruses revealed Ab-MLV that retained the original mutations. Taken together, these data indicate that the 54-56A, 85-87A, 60-62A, and G2A mutants are highly compromised with respect to pre-B-cell transformation.
Cells expressing the G2A mutant but not the 54-56A, 60-62A, and 85-87A mutants activate Erk.
Studies examining several gag deletion mutants revealed that mutations affecting MA sequences result in a decreased ability of v-Abl to activate Erk phosphorylation (34). Since Erk is a key component of the mitogen-activated protein kinase pathway important for transformation (reviewed in references 27 and 35), the gag mutants with alanine substitutions in MA sequences were tested for their ability to activate this pathway. 293T cells were transfected with viral plasmids and incubated under serum-starved conditions. Cell lysates were prepared and analyzed by Western blotting using anti-Abl, anti-Erk, and anti-phospho-Erk antibodies (Fig. 2). Consistent with the transformation data, cells expressing the 54-56A, 60-62A, and 85-87A mutants showed decreased levels of Erk phosphorylation, whereas the levels of Erk phosphorylation in cells expressing the 69-71A and 111-115A mutants were comparable to that found following expression of wild-type v-Abl. When densitometric quantitation was used to normalize phospho-Erk levels with v-Abl and total Erk levels, a more than twofold defect in Erk1 phosphorylation in cells expressing the 54-56A, 60-62A, and 85-87A mutants was observed; for Erk2 phosphorylation, about 11-fold, 3-fold, and 6-fold defects were observed with these mutants, respectively. Interestingly, the G2A mutant that lacks the myristoylation site was able to activate Erk to wild-type levels under these conditions, suggesting that a glycine-to-alanine substitution at this position affects v-Abl signaling to pathways other than the mitogen-activated protein kinase pathway. Thus, it is likely that transformation defects associated with the G2A mutant and the 54-56A, 60-62A, or 85-87A mutant result from defects in the activation of different cellular pathways.
FIG. 2.
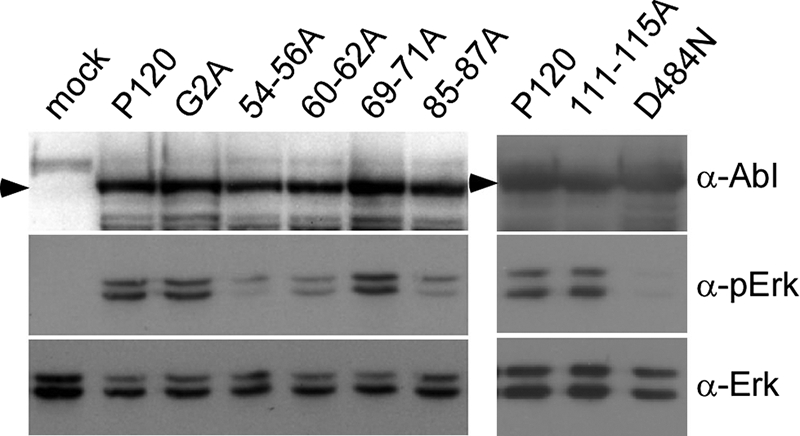
Some Ab-MLV gag mutants with alanine substitutions in MA sequences have defects in Erk activation. 293T cells were transiently transfected with plasmids expressing wild-type virus, the different gag mutants, or the kinase-defective D484N mutant (20). The cells were incubated in serum-free medium, and lysates were prepared and analyzed by Western blotting using the indicated antibodies. Arrowheads indicate the positions of the v-Abl proteins. The data shown are representative of the results of at least three independent experiments.
The G2A, 54-56A, 60-62A, and 85-87A mutants do not support growth and viability of pre-B cells.
Since the transformation-defective alanine mutants showed defects in signaling in 293T cells, the mutants were tested for the ability to signal in a pre-B-cell setting by use of temperature-sensitive 7C411 cells, pre-B cells that have been transformed with the Ab-MLV(P70/H590) temperature-sensitive strain (5). Cells superinfected with the wild-type virus or one of the mutants were derived using viruses expressing GFP. The infected cells were recovered by sorting for GFP-positive cells. All of the 7C411 derivatives grew well at the permissive temperature (34°C) and were readily subcultured. When the cells were shifted to the nonpermissive temperature (39.5°C), cells expressing wild-type virus or the 111-115A mutant grew rapidly and maintained high viability (Fig. 3). In contrast, cells expressing the G2A, 54-56A, 60-62A, and 85-87A mutants did not grow and displayed reduced viability at the nonpermissive temperature. These results indicate that the defective mutants fail to activate signaling pathways required to support growth and viability of pre-B cells.
FIG. 3.
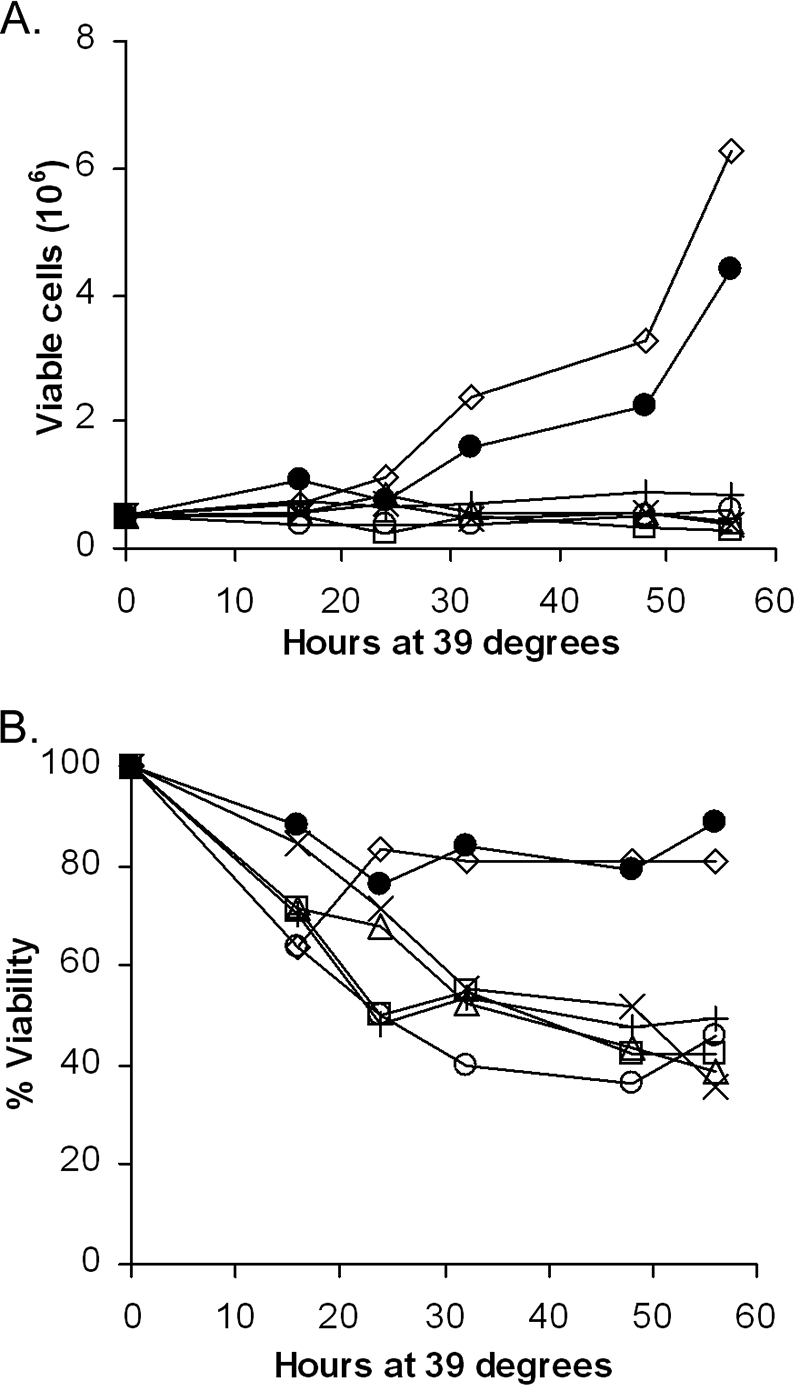
Transformation-defective alanine mutants do not support the growth and survival of 7C411 cells at the nonpermissive temperature. Temperature-sensitive 7C411 cells (5) that had been superinfected with wild-type virus or one of several of the alanine mutants were assayed for growth (A) and viability (B) at the nonpermissive temperature (39.5°C). (A) The growth of the cells was determined by counting the number of live cells by trypan blue exclusion using a hemocytometer after shifting the cells to the nonpermissive temperature. (B) Viable cells were identified by trypan blue staining, and the percent viability was calculated. Symbols represent the following: ×, uninfected; •, wild type; ○, 54-56A; Δ, 60-62A; □, 85-87A; ⋄, 111-115A; +, G2A. The data are representative of the results of at least three independent experiments.
The v-Abl proteins expressed by the transformation mutants show aberrant localization.
Earlier results (34) revealed a role for Gag sequences in localization of the protein. To determine whether the G2A and the alanine substitution mutations affected this parameter, the subcellular localization of the v-Abl proteins was examined in a pre-B-cell setting by use of the superinfected 7C411 cells described above. 7C411 cells expressing two deletion mutants affecting MA sequences, Δ35-81 and Δ82-133, previously shown to be highly compromised for transformation (34), were also studied. These mutants were similar to the alanine substitution mutants studied here with respect to transformation phenotype and Erk activation (34). After being stained with anti-Abl antibodies, the preparations were stained with DAPI and examined using a fluorescent microscope. As expected, the wild-type v-Abl protein was found at the plasma membrane and was also evenly distributed in the cytoplasm (3, 34). In contrast, the 7C411 cells expressing the Δ35-81, Δ82-133, 54-56A, 60-62A, and 85-87A mutants showed a distinct staining pattern, with bright condensed, fluorescent foci in the perinuclear region (Fig. 4A). The common pattern displayed by all of these mutants suggests that MA strongly affects trafficking of the protein through the cells. 7C411 cells expressing the G2A mutant showed a diffuse staining pattern, with expression throughout the cytoplasm and in the nucleus. Interestingly, when v-Abl localization was examined in the 60-62A and G2A lymphoid transformants that retained the original mutation, a pattern distinct from that displayed in 7C411 cells expressing these viruses was observed. The 60-62A transformant showed a staining pattern similar to that of the wild type (Fig. 4B). The v-Abl protein expressed by the G2A transformant was excluded from the nucleus in these cells but showed no signs of plasma membrane localization (Fig. 4B). These results reinforce the idea that the aberrant localization and the transformation defects are mechanistically related.
FIG. 4.

Alterations affecting MA result in aberrant localization of v-Abl proteins in pre-B cells. (A) 7C411 cells were superinfected with wild-type virus or one of several gag mutants, expanded at 34°C, the permissive temperature for 7C411 cells, and stained with anti-Abl antibodies and DAPI. The anti-Abl antibody used recognizes an epitope in the carboxyl terminus of the protein and does not react with the P70/H590 protein present in the 7C411 cells. The images shown are representative of the results of at least three experiments in which at least 250 cells were observed. (B) Localization of v-Abl proteins in pre-B-cell transformants that arose following infection with wild-type virus, the 60-62A mutant, or the G2A mutant was examined by immunofluorescent staining using anti-Abl antibodies. The Ab-MLV provirus in the 60-62A and G2A transformants analyzed here retained the original mutation present in the virus used to infect the cells. The cells were visualized using a fluorescence microscope. The images shown are representative of the results of at least two experiments in which at least 250 cells were observed.
v-Abl proteins compromised for transformation overlap in staining with vimentin.
To understand the nature of the aberrant localization more fully, additional studies to determine the subcellular compartments in which the mutant v-Abl proteins localize were conducted. 7C411 derivatives that express wild-type v-Abl, and the alanine substitution mutants, along with one of the deletion mutants (Δ82-133), were costained with antibodies against the v-Abl protein and antibodies against various cellular markers, including GM130 (Golgi), LAMP3 (multivesicular bodies), γ-tubulin (centrosomes), EEA1 (early endosomes), and calnexin (endoplasmic reticulum). Examination of the preparations showed that wild-type v-Abl was evenly present throughout the cytoplasm and did not specifically localize with any of the markers tested. In contrast, the v-Abl proteins expressed by the transformation-defective alanine substitution mutants or Δ82-133 did not colocalize with any of the cellular markers tested (Fig. 5A and B and data not shown). In contrast, when cells were stained with antibodies directed against vimentin, an intermediate filament protein, colocalization was observed with v-Abl protein encoded by the alanine substitution mutants and with the Δ82-133 v-Abl protein in about 80% of the cells (Fig. 5C), indicating an interaction with elements of the cytoskeleton. The results may reflect accumulation of mutant v-Abl proteins at an intermediate point during their transit to the plasma membrane due to the lack of proper targeting signals.
FIG. 5.

Δ82-133 and 85-87A v-Abl partially overlap with vimentin. Wild-type, 54-56A, 60-62A, Δ82-133, or 85-87A (panel C only) v-Abl was expressed in 7C411 cells. Localization of the v-Abl proteins was examined by costaining with antibodies that recognize a determinant in the carboxyl terminus of v-Abl and antibodies against various subcellular markers as indicated. Anti-mouse secondary antibody conjugated with Alexa 647 (red) was used for anti-Abl antibodies, and anti-rabbit secondary antibody conjugated to Alexa 568 (green) was used for antibodies against subcellular markers. The preparations were visualized using a spinning disc confocal microscope. The images shown are representative of data from at least two experiments in which 50 to 200 cells were observed.
54-56A and 85-87A mutations result in aberrant localization of Gag in the absence of abl-derived sequences.
To determine whether Gag plays a dominant role during trafficking of the entire v-Abl protein, mutants were constructed that express the Gag moiety normally present in the v-Abl protein but lack all abl-derived sequences. To facilitate analyses of these proteins in pre-B cells, a pre-B-cell line was generated by transforming lymphoid cells with the Δ134-215 mutant, a virus that readily transforms cells (34). This cell line made it possible to examine the Gag proteins with an antibody directed against an epitope in p12. This epitope is missing from the v-Abl protein encoded by the Δ134-215 mutant but present in the gag mutants under study. Immunofluorescent staining revealed that WT-Gag, a molecule that lacks all abl-derived sequences, was found at the plasma membrane and showed an even distribution in the cytoplasm, a pattern similar to that displayed by wild-type v-Abl protein (Fig. 6). In contrast, the 54-56A-Gag and the 85-87A-Gag proteins displayed localization patterns similar to those shown by the full-length 54-56A and 85-87A v-Abl proteins in these cells. These results highlight the fact that the Gag moiety retained in v-Abl plays a major role during trafficking of the full-length v-Abl protein.
FIG. 6.

MA sequences play a dominant role in Gag localization in the absence of abl-derived sequences. Full-length v-Abl proteins and Gag proteins containing MA, p12, and the CA residues normally found in v-Abl but lacking abl-derived sequences were expressed in pre-B cells transformed by the Δ134-215 mutant. The Δ134-215 mutant encodes a v-Abl protein lacking p12 sequences (34). Localization of the full-length v-Abl and Gag proteins was monitored using an anti-Gag antibody that recognizes an epitope in the p12 region of the protein but does not react with Δ134-215 v-Abl. The cells were visualized using a fluorescence microscope. The images shown are representative of the results of at least three experiments in which at least 100 cells were observed.
DISCUSSION
Our studies reveal that residues within MA that play an important role in Mo-MLV replication (8, 28) are also important for Ab-MLV-mediated transformation. Alteration of these residues results in aberrant localization of v-Abl proteins and loss of the characteristic plasma membrane localization of the wild-type P120 protein. In the case of the alanine substitution mutants and deletion mutants that lack portions of MA, the v-Abl proteins form distinct foci in pre-B cells. Colocalization of a significant fraction of these proteins with vimentin suggests that the mutant v-Abl proteins are tethered to cytoskeletal structures, a pattern indicative of defects in intracellular trafficking of the molecule. Intracellular transport of various cargos that include Gag occurs through the active rearrangements of cytoskeletal proteins (reviewed in reference 13), and our experiments suggest that this function is conserved in v-Abl and that an intact MA is important for these events. Indeed, MA may influence trafficking of v-Abl and the Mo-MLV-encoded pr65Gag protein in similar ways. This interpretation does not eliminate the possibility that MA plays additional roles in transformation and in pr65Gag function during Mo-MLV replication.
The v-Abl protein encoded by the G2A mutant is also aberrantly localized, but in contrast to the proteins encoded by the other transformation-defective mutants, displays a diffuse localization throughout the cell, indicating that the absence of myristoylation affects localization in a manner that is distinct from that caused by the other mutations studied here. Previous work analyzing localization in BaF3 cells that express v-Abl with a glycine-to-aspartic acid change at this position (3) also documented a diffuse expression pattern, but the molecule was not detected in the nucleus. This difference may reflect the fact that only clones of BaF3 cells that were rendered factor independent were examined in the earlier study. Consistent with this interpretation, the single transformant that arose following G2A infection expressed a v-Abl protein that was excluded from the nucleus but still exhibited a diffuse pattern of expression in the cytoplasm. Our earlier results (34) have shown that an Ab-MLV mutant encoding a nuclear kinase-active v-Abl does not transform cells, and perhaps this aspect of the G2A phenotype may influence transformation. In addition, the inability of v-Abl proteins lacking myristoylation to reach the plasma membrane likely compromises their ability to activate pathways that are critical for transformation. For example, Ras activation, an obligate step in v-Abl-mediated transformation (24), occurs at the plasma membrane.
In contrast to the alanine substitution v-Abl proteins, the v-Abl proteins encoded by the G2A mutant were able to stimulate phosphorylation of Erk to levels similar to that seen with wild-type v-Abl, at least in 293T cells. Erk is typically activated by Ras (reviewed in references 7 and 10) and has been shown to be important for the proliferative response that accompanies Ab-MLV transformation (1). However, the inability of the G2A mutant to transform indicates that activation of Erk is not sufficient to stimulate growth in primary cells or in the 7C411 transformant. Alternatively, work in a variety of systems has revealed that Erk phosphorylation affects cells in various ways, in part reflecting the localization of Erk (reviewed in references 7 and 10). This feature of Erk, coupled with the altered localization of the G2A v-Abl protein, may indicate that the G2A-encoded protein interacts with the Erk pathway in a manner different from that occurring in cells expressing wild-type v-Abl.
The importance of v-Abl localization for transformation is emphasized by analysis of the two pre-B-cell transformants that were isolated following infection with the transformation-defective mutants. These two cell lines contained proviruses that retained the mutation; in both cases, immunofluorescent analyses revealed that localization of the v-Abl protein had changed. The transformant derived from the 60-62A mutant displayed a localization pattern that was indistinguishable from that displayed by the wild-type protein. As discussed above, the v-Abl protein in the G2A-derived cell line retained the diffuse localization characteristic of most cells expressing this molecule, but the molecule was now excluded from the nucleus. In our studies, major changes that affect the size of the protein or its ability to react with an antibody specific for an epitope in the carboxyl terminus were not detected (data not shown). Thus, changes similar to those observed in gag mutant studies by others (17, 18) did not occur in these mutants. These transformants likely arose through selection for a cellular mutation that allows the altered v-Abl protein to function adequately to mediate transformation. Additional analyses of these cell lines to understand the nature of these compensatory changes should reveal important control points in the transformation process.
The crystal structure of Mo-MLV MA (22) suggests ways in which the mutations in MA sequences affect v-Abl function. The Mo-MLV MA protein has an N-terminal extension consisting of the first eight residues, including the myristoyl group, and contains five helices (22). Although the region containing residues 111-115 was not included in the crystal structure, the regions affected by the other mutations were evaluated. The transformation-defective 60-62A and the 85-87A mutants affect sequences within two of the helices, raising the possibility that these changes affect the tertiary structure of the molecule. Residues 54-56 and 69-71 lie within the loops that connect helices, but the phenylalanine residue at position 56 is important for a stacking interaction with the proline at position 8. The 54-56A mutant was compromised with respect to transformation, while the 69-71A mutant was similar to the wild type in its ability to transform cells, further supporting the hypothesis that the tertiary structure of MA is important for v-Abl function. However, all of the altered v-Abl proteins retain kinase activity, suggesting that this portion of the molecule is properly folded. One interpretation of these data is that the structure of the Gag moiety is not strongly influenced by the rest of the protein. This hypothesis is further supported by the observation that the Gag moiety expressed independently of v-Abl sequences localizes in a pattern similar to that seen with the full-length protein.
Earlier analyses (8) revealed that Mo-MLV expressing an MA protein with alanine substitutions in residues 54-56, 60-62, and 85-87, modifications that affect Ab-MLV transformation, was compromised for virus replication. In contrast, alanine substitutions in residues 69-71 and 111-115, changes that did not affect transformation by Ab-MLV, did not impair Mo-MLV replication (8). In Mo-MLV, defective replication correlated well with the ability of MA proteins to interact with the IQGAP1 protein (8), a molecule that could play a role in the intracellular trafficking of Gag because of its role in cytoskeletal regulation (reviewed in reference 2). Although the aberrant subcellular localization of the 54-56A, 60-62A, and 85-87A v-Abl proteins and their counterparts that lack abl-derived sequences is consistent with this hypothesis, interaction between v-Abl protein and IQGAP1 could not be demonstrated (data not shown). The failure to detect such interaction may reflect an inability to detect IQGAP1 interaction with Pr65 Gag (J. Leung and S. Goff, personal communication). Thus, while the role of Gag is conserved in Ab-MLV and Mo-MLV, this function may not involve IQGAP1.
Our earlier experiments indicated that sequences in the Gag portion of v-Abl suppress nuclear localization signals present in the v-Abl-derived portion of the protein (33, 34). A second important way in which Gag residues influence localization was revealed in this work and involves the role of MA residues in orchestrating proper trafficking of the protein. Because the Gag moiety expressed in the absence of abl-derived sequences is localized in a pattern that resembles that of the full-length molecule, Gag plays a dominant role in localization and trafficking of the protein. Both the Gag proteins and the altered v-Abl proteins appear to accumulate at an intermediate point in transport within the cell. Transport of Gag involves cytoskeletal interactions (reviewed in reference 13), and colocalization with vimentin suggests that the altered v-Abl proteins are being retained on cytoskeletal structures. Interactions with cellular proteins that regulate the cytoskeleton, including those known to interact with Mo-MLV MA, such as IQGAP1 (8) and Kif4 (6, 30), may be involved. The mutants developed here should provide valuable tools to assess the functional significance of these interactions.
Many v-onc-containing retroviruses encode Gag-v-Onc fusion proteins that are similar to the v-Abl protein (16). The vast majority of these proteins display constitutive kinase activity; in cases that have been studied, these molecules display localization similar to that seen with v-Abl, a property that is likely important for their ability to transform cells. The critical role of Gag sequences in Ab-MLV transformation suggests that gag sequences may influence transformation in a similar fashion in a wide range of rapidly transforming retroviruses and likely function in part in a manner analogous to their role in the replication-competent viruses that gave rise to the Gag-v-Onc-containing retroviruses.
Acknowledgments
We are grateful to Juliana Leung and Stephen Goff for sharing data on Mo-MLV alanine substitution mutants prior to publication and for useful discussion.
This work was supported by grants CA24220 and CA33771 from the National Cancer Institute.
Footnotes
Published ahead of print on 26 March 2008.
REFERENCES
- 1.Baughn, L. B., and N. Rosenberg. 2005. Disruption of the Shc/Grb2 complex during Abelson virus transformation affects proliferation, but not apoptosis. J. Virol. 792325-2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Briggs, M. W., and D. B. Sacks. 2003. IQGAP1 proteins are integral components of cytoskeletal regulation. EMBO Rep. 4571-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daley, G. Q., R. A. Van Etten, P. K. Jackson, A. Bernards, and D. Baltimore. 1992. Nonmyristoylated Abl proteins transform a factor-dependent cell line. Mol. Cell. Biol. 121864-1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Déjardin, J., G. Bompard-Marechal, M. Audit, T. J. Hope, M. Sitbon, and M. Mougel. 2000. A novel subgenomic murine leukemia virus RNA transcript results from alternative splicing. J. Virol. 743709-3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engelman, A., and N. Rosenberg. 1990. Temperature-sensitive mutants of Abelson murine leukemia virus deficient in protein tyrosine kinase activity. J. Virol. 644242-4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim, W., Y. Tang, Y. Okada, T. A. Torrey, S. K. Chattopadhyay, M. Pfleiderer, F. G. Falkner, F. Dorner, W. Choi, N. Hirokawa, and H. C. Morse III. 1998. Binding of murine leukemia virus Gag polyproteins to KIF4, a microtubule-based motor protein. J. Virol. 726898-6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kolch, W. 2005. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 6827-837. [DOI] [PubMed] [Google Scholar]
- 8.Leung, J., A. Yueh, F. S. Appah, Jr., B. Yuan, K. de los Santos, and S. P. Goff. 2006. Interaction of Moloney murine leukemia virus matrix protein with IQGAP. EMBO J. 252155-2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mainville, C. A., K. Parmar, I. Unnikrishnan, L. Gong, G. D. Raffel, and N. Rosenberg. 2001. Temperature-sensitive transformation by an Abelson virus mutant encoding an altered SH2 domain. J. Virol. 751816-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marshall, C. J. 1995. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80179-185. [DOI] [PubMed] [Google Scholar]
- 11.Mayer, B. J., P. K. Jackson, R. A. Van Etten, and D. Baltimore. 1992. Point mutations in the abl SH2 domain coordinately impair phosphotyrosine binding in vitro and transforming activity in vivo. Mol. Cell. Biol. 12609-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muller, A. J., J. C. Young, A. M. Pendergast, M. Pondel, N. R. Landau, D. R. Littman, and O. N. Witte. 1991. BCR first exon sequences specifically activate the BCR/ABL tyrosine kinase oncogene of Philadelphia chromosome-positive human leukemias. Mol. Cell. Biol. 111785-1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Naghavi, M. H., and S. P. Goff. 2007. Retroviral proteins that interact with the host cell cytoskeleton. Curr. Opin. Immunol. 19402-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parmar, K., R. C. Huebner, and N. Rosenberg. 1991. Carboxyl-terminal determinants of Abelson protein important for lymphoma induction. J. Virol. 656478-6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parmar, K., and N. Rosenberg. 1996. Ras complements the carboxyl terminus of v-Abl protein in lymphoid transformation. J. Virol. 701009-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petropoulos, C. 1997. Retroviral taxonomy, protein structures, sequences, and genetic maps, p. 757-805. In J. M. Coffin, S. E. Hughes, and H. E. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 17.Prywes, R., J. G. Foulkes, N. Rosenberg, and D. Baltimore. 1983. Sequences of the A-MuLV protein needed for fibroblast and lymphoid cell transformation. Cell 34569-579. [DOI] [PubMed] [Google Scholar]
- 18.Prywes, R., J. Hoag, N. Rosenberg, and D. Baltimore. 1985. Protein stabilization explains the gag requirement for transformation of lymphoid cells by Abelson murine leukemia virus. J. Virol. 54123-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Radfar, A., I. Unnikrishnan, H. W. Lee, R. A. DePinho, and N. Rosenberg. 1998. p19(Arf) induces p53-dependent apoptosis during Abelson virus-mediated pre-B cell transformation. Proc. Natl. Acad. Sci. USA 9513194-13199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raffel, G. D., K. Parmar, and N. Rosenberg. 1996. In vivo association of v-Abl with Shc mediated by a non-phosphotyrosine-dependent SH2 interaction. J. Biol. Chem. 2714640-4645. [DOI] [PubMed] [Google Scholar]
- 21.Rees-Jones, R. W., and S. P. Goff. 1988. Insertional mutagenesis of the Abelson murine leukemia virus genome: identification of mutants with altered kinase activity and defective transformation ability. J. Virol. 62978-986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riffel, N., K. Harlos, O. Iourin, Z. Rao, A. Kingsman, D. Stuart, and E. Fry. 2002. Atomic resolution structure of Moloney murine leukemia virus matrix protein and its relationship to other retroviral matrix proteins. Structure 101627-1636. [DOI] [PubMed] [Google Scholar]
- 23.Rosenberg, N., and O. N. Witte. 1988. The viral and cellular forms of the Abelson (abl) oncogene. Adv. Virus Res. 3539-81. [DOI] [PubMed] [Google Scholar]
- 24.Sawyers, C. L., J. McLaughlin, and O. N. Witte. 1995. Genetic requirement for Ras in the transformation of fibroblasts and hematopoietic cells by the Bcr-Abl oncogene. J. Exp. Med. 181307-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scher, C. D., and R. Siegler. 1975. Direct transformation of 3T3 cells by Abelson murine leukaemia virus. Nature 253729-731. [DOI] [PubMed] [Google Scholar]
- 26.Schiff-Maker, L., M. C. Burns, J. B. Konopka, S. Clark, O. N. Witte, and N. Rosenberg. 1986. Monoclonal antibodies specific for v-abl- and c-abl-encoded molecules. J. Virol. 571182-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shore, S. K., R. V. Tantravahi, and E. P. Reddy. 2002. Transforming pathways activated by the v-Abl tyrosine kinase. Oncogene 218568-8576. [DOI] [PubMed] [Google Scholar]
- 28.Soneoka, Y., S. M. Kingsman, and A. J. Kingsman. 1997. Mutagenesis analysis of the murine leukemia virus matrix protein: identification of regions important for membrane localization and intracellular transport. J. Virol. 715549-5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swanstrom, R., and J. Wills. 1997. Synthesis, assembly and processing of viral proteins, p. 263-334. In J. Coffin, S. E. Hughes, and H. E. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory Press, Plainview, NY. [PubMed]
- 30.Tang, Y., U. Winkler, E. O. Freed, T. A. Torrey, W. Kim, H. Li, S. P. Goff, and H. C. Morse III. 1999. Cellular motor protein KIF-4 associates with retroviral Gag. J. Virol. 7310508-10513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Parijs, L., Y. Refaeli, J. D. Lord, B. H. Nelson, A. K. Abbas, and D. Baltimore. 1999. Uncoupling IL-2 signals that regulate T cell proliferation, survival, and Fas-mediated activation-induced cell death. Immunity 11281-288. [DOI] [PubMed] [Google Scholar]
- 32.Warren, D., D. S. Griffin, C. Mainville, and N. Rosenberg. 2003. The extreme carboxyl terminus of v-Abl is required for lymphoid cell transformation by Abelson virus. J. Virol. 774617-4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wen, S. T., P. K. Jackson, and R. A. Van Etten. 1996. The cytostatic function of c-Abl is controlled by multiple nuclear localization signals and requires the p53 and Rb tumor suppressor gene products. EMBO J. 151583-1595. [PMC free article] [PubMed] [Google Scholar]
- 34.Yi, C. R., and N. Rosenberg. 2007. Gag influences transformation by Abelson murine leukemia virus and suppresses nuclear localization of the v-Abl protein. J. Virol. 819461-9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zou, X., and K. Calame. 1999. Signaling pathways activated by oncogenic forms of Abl tyrosine kinase. J. Biol. Chem. 27418141-18144. [DOI] [PubMed] [Google Scholar]