

Abstract
Small molecule tyrosine kinase inhibitors, such as imatinib, are effective therapies for BCR-ABL-mediated human leukemias. However, clinical drug resistance occurs, which warrants development of alternative and/or complementary therapeutic strategies to target critical downstream signaling molecules. We recently demonstrated that disrupting 14-3-3/ligand association by a peptide-based 14-3-3 competitive antagonist R18 induces significant apoptosis, partially through reactivation of AKT-inhibited proapoptotic FOXO3a, in FGFR1 fusion-transformed hematopoietic cells. Here, we report that targeting 14-3-3 by R18 effectively induced significant apoptosis in Ba/F3 and K562 cells expressing BCR-ABL, similarly through liberation and reactivation of FOXO3a. Moreover, R18 sensitized BCR-ABL-transformed cells to inhibition with MEK1 inhibitor U0126, Bcl-2 inhibitor GX15-070, or mTOR inhibitor rapamycin. Treatment with these reagents potentiated R18-induced reactivation of proapoptotic FOXO3a with enhanced expression of downstream transcription targets p27kip1 and Bim1. Furthermore, R18-induced apoptotic cell death in cells expressing diverse imatinib-resistant BCR-ABL mutants, including T315I. This inhibition was enhanced by R18 in combination with U0126 and rapamycin. Thus, our findings suggest that targeting 14-3-3 may potentiate the effects of conventional therapy for BCR-ABL-associated hematopoietic malignancies, and overcome drug resistance.
Keywords: BCR-ABL, chronic myelogenous leukemia (CML), peptide-based 14-3-3 antagonist R18, BCR-ABL imatinib-resistant mutations
Introduction
Small molecule tyrosine kinase inhibitors, such as imatinib (Gleevec), are effective therapies in the clinical treatment of BCR-ABL-positive chronic myelogenous leukemia (CML) and TEL-PDGFβR associated chronic myelomonocytic leukemia.1 However, the emergence of molecular resistance to imatinib poses a significant clinical problem in treatment of BCR-ABL-positive CML patients, which is often caused by point mutations in BCR-ABL gene.2,3 Identification of the spectrum of imatinib-resistant BCR-ABL mutations has led to rapid development of new generation of small molecule ABL inhibitors with distinct mechanism of action against imatinib-resistant cell lines. The clinical activity of these agents such as AMN107 and SKI606 is currently evaluated in ongoing Phase I/II clinical trials, while dasatinib, a Src/ABL dual inhibitor, has received FDA approval for clinical treatment of imatinib-resistant CML patients. However, although these agents in general are very active in treatment of imatinib-resistant CML, they still fail to inhibit some BCR-ABL imatinib-resistant mutants including T315I, which is among the most common BCR-ABL mutations identified in imatinib-resistant CML patients.4,5 Therefore, it is of interest to develop alternative and/or complementary therapeutic strategies to target critical signaling molecules of aberrant signaling pathways activated by leukemogenic tyrosine kinases, which may attenuate their transforming potential and overcome drug resistance.
BCR-ABL has been demonstrated to mediate hematopoietic transformation by providing prosurvival and proliferative signaling through activation of PI3K/AKT and Ras/Raf/MAPK pathways.6-8 We have recently shown that constitutively activated ZNF198-FGFR1 fusion tyrosine kinase, which is associated with human t(8;13)(p11;q12) 8p11 stem cell myeloproliferative disorder,9 activates the AKT and MAPK pathways in hematopoietic cells, and 14-3-3 proteins integrate prosurvival signals through sequestering the proapoptotic FOXO3a and BAD downstream of AKT and MAPK.10 Moreover, disrupting 14-3-3/ligand association by a peptide-based 14-3-3 competitive antagonist R18 induces apoptosis by, in part, disrupting the interaction of 14-3-3/FOXO3a but not 14-3-3/BAD in ZNF198-FGFR1-transformed cells.10
Here, we report that targeting 14-3-3 by R18 similarly induces significant apoptosis in hematopoietic cells expressing BCR-ABL, through liberation and reactivation of FOXO3a. Moreover, R18 sensitizes BCR-ABL-transformed cells to inhibition with anticancer agents targeting prosurvival signaling effectors in parallel to AKT-inhibited FOXO3a, including MEK1 inhibitor U0126, Bcl-2 inhibitor GX15-07011 and mTOR inhibitor rapamycin. Targeting 14-3-3 also induces apoptotic cell death in cells expressing diverse imatinib-resistant BCR-ABL mutants, including T315I, and this apoptosis is further enhanced in combination of U0126 and rapamycin treatment.
Materials and methods
DNA constructs and reagents
Native and mutant BCR-ABL cDNA constructs were subcloned into pMSCV-neo or -puro derived Gateway destination vectors as previously described.12 The plasmids of pREV-TRE-Hyg-YFP-R18 dimer or mutant, pECFP-R18 dimer or mutant were described.10 GX15-070 was provided by Gemin X Biotechnologies Inc.(Montreal, Quebec, Canada).
Retroviral infections, proliferation and apoptosis assays
Doxycycline (Dox)-inducible R18 dimer or mutant expressing TonBaF cell lines were described.10 Cell lines inducibly expressing R18 dimer or mutant were transduced by retroviral supernatant carrying pMSCV-puro vectors encoding BCR-ABL, followed by antibiotic selection. MTT assay and apoptosis assays were described.10
Immunoprecipitation and western blot
The immunoprecipitation and immunoblotting were performed as described.10 Applied antibodies included antibodies against BAD, phospho-BAD (S112), β-actin, p44/42 ERK, phospho-p44/42 ERK (Thr202/Tyr204), c-Abl, AKT, phospho-AKT (Cell Signaling Technology Inc., Danvers, MA, USA); antibodies against p27, GFP, 14-3-3β (Santa Cruz Biotechnology, Santa Cruz, CA, USA); antibodies against Bim1 (Affinity Bio Reagents, Golden, CO, USA) and antibodies against phospho-tyrosine (clone 4G10), FOXO3a and phospho-FOXO3a (Thr32) (Upstate, Lake Placid, NY, USA).
Purification of recombinant TAT-YFP-R18 fusion proteins
In brief, the expressed fusion protein was purified by sonication of high expressing BL21(DE3)pLysS cells obtained from 250 ml of culture with IPTG-induction. Cellular lysates were resolved by centrifugation and loaded onto a Ni-NTA column in 20 mm imidazole. After a step of two times washing, the protein was eluted with 250 mm imidazole. TAT fusion proteins were desalted on a PD-10 column into phosphate-buffered saline (PBS) and the purification efficiency was examined by silver staining and western blotting.
Results
Induced expression of the 14-3-3 antagonist, R18, induces apoptosis in hematopoietic cells transformed by BCR-ABL
We generated hematopoietic TonBaF cell lines inducibly expressing a dimeric version of R18 combining two R18 peptides (R18 dimer; Figure 1a), or a mutant form of R18 (R18 mutant, Figure 1a) that contains one R18 peptide with two substitutions D12K and E14K that abolish binding ability to 14-3-3.10 These cell lines were used to generate TonBaF cell lines that express either R18 dimer or R18 mutant in an inducible manner with stable expression of BCR-ABL (Figure 1b, right). The resulting cell lines were cultured in the presence or absence of Dox, followed by analysis of R18-induced apoptosis, in the absence or presence of interleukin-3 (IL-3). Expression of YFP-R18 dimer induced significant apoptosis in cells stably transformed by BCR-ABL, which was assessed as the fraction of annexin V/YFP double-positive cells in a dose-dependent manner (Figure 1b, left). In contrast, R18 mutant failed to induce significant apoptosis in these cells. Moreover, in the presence of IL-3, R18 dimer induced much less apoptosis in the control parental TonBaF cells (Figure 1b, left), suggesting that the cells stably transformed by BCR-ABL are more sensitive to apoptosis induced by R18 expression.
Figure 1.
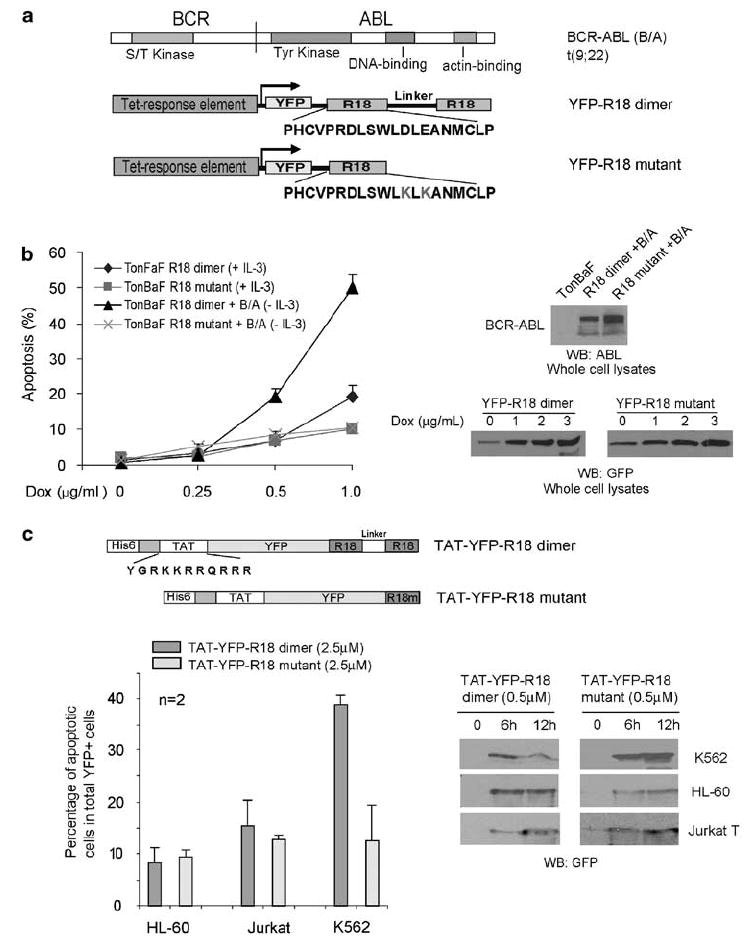
Inducible expression of a 14-3-3 antagonist R18 induces apoptosis in hematopoietic cells transformed by BCR-ABL. (a) Schematic diagram of BCR-ABL, as well as structure and amino-acid sequences of YFP-tagged R18 dimer and mutant. R18 dimer construct contains two R18 motifs separated by a linker of 11 amino acids. (b) TonBaF cells transformed by BCR-ABL are more sensitive to R18 dimer-induced apoptosis compared with control TonBaF-R18 dimer cells. Cells were stained with PE-conjugated anti-annexin V reagent and analyzed by FACS for apoptotic population that is characterized as the fraction of annexin V/YFP double-positive cells in total YFP-positive cells (%). (c) Upper: schematic diagram of structure of TAT-YFP-R18 dimer and -R18 mutant. Lower: TAT-YFP-R18 dimer transduces into and induces significant apoptosis in human leukemia K562 cells, but not in control HL-60 and Jurkat-T cells.
We have also generated bacterial expression plasmids to express fusion protein TAT-YFP-R18 proteins, containing a N-terminal 6-histidine leader followed by the 11-amino acid-TAT protein transduction domain (YGRKKRRQRRR) and the YFP-tagged R18 dimer or mutant (Figure 1c).10 As shown in Figure 1c, purified TAT-YFP-R18 dimer and mutant effectively crossed the cell membrane and transduced into human leukemia cell lines, including K562 that express BCR-ABL, as well as HL-60 and Jurkat-T cells. Moreover, treatment with TAT-YFP-R18 dimer effectively induced apoptosis in BCR-ABL-transformed K562 cells, but not in HL-60 and Jurkat-T cells that are not transformed by any constitutively activated tyrosine kinase. Apoptosis was assessed as the fraction of annexin V+/YFP+ cells, compared with cells treated with TAT-YFP-R18 mutant.
Taken together, these data suggest that the constitutively activated BCR-ABL-transformed human leukemia cells are more sensitive to the R18-induced apoptosis, whereas R18 has minimal nonspecific cytotoxicity in human leukemia cells that are not transformed by leukemogenic fusion/mutant tyrosine kinases. We also observed that induced R18 expression disrupted the interaction between 14-3-3 and FOXO3a, but not 14-3-3/BCR-ABL association as well as BCR-ABL kinase activity in Ba/F3 cells (Supplementary Figure 1).
Induced R18 expression enhances apoptosis induced by MEK1 inhibitor U0126 or a Bcl-2 inhibitor GX15-070 in TonBaF cells expressing BCR-ABL
Our data (Supplementary Figure 1) and previous publication10 suggest a model that targeting 14-3-3 by R18 induces apoptosis in BCR-ABL or FGFR1 fusion transformed cells, partially through liberation and reactivation of FOXO3a, but not BAD, from 14-3-3 binding and inhibition. Thus, we hypothesize that targeting 14-3-3 by R18 may potentiate the inhibition of BCR-ABL-transformed cells by in parallel targeting the MEK1/ERK pathway or blocking the downstream Bcl-2 prosurvival pathway (Figure 2a).
Figure 2.
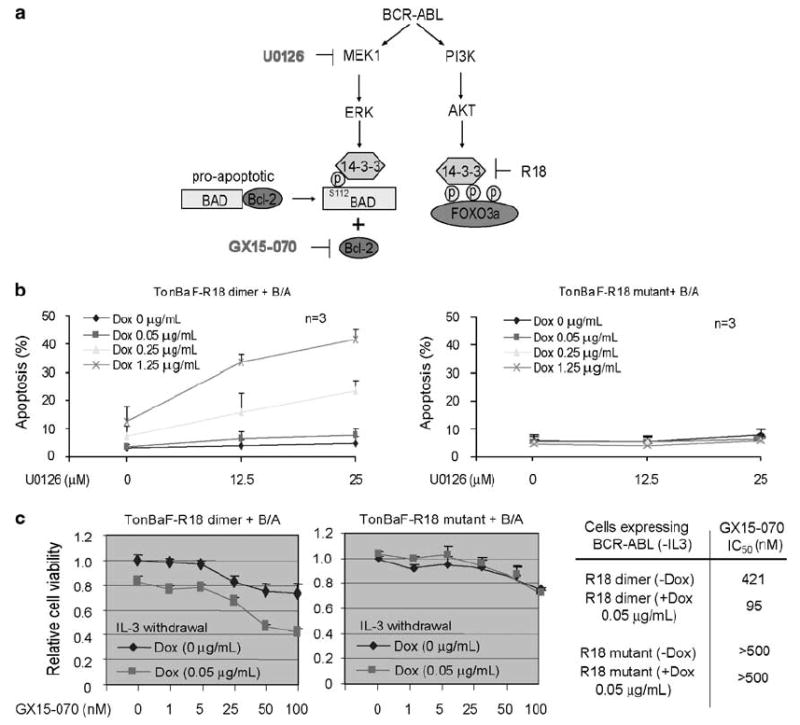
Targeting 14-3-3 by R18 enhances MEK1 inhibitor U0126 or Bcl-2 inhibitor GX15-070-induced apoptosis in BCR-ABL-transformed TonBaF cells. (a) Proposed model of targeting 14-3-3 by R18 to induce apoptosis through reactivation FOXO3a and sensitize BCR-ABL-dependent transformation signaling to inhibition with U0126 or GX15-070. (b) Cells were treated with indicated concentrations of doxycycline in the absence or presence of increasing concentrations of U0126, followed by annexin V staining and FACS analysis for apoptotic population. (c) The cells were treated with increasing concentrations of GX15-070 in the absence or presence of Dox. The relative cell viability determined by MTT assay was normalized to cells without treatment with both doxycycline and GX15-070. Right: cellular IC50 was determined for each cell line treated with GX15-070 in the presence or absence of Dox.
TonBaF cells stably expressing BCR-ABL and inducibly expressing R18 dimer or mutant (Supplementary Figure 2a) were cultured in the absence of IL-3 and presence of increasing doses of U0126, with or without Dox treatment at increasing concentrations. We observed that although treatment with the MEK1 inhibitor U0126 at 12.5 and 25 μm was sufficient to abolish ERK phosphorylation and activation (Supplementary Figure 2b), U0126 as a single agent was insufficient to induce significant apoptosis in BCR-ABL-transformed cells (Figure 2b). In contrast, induced R18 expression enhanced the proapoptotic activity of MEK1 inhibitor U0126 in TonBaF cells transformed by BCR-ABL, compared with R18 mutant. The enhanced apoptosis was assessed as the increased fraction of annexin V/YFP double-positive cells in a dose-dependent manner (Figure 2b), and enhanced cleavage of poly(ADP-ribose) polymerase (PARP) (Supplementary Figure 2c), compared to cells treated with R18 and U0126 alone.
GX15-070 (Gemin X Biotechnologies Inc.) is a small molecule pan-Bcl-2 inhibitor that targets the Bcl-2 family of proteins, which is currently being studied in clinical trials for the treatment of patients with chronic lymphocytic leukemia and solid tumors. We observed that GX15-070 treatment inhibited cell growth of TonBaF cells expressing BCR-ABL in a dose-dependent manner as assessed in an MTT assay, and induced expression of YFP-R18 dimer, but not R18 mutant, significantly enhanced GX15-070 inhibitory effects (Figure 2c, left and middle). The cellular IC50 of GX15-070 was decreased more than 4-fold in cells with R18 induction compared with cells not expressing R18 (Figure 2c, right).
R18 enhances the mTOR inhibitor rapamycin-induced apoptotic cell death in BCR-ABL-transformed TonBaF cells
We also hypothesize that simultaneously targeting two separate signaling effectors mTOR and FOXO3a downstream of AKT in parallel may result in enhanced abrogation of the prosurvival PI3K/AKT pathway, and achieve synergistic inhibition of leukemia cells (Figure 3a). We treated the cells with rapamycin at 1 and 4 nm, which were sufficient to inhibit the phosphorylation and activation of mTOR downstream kinase p70 S6 kinase (Supplementary Figure 2d), but insufficient to induce significant apoptosis in BCR-ABL-transformed cells as a single agent (Figure 3b). However, R18 enhanced the proapoptotic activity of rapamycin in TonBaF cells transformed by BCR-ABL, with enhanced annexin V staining (Figure 3b) and PARP cleavage (Supplementary Figure 2e), compared with treatment with rapamycin alone.
Figure 3.
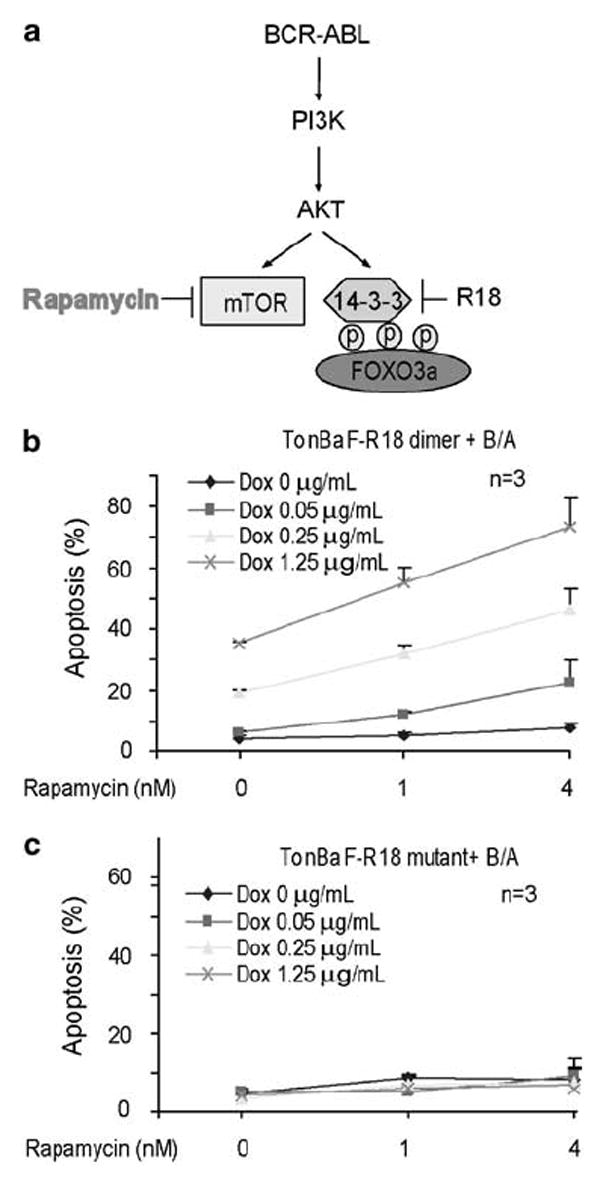
Targeting 14-3-3 by R18 potentiates mTOR inhibitor rapamycin-induced apoptotic cell death TonBaF cells expressing BCR-ABL. (a) Proposed model of simultaneous inhibition of targeting 14-3-3 by R18 and mTOR by rapamycin to induce synergistic apoptosis in BCR-ABL-transformed cells. (b) Cells were treated with indicated concentrations of Dox in the absence or presence of increasing concentrations of rapamycin, followed by annexin V staining and FACS analysis for apoptosis.
U0126, rapamycin or GX15-070 potentiate R18-induced reactivation of FOXO3a with enhanced expression of FOXO3a transcription targets Bim1 and p27kip1
We next found that combined treatment with R18 and U0126 (Figure 4a), GX15-070 (Figure 4b) or rapamycin (Figure 4c) results in enhanced upregulation of FOXO3a transcriptional targets, such as the cyclin-dependent kinase inhibitor p27kip1 and the proapoptotic Bcl-2 family member Bim1, including Bim1 L (long isoform) and Bim1 S (short isoform). p27kip1 and Bim1 have been implicated in regulation of cell cycle and apoptosis. These data suggest the potential molecular mechanism by which combined treatment with R18 and U0126, rapamycin or GX15-070 results in enhanced inhibition of BCR-ABL-expressing cells.
Figure 4.
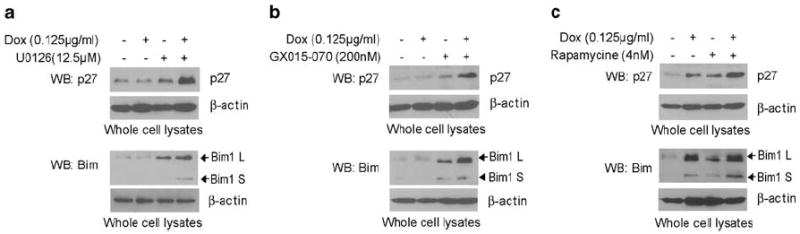
U0126, GX15-070 or rapamycin potentiate R18-induced reactivation of FOXO3a with enhanced expression of FOXO3a transcription targets Bim1 and p27kip1. TonBaF-R18 cells expressing BCR-ABL were treated with (a) U0126, (b) GX15-070 or (c) rapamycin in the presence and absence of Dox at indicated concentrations.
Targeting 14-3-3 by R18 alone, and in combination with U0126 or rapamycin induce apoptosis in cells transformed by diverse imatinib-resistant BCR-ABL mutants
We next evaluated the effects of targeting 14-3-3 on overcoming drug resistance. We generated distinct TonBaF-R18 dimer cell lines stably expressing individual clinically identified, imatinib-resistant BCR-ABL mutants, including E255V, M351T, G250E, F317L, T315I, A380T and F486S. Among these mutants, T315I is a potent imatinib-resistant BCR-ABL mutant, which is also resistant to the second generation ABL tyrosine kinase inhibitors including AMN107.4 We observed that induction of R18-induced apoptosis in a dose-dependent manner in cells expressing BCR-ABL T315I mutant, and the R18 proapoptotic activity was significantly enhanced in combination of either U0126 or rapamycin (Figures 5a and b, left, respectively). Moreover, U0126 or rapamycin enhanced R18-induced apoptosis in cells expressing diverse BCR-ABL imatinib-resistant mutants (Figures 5a and b, right, respectively).
Figure 5.
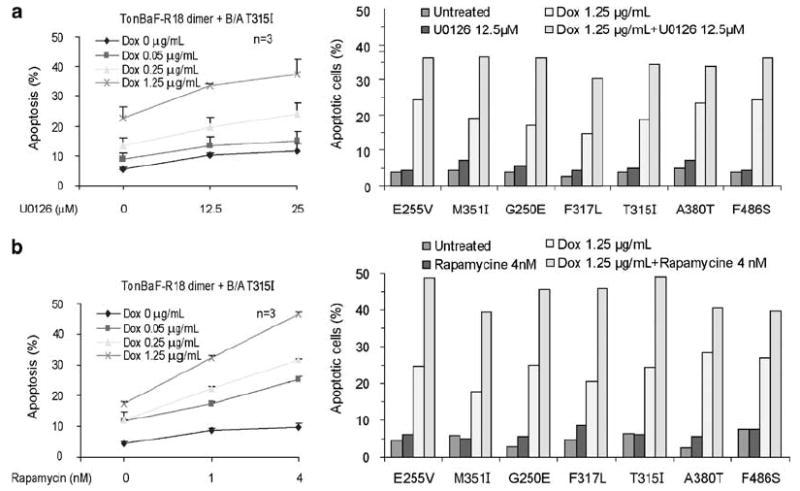
Combined treatment with R18 expression and U0126 (a) or rapamycin (b) induces enhanced apoptosis in TonBaF cell lines expressing distinct imatinib-resistant BCR-ABL mutants.
Discussion
Emerging data have shown that clinical emergence of imatinib-resistant mutations in BCR-ABL confers drug resistance and results in a relapse of the disease. Although new molecules were rapidly developed to tackle the imatinib-resistant mutants of BCR-ABL, few agents have activity against the T315I mutant. Here, we show that targeting 14-3-3 by a peptide-based, competitive pan-14-3-3 inhibitor R18 alone, and in combination with other agents such as U0126 or rapamycin induce apoptosis in cells transformed by native BCR-ABL or diverse imatinib-resistant mutants, including T315I. Our findings suggest that targeting 14-3-3 may represent a helpful strategy to potentiate the effects of conventional therapy for BCR-ABL-associated hematopoietic malignancies, and overcome drug resistance.
14-3-3 interacts with both BCR and c-ABL, as well as BCR-ABL.13,14 The exact role of 14-3-3-binding in regulation of BCR-ABL is still unknown. 14-3-3 was implied to link specific signaling protein components to BCR-ABL to mediate transformation signals or regulate BCR-ABL activation via protein–protein interaction.15 Interestingly, R18 as a competitive antagonist of 14-3-3 is insufficient to dissociate BCR-ABL from 14-3-3 binding (Supplementary Figure 1a). Since the phosphorylation levels of BCR-ABL were not altered by expression of R18 (Supplementary Figure 1a), this difference might be due to the relatively high binding affinity of 14-3-3 for BCR-ABL such that R18 is able to compete with FOXO3a for 14-3-3 (Supplementary Figures 1b and c), but insufficient to disrupt 14-3-3/BCR-ABL association. These data suggest that R18 induces apoptosis in BCR-ABL-transformed cells probably in part through liberation and reactivation of FOXO3a that is downstream of AKT, but not by disrupting 14-3-3/BCR-ABL association or attenuating BCR-ABL tyrosine kinase activity.
Moreover, targeting 14-3-3 by R18 alone or in combination with U0126 or rapamycin effectively induced apoptosis in cells expressing a spectrum of imatinib-resistant BCR-ABL mutants, including T315I (Figure 5), suggesting that targeting 14-3-3 may present an alternative solution to the clinical problem of drug-resistance in BCR-ABL-associated hematopoietic malignancies.
Supplementary Material
Supplementary Information accompanies the paper on the Leukemia website (http://www.nature.com/leu)
Acknowledgments
This work was supported in part by NIH grant CA120272 (J Chen), the Leukemia and Lymphoma Society (J Chen) and the Golfer Against Cancer Foundation (J Chen and S Lonial). J Chen is a Georgia Cancer Coalition Distinguished Cancer Scholar.
References
- 1.Gilliland DG, Jordan CT, Felix CA. The molecular basis of leukemia. Hematology (Am Soc Hematol Educ Program) 2004:80–97. doi: 10.1182/asheducation-2004.1.80. [DOI] [PubMed] [Google Scholar]
- 2.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 3.Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell. 2003;112:831–843. doi: 10.1016/s0092-8674(03)00190-9. [DOI] [PubMed] [Google Scholar]
- 4.Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129–141. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 5.Kimura S, Ashihara E, Maekawa T. New tyrosine kinase inhibitors in the treatment of chronic myeloid leukemia. Curr Pharm Biotechnol. 2006;7:371–379. doi: 10.2174/138920106778521532. [DOI] [PubMed] [Google Scholar]
- 6.Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18:189–218. doi: 10.1038/sj.leu.2403241. [DOI] [PubMed] [Google Scholar]
- 7.Kawauchi K, Ogasawara T, Yasuyama M, Ohkawa S. Involvement of Akt kinase in the action of STI571 on chronic myelogenous leukemia cells. Blood Cells Mol Dis. 2003;31:11–17. doi: 10.1016/s1079-9796(03)00070-6. [DOI] [PubMed] [Google Scholar]
- 8.Sattler M, Mohi MG, Pride YB, Quinnan LR, Malouf NA, Podar K, et al. Critical role for Gab2 in transformation by BCR/ABL. Cancer Cell. 2002;1:479–492. doi: 10.1016/s1535-6108(02)00074-0. [DOI] [PubMed] [Google Scholar]
- 9.Xiao S, Nalabolu SR, Aster JC, Ma J, Abruzzo L, Jaffe ES, et al. FGFR1 is fused with a novel zinc-finger gene, ZNF198, in the t(8;13) leukaemia/lymphoma syndrome. Nat Genet. 1998;18:84–87. doi: 10.1038/ng0198-84. [DOI] [PubMed] [Google Scholar]
- 10.Dong S, Kang K, Gu T, Kardar S, Fu H, Lonial S, et al. 14-3-3 integrates pro-survival signals mediated by the AKT and MAPK pathways in ZNF198-FGFR1 transformed hematopoietic cells. Blood. 2007;110:360–369. doi: 10.1182/blood-2006-12-065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perez-Galan P, Roue G, Villamor N, Campo E, Colomer D. The BH3-mimetic GX15-070 synergizes with bortezomib in mantle cell lymphoma by enhancing Noxa-mediated activation of Bak. Blood. 2007;109:4441–4449. doi: 10.1182/blood-2006-07-034173. [DOI] [PubMed] [Google Scholar]
- 12.Chen J, Williams IR, Lee BH, Duclos N, Huntly BJ, Donoghue DJ, et al. Constitutively activated FGFR3 mutants signal through PLCgamma-dependent and -independent pathways for hematopoietic transformation. Blood. 2005;106:328–337. doi: 10.1182/blood-2004-09-3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu H, Subramanian RR, Masters SC. 14-3-3 proteins: structure, function, and regulation. Annu Rev Pharmacol Toxicol. 2000;40:617–647. doi: 10.1146/annurev.pharmtox.40.1.617. [DOI] [PubMed] [Google Scholar]
- 14.Porter GW, Khuri FR, Fu H. Dynamic 14-3-3/client protein interactions integrate survival and apoptotic pathways. Semin Cancer Biol. 2006;16:193–202. doi: 10.1016/j.semcancer.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Reuther GW, Fu H, Cripe LD, Collier RJ, Pendergast AM. Association of the protein kinases c-Bcr and Bcr-Abl with proteins of the 14-3-3 family. Science. 1994;266:129–133. doi: 10.1126/science.7939633. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information accompanies the paper on the Leukemia website (http://www.nature.com/leu)