

Summary
GCET2 (Germinal centre B-cell expressed transcript 2; also named HGAL) is a newly cloned gene that has been shown to be a useful marker for germinal centre (GC) B cells and GC B-cell derived malignancies, including follicular lymphomas and germinal centre B cell-like diffuse large B-cell lymphomas (GCB-DLBCLs), and is a useful prognosticator for DLBCLs. We report here the biochemical and biological properties of GCET2, which may help to determine its role in the GC reaction. GCET2 is constitutively localised in the plasma membrane but is excluded from lipid rafts. GCET2 does not have a transmembrane domain, and its membrane localisation is mediated by myristoylation and palmitoylation. GCET2 has five conserved putative tyrosine phosphorylation sites, and it can be phosphorylated following pervanadate treatment in B cells. By serially mutating the five tyrosines, the third and fourth tyrosines were found to be essential for GCET2 phosphorylation. GCET2 was phosphorylated when co-transfected into COS7 cells with protein tyrosine kinases (PTKs) LYN, LCK or SYK, and therefore it could be a substrate of these kinases in B cells. The third tyrosine site (107YENV) of GCET2 is a consensus GRB2 binding site, and GCET2 was found to associate with GRB2 through the third tyrosine following phosphorylation. Our data suggests that GCET2 may be an adaptor protein in GC B cells that transduces signals from GC B-cell membrane to the cytosol via its association with GRB2.
Keywords: germinal centre, adaptor protein, B-cell signalling
Diffuse large B-cell lymphoma (DLBCL) can be divided into two major subtypes based on their gene expression profiles (Alizadeh et al, 2000; Husson et al, 2002; Rosenwald & Staudt, 2003). One is the germinal centre B cell-like DLBCL (GCB-DLBCL) with high expression of genes in the normal GC B-cell signature, and the other is the activated B cell-like DLBCL (ABC-DLBCL) with high expression of genes that are normally induced during activation of peripheral blood B cells. These two subtypes are clinically different, with the GCB-DLBCL having better prognosis than the ABC-DLBCL (Alizadeh et al, 2000; Husson et al, 2002; Ramsay, 2002; Rosenwald et al, 2002; Rosenwald & Staudt, 2003).
A group of genes are selectively expressed in the normal GC B cells and GCB-DLBCLs, including many known markers of GC differentiation, such as BCL6, CD10, CD38, transcriptional factor A-MYB and OGG1, as well as a number of uncharacterised genes represented as expressed sequence tags (ESTs) (Kelsoe, 1996a,b; Alizadeh et al, 2000; Lossos et al, 2003; Pan et al, 2003). The molecular mechanisms underlying the GC reaction are not well-defined. Cloning and characterising the new genes that are restricted to GC B cells is one useful approach to investigate the GC reaction.
GCET2 (Germinal centre B-cell expressed transcript 2) is a recently cloned gene that is highly expressed in normal GC B cells and GC B-cell derived lymphomas, including follicular lymphoma (FL) and GCB-DLBCL (Lossos et al, 2003; Pan et al, 2003; Natkunam et al, 2005). GCET1, GCET2 and BCL6 are among the most representative genes in the GC B-cell signature (Rosenwald et al, 2002; Lossos et al, 2003; Pan et al, 2003) for the classification of DLBCLs. GCET2 is a homologue of the mouse gene M17 (Christoph et al, 1994), a mouse GC B-cell expressed transcript. In the mouse, we detected two M17 isoforms encoding two proteins of 181 and 159 amino acids respectively. GCET2 and M17 have similar exon patterns and the shorter isoform of M17 lacks the second exon due to alternative splicing. Human GCET2 has only one translated protein of 178 amino acids, even though Northern blotting showed two major transcripts of about 1·5 kb and 3·3 kb, which results from the use of different polyadenylation sites (Lossos et al, 2003; Pan et al, 2003).
Analysis of the GCET2 protein (Fig 1) revealed five conserved putative tyrosine phosphorylation sites but no functional domains that may have enzymatic activity or may mediate protein–protein interactions. GCET2 also has a putative myristoylation site (¹MGNS) and a putative palmitoylation site (43CFC), which may mediate its membrane attachment. These features suggest that GCET2 may be a novel membrane-associated adaptor protein that transduces signals from the cell surface to the cytosol. Adaptors are essential for B-cell proliferation and differentiation (Sosa, 1996; Pawson & Scott, 1997; Peterson et al, 1998; Wienands, 2000; Kurosaki, 2002; Janssen & Zhang, 2003; Janssen et al, 2003; Togni et al, 2004). They mediate intermolecular interactions within a signal transduction pathway but lack intrinsic enzymatic or transcriptional activity. We hypothesise that, following B-cell activation, GCET2 is phosphorylated by protein tyrosine kinases (PTKs), and it then recruits cytosolic proteins to the cell membrane using its phosphorylated tyrosines as docking sites. Eventually, a signalling complex is formed on the inner plasma membrane to activate downstream pathways.
Fig 1.

Alignment of germinal centre B-cell expressed transcript 2 (GCET2) with M17. M17 is a mouse germinal centre B-cell expressed gene and is 57% identical to GCET2. M17-L is an M17 isoform that was cloned by our laboratory from the mouse spleen and the mouse B-cell line, A20. The second exon is absent from the previously described shorter form of M17. The bolded and underlined sequences of GCET2 are the myristoylation site (¹MGNS) and the palmitoylation site (43CFC) respectively. The bolded sequences of GCET2 are the predicated tyrosine phosphorylation sites and all five are conserved in GCET2 and M17.
Materials and methods
Cells, antibodies and vectors
The human FL cell line, DHL16 (Epstein et al, 1978), and human Burkitt lymphoma cell line, Daudi(Klein et al, 1968), were cultured in RPMI 1640 medium with 10% fetal bovine serum (FBS) (Invitrogen Corporation, Carlsbad, CA, USA), penicillin (100 units/ml) and streptomycin (100 µg/ml) (Invitrogen). The mouse B cell line, A20 (Kim et al, 1979), a BALB/c B-cell lymphoma line, was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and was cultured in RPM1640 medium supplemented with 0·05 mmol/l 2-mercaptoethanol and 10% FBS and penicillin/streptomycin. The green monkey kidney cell line, COS7 (Gluzman, 1981), was cultured in DMEM supplied with 10% FBS and penicillin/streptomycin.
Mouse monoclonal antibodies (MAb) to human GRB2, B-cell linker protein (BLNK) and LYN were ordered from Santa Cruz Biotechnology (Santa Cruz, CA, USA), mouse anti-phospho-tyrosine MAb, 4G10 from Upstate Company (Waltham, MA, USA) and mouse anti-V5 from Invitrogen.
The vectors used were kindly provided to us by the following investigators: pMIG vector (Dr Thomas Mitchell, University of Louisville School of Medicine); SYK in pEF-M, ZAP70 in pcDNA3 and LCK in pEF (Dr Marianne Mollenauer, University of California); FLAG-tagged BTK in pcDNA3 (Dr Andrew C. Chan, Genentech Company); LYN in pcDNA3 (Dr Tom Smithgall, University of Pittsburgh) and pMIN (Dr Joyce Solheim, University of Nebraska Medical Center).
Plasmid construction
Cloning of GCET2 into pcDNA3.1 vector
The full-length open-reading-frame sequence (ORF) of GCET2 was amplified using the following primer pair:
Forward primer: 5′-CACCATGGGAAATTCTCTGCTGAGAG-3′
Reverse primer: 5′-TAAATGGGAAAACTGAGTCACAG-3′
The amplified product was electrophoresed in a 1% agarose gel, and the specific band was purified using the QIAquick gel extraction kit (Qiagen, Valencia, CA, USA). The purified PCR product was cloned into the pcDNA3.1 vector using pcDNA Directional TOPO Expression Kit (Invitrogen) according to the manufacturer’s directions. In this vector, GCET2 has a C-terminal V5 tag and a 6-histidine (His) tag. The insert was verified by sequencing.
Subcloning of GCET2 into pMIG vector
The DNA fragment with full-length GCET2 ORF and V5 and 6His tags in pcDNA3.1 vector was recovered using restriction endonucleases BamHI and PmeI, and the pMIG vector was linearised with BglII and PmeI. The purified GCET2 fragment was ligated into the linearised pMIG vector to form a new pMIG-GCET2 vector. The pMIG vector is an MSCV-based retroviral vector containing EGFP as an expression marker downstream of the internal ribosomal entry site (IRES).
Mutagenesis of GCET2
Five independent mutations of the tyrosine residues of GCET2 (M1 to M5) were separately introduced to the pcDNA3. 1-GCET2 vector using the QuikChange site-directed mutagenesis kit (Stratagene, Cedar Creek, TX, USA), according to the manufacturer’s instructions. For convenience of selection, a novel restriction enzyme site was introduced for each construct. The following oligonucleotides and their complementary sequences were used (base alterations are underlined):
M1, ATCCAGGACAATGTCGACCAGACCTTCTCAGAGGAGCTG
M2, CTCAGAGGAGCTCTGCTTTACCCTCATCAATCATC
M3, GAACTCTGCTGAAGAGTTCTTTGAGAATGTTCCCTGC
M4, GGAGGAACTGAGACTGAATTCTCACTTCTACATATG
M5, ATCCCCAGAAGATGAATTCGAACTTCTCATGCCTC
Based on the five single mutations, multiple combinations of tyrosine mutants were generated using both the QuikChange site-directed mutagenesis kit and appropriate restriction enzyme digestions. Furthermore, an individual mutation in the putative myristoylation site was constructed by replacing the glycine (G) codon at the second amino acid position of GCET2 by alanine (A), and we named this construct pcDNA3.1-GCET2-G2A. The primer used was (mutated base underlined):
GCET2 (G2A) Forward: 5′-CACCATGGCAAATTCTCTGCTGAGAG-3′
Two separate mutations in the putative palmitoylation sites of GCET2 were introduced into plasmids pcDNA3.1-GCET2 and pcDNA3.1-GCET2-G2A respectively. The primers used were (mutated bases underlined):
M6-forward, CATATCGCTGAAGGCGCCTTCGCCCTTCCATGG
M6-reverse, CCATGGAAGGGCGAAGGCGCCTTCAGCGATATG.
All mutations were confirmed by restriction enzyme digestion and sequencing.
Transfection of Cos7 cells
COS7 cells were seeded onto 6-well plates (Becton Dickinson Labware, Franklin Lakes, NJ, USA) at a density of 0·5 × 106 cells/well and cultured for 24 h. For the preparation of transfection medium, 5 µg plasmid were added into 500 µl OPTI MEM I (Invitrogen) in one tube, and 10 µl Lipofection 2000 (Invitrogen) were added into 500 µl OPTI MEM I in another tube. Both tubes were kept at room temperature for 5 min, and the contents mixed together followed by 20 min incubation at room temperature. The culture medium was removed from the 6-well plates and 1 ml of the transfection solution was applied to each well. After 4 h, transfection medium was removed and fresh culture medium was added, and 24 h later, the cells were harvested for experiments (Fessart et al, 2005).
Transduction of B-cell lines
The packaging cell line, Phoenix A, was seeded in a 10 cm cell culture dish at a density of 2·5 × 106 cells/dish. After 24 h, 10 µg pMIG-GCET2 vector and 20 µl Lipofection 2000 were used for transfection as described above. 48 h later, the supernatant containing the viruses was harvested and briefly centrifuged at 1800 g for 5 min to eliminate possible contamination of cells and cellular debris. 5 × 106 DHL16 cells or Daudi cells were mixed with 5 ml supernatant containing 10 µmol/l polybrene (Specialty Media, Phillipsburg, NJ, USA). Thereafter, 6-well plates were centrifuged at 400 g at room temperature for 2 h. Cells were cultured and sorted using EGFP as a selection marker (Koonpaew et al, 2004).
Separation of cellular membrane and cytosol
5 × 107 DHL16 cells were collected and washed once with phosphate-buffered saline (PBS), followed by another wash in TEAS buffer (10 mmol/l triethanolamine pH 7.4, 1 mmol/l EDTA and 250 mmol/l sucrose) (Cavenagh et al, 1996). Cells were then resuspended in four volumes of TEAS buffer and disrupted on ice with 10 passes in a Dounce homogenizer (Bellco Glass Inc., Vineland, NJ, USA). The resulting homogenate was centrifuged at 1000 × g for 10 min to obtain a post-nuclear supernatant. This supernatant was further centrifuged at 200 000 g for 1 h (SW 41 rotor, Beckman Instruments, Fullerton, CA, USA) to separate the cytosol (supernatant) and the plasma membranes (pellet). The membrane pellet was resuspended in the same volume of TEAS buffer as the supernatant. Equal volumes of both fractions were analysed by 12% sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and blotted with appropriate antibodies.
Isolation of lipid rafts
Lipid rafts were isolated using modified lysis conditions and centrifugation on discontinuous sucrose gradient (Zhang et al, 1998). In brief, 1 × 108 DHL16 cells were washed with ice-cold PBS and lysed for 30 min on ice in TNEV buffer (10 mmol/l Tris/HCl, pH 7·5, 150 mmol/l NaCl and 5 mmol/l EDTA) with 1% Triton X-100 and protease and phosphatase inhibitors. The cells in lysis buffer were further homogenised with ten strokes in a Wheaton Dounce homogenizer (Wheaton, Millville, NJ, USA). Cell nuclei and cellular debris were pelleted by centrifugation at 900 g for 10 min. One millilitre of cleared supernatant was mixed with 1 ml of 80% sucrose in TNEV and transferred to the bottom of a Beckman centrifuge tube. The diluted lysate was overlaid with 6 ml 30% sucrose in TNEV buffer and, finally, 3·5 ml 5% sucrose in TNEV buffer. The samples were centrifuged in an SW41 rotor at 200 000 g for 16–20 h at 4°C; 1-ml fractions were collected starting from the top of the gradient. Ten microlitres of each fraction were separated by 12% SDS-PAGE, and the appropriate antibodies were then used to detect the proteins in these fractions.
Metabolic labelling of DHL16 cells
DHL16 cells transduced with pMIG-GCET2 construct were used for labelling with [³H] myristate or [³H] palmitate (PerkinElmer Life Sciences, Inc., Boston, MA, USA). 4 × 107 cells were washed twice in cold FBS-free RPMI 1640 medium, and resuspended in the same medium at a final concentration of 1 × 107 cells/ml. After starvation for 2 h, cells were resuspended in RPMI 1640 medium with 5% dialysed FBS (Invitrogen), and myristic acid [9, 10-³H (N)] or palmitic acid, [9, 10-³H (N)] was added to the culture medium at a final specific radioactivity of 14·8 MBq/ml each. After incubation at 37°C for 4 h, cells were washed twice in cold PBS and lysed in 1 ml Triton X-100 lysis buffer with protease inhibitors. A total of 500 µl cell lysate was used for immunoprecipitation with 4 µg anti-V5. The immunoprecipitates were dissolved in loading buffer with or without 2-mercaptoethanol. Proteins were separated using 4–20% gradient SDS-PAGE gels, and the gels were fixed in SDS-PAGE Fixing Buffer [10% (v/v) glacial acetic acid, 30% (v/v) methanol] for 1 h. Gels were then treated with EN³HANCE™ Autoradiography Enhancer (PerkinElmer, Inc.) for 1 h under gentle agitation in a fume hood, and then immersed in cold water containing 1% glycerol and 10% polyethylene glycol (PEG, MW 8000) with gentle agitation for 30 min in order to precipitate the scintillators. Gels were dried using BioDesignWrap Gel Drying System (BioDesign Inc., Carmel, NY, USA) overnight in the fume hood, and the dried gels were exposed to Kodak BioMax XAR films (Eastman Kodak Company, Rochester, NY, USA) at −80°C for 4 weeks.
Confocal localisation of GCET2 in COS7 cells and DHL16 cells
1 × 105 COS7 cells were plated in a 6-well plate with autoclaved round coverslips (Thermo Fisher Scientific Inc., Waltham, Miami, FL, USA) and incubated overnight for cells to adhere. Cells in each well were transfected using 3 µg pcDNA-GCET2 vector and allowed to grow for 40 h. After three washes with cold PBS, cells were fixed and permeablised with 2% paraformaldehyde and 0·1% Triton X-100 for 10 min at room temperature. After treatment with blocking solution with 1% bovine serum albumin (BSA) for 10 min, cells were incubated in mouse anti-V5 primary antibody (5 µg/ml) for 1 h at room temperature. Cells were then incubated in fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG secondary antibody (10 µg/ml; Vector Laboratories, Burlingame, CA, USA) in the dark for 1 h. Cell nuclei were counter-stained using propidium iodide (1 µg/ml) for 1 min, and coverslips were mounted with mounting media containing Fluoromount and sealed with nail polish. The staining was analysed using a Zeiss LSM 410 confocal laser scanning microscope (CLSM; Geottinger, Germany).
pMIG-GCET2 transduced DHL16 cells were used for fluorescence staining and localisation of GCET2, and DHL16 cells without transduction were used as a negative control. DHL16 cells were harvested and washed twice in cold PBS and resuspended at a density of 1 × 105 cells/ml; 0·5 ml of the DHL16 cell suspension was centrifuged onto each slide at 400 g for 5 min using a Shandon Cytospin® 4 Cytocentrifuge (Thermo Electron Corporation, San Jose, CA, USA). After drying at room temperature for 30 min, cells were fixed with 2% paraformaldehyde for 10 min, and then permeablised and blocked in Permeablisation and Blocking solution for 5 min (0·1% cold fish skin gelatin, 0·1% Triton X-100, 0·05% Tween 20, BSA 1·0% and PBS pH 7.4). Slides were incubated in anti-V5 primary antibody (5 µg/ml) for 1 h at room temperature with gentle agitation, and in Texas Red conjugated horse anti-mouse IgG secondary antibody (7·5 µg/ml; Vector Laboratories) for 1 h at room temperature in the dark with gentle agitation. Slides were mounted with mounting media containing Fluoromount and sealed with nail polish. The staining was analysed using the Zeiss LSM 410 CLSM.
Treatment of B cells with pervanadate
5 × 107 DHL16 or Daudi B cells were harvested and washed twice in cold PBS, and resuspended in 1 ml PBS. Pervanadate was freshly prepared in each experiment as follows: 900 µl water was added to 100 µl sodium vanadate stock solution (100 mmol/l); 20 µl H2O2 was added to the sodium vanadate solution, followed by incubation at room temperature for 10 min to form pervanadate. B cells were treated with pervanadate at a final concentration of 200 µmol/l for 5 min at 37°C (Secrist et al, 1993; Radha et al, 2004).
Stimulation of B cells by BCR cross-linking
5 × 107 DHL16 or Daudi cells were harvested and washed twice in cold PBS, and the cells were stimulated with goat antihuman IgG or IgM F(ab)2 fragment (The Jackson Laboratory, Bar Harbor, ME, USA) at a concentration of 24 µg/ml at 37°C for 5 min. Cells were pelleted by centrifugation at 400 g at 4°C for 3 min, and lysed in an equal volume of cold Triton X-100 lysis buffer containing protease and phosphatase inhibitors (Sigma-Aldrich, St Louis, MO, USA).
Immunoprecipitation and immunoblotting
Cells were washed with ice-cold PBS and lysed in cell lysis buffer (0·5% Triton X-100, 150 mmol/l NaCl, 20 mmol/l Tris pH7.5 and 5 mmol/l EDTA) with protease and phosphatase inhibitors on ice for 20 min. Lysates were clarified by centrifugation and precleared with protein G-Sepharose. Lysates were subjected to immunoprecipitation with protein G-Sepharose-coupled antibodies at 4°C. The beads were washed twice in PBS and boiled in SDS sample buffer. The proteins were separated using 12% SDS- PAGE, and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA). Membranes were immersed in PBS with 5% milk for 1 h to block non-specific background. Membranes were incubated with the appropriate antibodies at room temperature for 1 h, followed by three washes with PBS. Membranes were incubated with horseradish peroxidase-labelled secondary antibodies (Amersham Biosciences Corp, Piscataway, NJ, USA) for 1 h, and then washed three times with PBS. An enhanced chemiluminescence (ECL) Advance Western Blotting Detection Kit (Amersham) was used to visualise immunoreactive proteins.
Results
GCET2 is constitutively localised on the cell membrane
The GCET2 protein does not contain a transmembrane sequence (Fig 1), but analysis of the amino acid sequences of both human GCET2 and murine M17 revealed two conserved cysteine residues, 43CFC in GCET2 and 46CSC in M17 (longer isoform). These cysteines are possible sites for palmitoylation of membrane-associated proteins. Analysis with the Myristoylator Program (http://us.expasy.org/tools/myristoylator) showed that GCET2 has a possible myristoylation sequence (¹MGNS). Therefore, GCET2 may attach to the cell membrane through the putative palmitoylation sites and/or the myristoylation site.
In order to determine whether GCET2 is a cell membrane-associated protein, we separated the cell membrane and the cytosol of pMIG-GCET2 transduced DHL16 cells, and found that both the endogenously expressed and the exogenously expressed GCET2 are constitutively localised in the cell membrane rather than the cytosol (Fig 2A). We also isolated the rafts using sucrose gradient centrifugation, and found that GCET2 was excluded from the rafts both before and after pervanadate treatment (Fig 2B).
Fig 2.
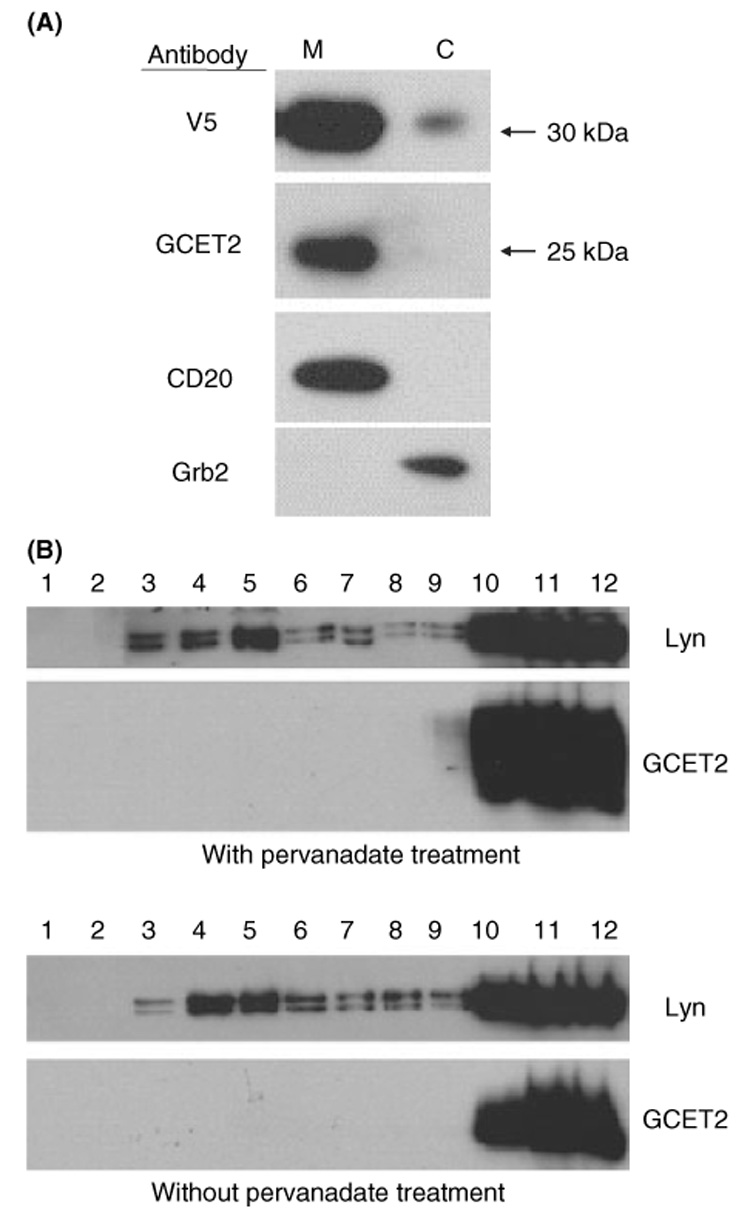
Germinal centre B-cell expressed transcript 2 (GCET2) is localised in the cell membrane but is excluded from the lipid rafts. (A) GCET2 is constitutively localised in the plasma membrane. The cell membrane (M) and the cytosol (C) were separated from DHL16 cells transduced with pMIG-GCET2. The upper panel was blotted using anti-V5 antibody to detect the exogenously expressed GCET2, and the second panel was blotted using anti-GCET2 antibody to detect the endogenous GCET2. CD20 and GRB2 were used as markers for the plasma membrane and cytosolic fractions, respectively. (B) GCET2 is excluded from the lipid rafts of cell membrane. GCET2 is not detected in the fractions 4–6 of the sucrose gradient, which represent the lipid rafts, before or after pervanadate treatment. LYN, known to be localised in the lipid rafts, was used as the positive control.
The CLSM was used to visualise the intracellular expression of GCET2. COS7 cells were transfected with GCET2, which was tagged with V5 in the c-terminus, and mouse anti-V5 MAb was used as primary antibody followed by staining using FITC-conjugated goat anti-mouse IgG secondary antibody. Two control samples were also included: in one sample, COS7 cells were not transfected with GCET2, and were stained using primary and secondary antibodies; in another sample, GCET2-transfected COS7 cells were incubated in PBS instead of primary antibody. Both of these two controls were negative for green fluorescence while in transfected cells, GCET2 had a clear plasma membrane association with some internal distribution (Fig 3A–C).
Fig 3.
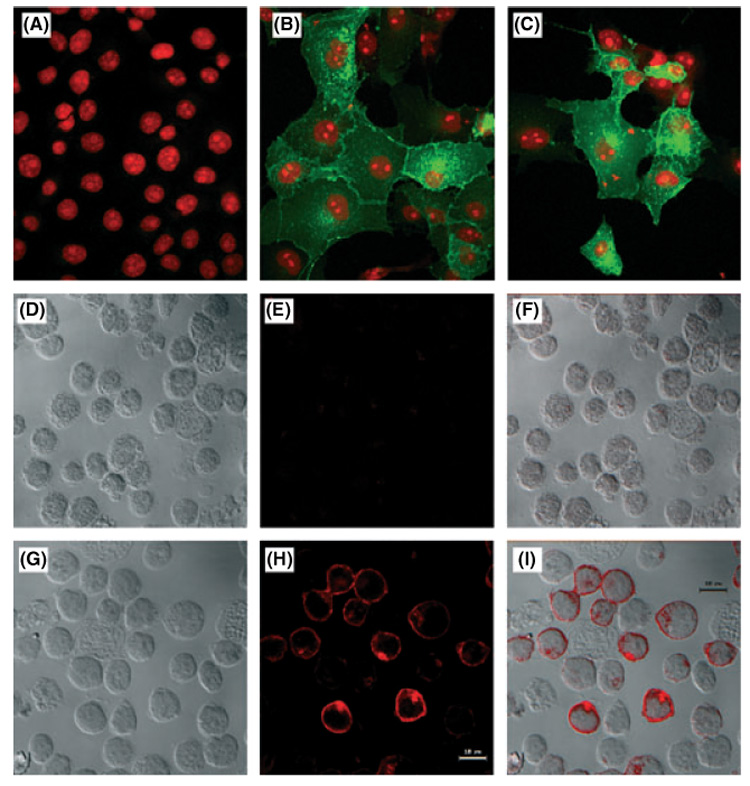
Localisation of germinal centre B-cell expressed transcript 2 (GCET2) in COS7 and B cells by confocal microscopy. (A) Localisation of GCET2 in COS7 cells. This figure shows the negative control. (B) Localisation of GCET2 in COS7 cells by confocal analysis. Wild-type GCET2 was mainly localised in the cell membrane with some distribution in the cytosol, probably in the Golgi apparatus and the membrane of cytosolic organelles. (C) Localisation of wild-type GCET2 in COS7 cells. (D) Localisation of GCET2 in DHL16 cells. DHL16 cells without pMIG-GCET2 transduction were used as negative control. Cells were stained using mouse anti-V5 monoclonal primary antibody and Texas red conjugated horse anti-mouse IgG secondary antibody. This picture is the Differential Interface Contrast (DIC) of control DHL16 B cells. (E) Fluorescence of control DHL16 cells. The cells were totally negative for red signals. (F) Combination (overlay) of G and H. (G) DIC of pMIG-GCET2 transduced B cell. (H) Red fluorescence of pMIG-GCET2 transduced B cells. Red signals were predominantly in the cell membrane and the Golgi area. (I) Combination (overlay) of J and K.
pMIG-GCET2-transduced DHL16 cells were also used to localise the expression of GCET2, and Texas Red, instead of FITC, was conjugated anti-mouse IgG secondary antibody because DHL16 cells express GFP from the pMIG-GCET2 vector. In DHL16 cells, the red fluorescence signal was predominantly at the cell membrane with some Golgi localisation, and these results were consistent to those obtained from COS7 cells (Fig 3D–I).
GCET2 attaches to the plasma membrane by myristoylation and palmitoylation
Structural analysis predicated one potential myristoylation site and one potential palmitoylation site on GCET2. We performed metabolic labelling experiments with myristic acid [9, 10-³H (N)] and palmitic acid [9, 10-³H (N)] on pMIG-GCET2-transduced DHL16 cells. The labelled cells were then lysed and immunoprecipitated using anti-V5. We found that GCET2 was both myristoylated and palmitoylated (Fig 4A and B).
Fig 4.
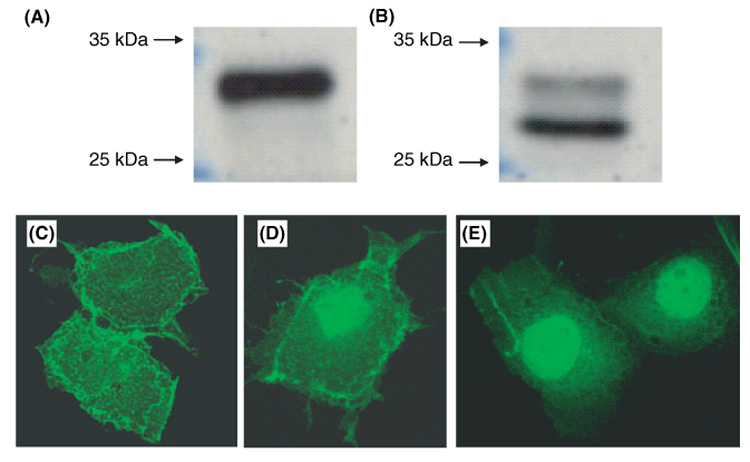
Germinal Centre B-cell Expressed Transcript 2 (GCET2) membrane localisation is mediated through myristoylation and palmitoylation. DHL16 cells were labelled with myristic acid [9, 10-³H (N)] or palmitic acid [9, 10-³H (N)]. GCET2 was both myristoylated (A) and palmitoylated (B). Myristoylation is a irreversible process that modifies one amino acid (Glycine-2) of GCET2, while palmitoylation is a reversible process that modifies two amino acids of GCET2 (Cysteine-46 and -48), and GCET2 in the lower molecular weight band (B) may represent the form that is not fully palmitoylated. (C) COS7 cells transfected with wild-type pcDNA3.1-GCET2 with V5 tag. COS7 cells were stained using FITC-labelled anti-V5 antibody. GCET2 protein is predominantly localised on the cell membrane; D. COS7 cells were transfected with GCET2 mutated in the myristoylation site. GCET2 had increased distribution in the nuclei; E. COS7 cells were transfected with GCET2 mutated in the myristoylation and the palmitoylation sites. GCET2 showed nuclear but not membranous distribution.
The COS7 cells were also transfected with mutated GCET2 in the myristoylation site or both of the myristoylation site and the palmitoylation site, and the localisations of GCET2 in the transfected COS7 were determined under fluorescence microscopy. With mutation of the myristoylation site, GCET2 had increased distribution into the nuclei (Fig 4C and D), and GCET2 showed nucleus but not membrane distribution after mutating both myristoylation site and palmitoylation site (Fig 4E). Taken together, all our data indicated that the membrane association of GCET2 was due to these two modifications.
GCET2 can be tyrosine-phosphorylated in B cells
The GCET2 protein sequence shows five putative tyrosine phosphorylation sites, of which all are conserved in the mouse M17 protein (Fig 1). To determine whether GCET2 can be phosphorylated in B cells, we cloned V5-tagged GCET2 into a pcDNA3 vector and a pMIG vector. Daudi cells and DHL16 cells were transiently transfected using pcDNA3-GCET2 or permanently transduced using pMIG-GCET2. Pervanadate, an irreversible protein-tyrosine phosphatase inhibitor, was used to induce tyrosine phosphorylation. In these two B-cell lines, GCET2 was phosphorylated after pervanadate treatment. The endogenous GCET2 migrated to the 25 kD position in SDS-PAGE gel, whereas the V5-tagged GCET2 migrated to the 35 kD position. The phosphorylated protein formed a higher molecular weight smear extending up to 40 kD (Fig 5A and B). Our data indicated that GCET2 can be phosphorylated in B cells after pervanadate treatment.
Fig 5.
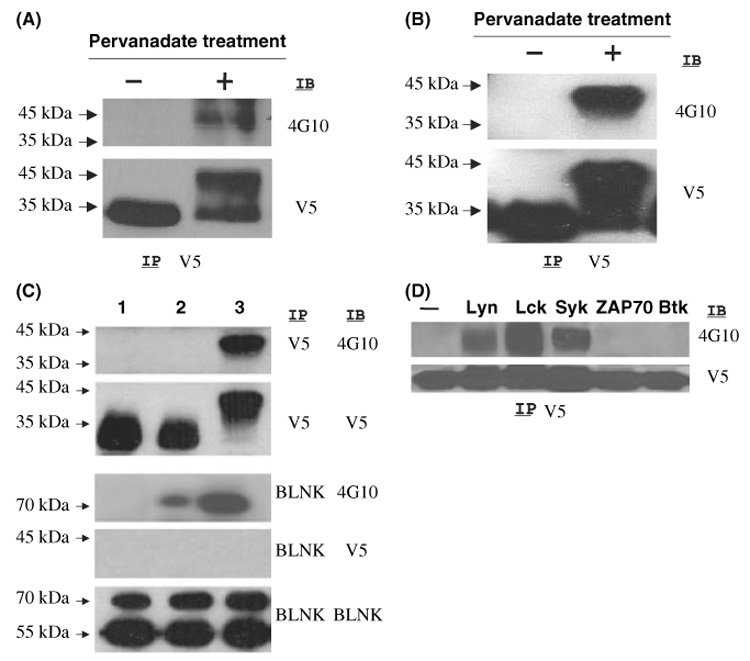
Germinal centre B-cell expressed transcript 2 (GCET2) is phosphorylated after PV Treatment of DHL16 Cells and Daudi Cells. (A) V5 tagged GCET2 was transiently transfected into DHL16 cells. Upon pervanadate treatment, phosphorylated GCET2 migrated to a band of higher molecular weight that reacted with 4G10. (B) GCET2 was phosphorylated in Daudi cells after pervanadate treatment. (C) BCR cross-linking cannot induce GCET2 phosphorylation in DHL16 cells. Both GCET2 and BLNK are phosphorylated upon pervanadate treatment, but BCR cross-linking can only induce BLNK phosphorylation. GCET2 cannot be co-precipitated with BLNK upon pervanadate treatment. Lane 1, DHL16 without treatment; 2, BCR cross-linking; 3, pervanadate treatment. (D) GCET2 is phosphorylated when co-transfected with LYN, LCK or SYK in COS7 cells. Anti-V5 antibody was used for immunoprecipitation (IP) followed by immunoblotting using the anti-phospho-tyrosine antibody, 4G10.
Adaptors are crucial in BCR signalling pathways, and we were interested in examining whether BCR cross-linking could induce GCET2 phosphorylation. BLNK, a central adaptor in BCR signalling, undergoes phosphorylation upon BCR cross-linking, and was used as a positive control for this experiment. We could not demonstrate GCET2 phosphorylation upon BCR cross-linking (Fig 5C). We also tried to induce GCET2 phosphorylation using other stimuli, including CD40 ligation, CD19 ligation, PMA and LPS, but none of them could induce GCET2 phosphorylation (data not shown).
GCET2 can be phosphorylated when co-transfected with LYN, LCK or SYK
Three major PTK families, SRC, SYK and TEC, are important for the activation of B cells through phosphorylation of their substrates. We transfected pcDNA3-GCET2 into the COS7 cells together with vectors expressing LCK, LYN, SYK, ZAP-70 or BTK to determine which PTKs could phosphorylate GCET2. LCK and LYN belong to the SRC family, and LCK is mainly expressed in T/NK cells, whereas LYN is highly expressed in B cells but not T cells. SYK and ZAP-70 belong to SYK family and mainly function in B cells and T cells respectively. BTK belongs to the TEC PTK family, and it is important for B-cell signalling. GCET2 is phosphorylated when co-transfected with LCK, LYN or SYK in COS7 cells, but not ZAP-70 or BTK (Fig 5D). These data indicate that GCET2 may be a substrate of LYN, LCK and SYK.
The third and fourth tyrosines are important for GCET2 phosphorylation
The GCET2 contains five putative tyrosine phosphorylation sites, which are highly conserved in mouse M17 protein, and GCET2 was phosphorylated in B cells after pervanadate treatment. It was not clear which of these five tyrosines could be phosphorylated. We constructed GCET2 mutants (Fig 6A) in its tyrosine sites, and examined the phosphorylation of these constructs after co-transfection into COS7 cells with LYN and SYK. Mutation in the first (M1) or the second tyrosine (M2) or both (M1+2) showed no significant decrease in phosphorylation levels (Fig 6B and C). GCET2 with a single mutation in the third or the fourth tyrosine (M3 or M4) showed significantly decreased phosphorylation compared with other single mutations and the wild type GCET2. Combined mutants in the first three tyrosines (M1+2+3), the first and the third (M1+3), the second and the third (M2+3), and the fourth and the fifth (M4+5) also revealed remarkable decrease in GCET2 phosphorylation (Fig 6B and C). We also constructed a truncated GCET2 without the fifth tyrosine (T5) and another truncated GCET2 without the fourth and the fifth tyrosines (T45) by removing the sequence after the 112th amino acid or the sequence after the 106th amino acid. After co-transfecting into COS7 cells with PTKs, T5 had no decrease of tyrosine phosphorylation compared with the wild-type GCET2, but only very weak tyrosine phosphorylation could be detected in T45 (Fig 6D). The mutant on the myristoylation site (G2A) also showed no decrease of tyrosine phosphorylation (Fig 6D). If both myristoylation site and palmitoylation site were mutated, no phosphorylation of GCET2 could be detected (data not shown). The data indicate that these two lipid modifications are important for GCET2 membrane localisation and phosphorylation.
Fig 6.

Constructs of germinal centre B-cell expressed transcript 2 (GCET2) mutants in the five conserved tyrosine sites. The tyrosines were mutated to phenylalanine. M1 to M5 represent the constructs with single mutations in the first to fifth tyrosine respectively, and M1+2 to M4+5 represent the constructs with mutation in the tyrosines indicated. W1 to W5 represent constructs with mutations of all tyrosines except the one indicated by ‘Y’. (B) Phosphorylation of GCET2 single mutants. Only the mutant in the third or fourth tyrosine has significantly decreased phosphorylation. (C) Phosphorylation of GCET2 combined mutants. All combined mutants that contain either the third or the fourth tyrosine have significantly decreased phosphorylation. (D) Phosphorylation of GCET2 truncated mutants and mutants on the lipid modification site. The truncated mutant that does not contain the fifth tyrosine (T5) has no decrease in tyrosine phosphorylation compared with the wild-type. Only very weak tyrosine phosphorylation can be detected in T45, the mutant that does not contain the fourth and the fifth tyrosines. The mutant on the myristoylation site (G2A) has no decrease of tyrosine phosphorylation. The phosphorylation of single GCET2 mutants is similar to the previous experiments, showing that only M3 and M4 have significant decreasing in phosphorylation levels. (E) GCET2 with one single tyrosine cannot be phosphorylated. No phosphorylation of W3, W4 or W5 was detected. Wild-type GCET2 co-transfected with LYN and SYK shows significantly increased phosphorylation after pervanadate treatment. IP, immunoprecipitation, IB, immunoblotting.
We also constructed five GCET2 mutants by mutating four of the five tyrosines and leaving only one tyrosine site intact, and we named these constructs from W1 to W5 (Fig 6A). No phosphorylation was detected on these constructs when cotransfected with PTKs in repeated experiments (data not shown). After transfection of these GCET2 mutants with LYN and SYK into COS7 cells, we also treated COS7 cells with pervanadate to maximise tyrosine phosphorylation of GCET2, but no phosphorylation was detected (Fig 6E).
Taken together, these data indicate that the third and the fourth tyrosines of GCET2 are important for its phosphorylation, and they require the presence of each other to be efficiently phosphorylated.
GCET2 associates with GRB2
The third tyrosine of GCET2 is within a sequence, YENV, which matches the consensus GRB2 binding site (YxN). In order to determine whether GRB2 binds to phosphorylated GCET2, pMIG-GCET2 transduced DHL16 cells were treated with pervanadate for 5 min, and the cell lysates were subjected to immunoprecipitation using anti-GRB2 or anti-V5 antibodies, followed by Western blot using anti-GRB2, anti-V5 and anti-phospho-tyrosine antibodies. GCET2 was phosphorylated following pervanadate treatment (Fig 7A), and a strong signal for GCET2 was detected in the immunoprecipitates using GRB2 antibody (Fig 7A), suggesting that they bound to each other after pervanadate treatment. Another experiment showed that phosphorylated GCET2 did not bind to BLNK following pervanadate treatment (Fig 5B).
Fig 7.
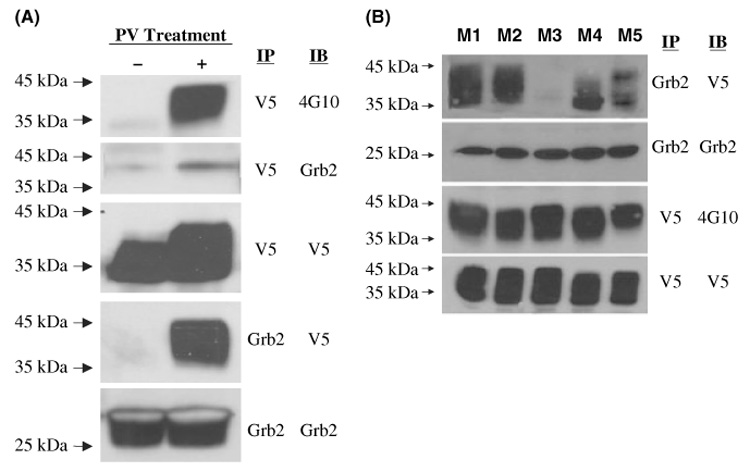
(A) Germinal centre B-cell expressed transcript 2 (GCET2) associates with GRB2 following pervanadate treatment. GCET2 is phosphorylated (first panel) and a weak signal of GRB2 is detected in the immunoprecipitates with V5 antibody (second panel). A strong signal of GCET2 is detected in the immunoprecipitates with GRB2 antibody (fourth panel). (B) The third tyrosine of GCET2 is important for its association with GRB2. Single mutants of GCET2 (M1 to M5) were transfected into COS7 cells together with LYN and SYK, and then anti-GRB2 antibody was used to precipitate GCET2. M3 has no association with GRB2. M1 and M2 had strong association to GRB2. M4 and M5 had slight decrease of association. IP, immunoprecipitation, IB, immunoblotting.
In order to determine which of the tyrosines on GCET2 bind to GRB2, we transfected the single mutants of GCET2 (M1 to M5) into COS7 cells together with PTKs, and then anti-GRB2 antibody was used to precipitate GCET2. In prior experiments, we found that all five single mutants can be phosphorylated when co-transfected with PTKs, but M3 and M4 (mutation of the third and the fourth tyrosine respectively) had significantly decreased phosphorylation levels compared with other mutants. This experiment showed that M3 had no association with GRB2 (Fig 7B). M1 and M2 had strong association with GRB2; M4 had some decrease of association that may be due to the decrease of phosphorylation of this mutant; and M5 also showed slightly decreased association although its tyrosine phosphorylation level showed no significant changes. Therefore, the data clearly showed that the third tyrosine of GCET2 is a GRB2 binding site.
Discussion
Germinal centres are specialised regions in secondary lymphoid organs where antigen-primed B cells proliferate, undergo isotype switching and affinity maturation and subsequently give rise to plasma cells and affinity-matured, long-lived memory B cells. The molecular mechanisms underlying the GC reaction are not well-defined. The GC reaction is associated with the expression of a number of unique genes, including known genes and uncharacterised genes, which are represented as ESTs. These genes are highly expressed in both normal GC B cells and GC B-cell derived lymphomas, including FL and GCB-DLBCL, but not in resting or in vitro activated peripheral blood B cells (Alizadeh et al, 2000; Husson et al, 2002; Ramsay, 2002; Rosenwald et al, 2002; Rosenwald & Staudt, 2003; Dave et al, 2004). One of the approaches to understand the GC reaction is to characterise this set of GC B-cell associated genes.
GCET2 (also named HGAL) was cloned from an EST in the GCB signature. The present study has demonstrated that GCET2 attaches to plasma membrane through myristoylation and palmitoylation. The third and the fourth tyrosines of GCET2 are essential for its phosphorylation, and GCET2 binds to GRB2 using its third phosphotyrosine. All these indicate that GCET2 may be a membrane-associated adapter protein that recruits cytosolic proteins to the cellular membrane to form a signal transduction complex.
Adapters are defined as proteins that lack both enzymatic and transcriptional activities but mediate protein–protein or protein–lipid interactions via various interaction domains. These domains include Src homology 2 (SH2), Src homology 3 (SH3), phosphotyrosine-binding (PTB), and pleckstrin-homology (PH) domains, as well as tyrosine-based signalling motifs (TBSMs). TBSMs comprise short peptide (4–6 amino acids) sequences containing a core tyrosine residue, which interacts with SH2 domains upon phosphorylation. The specificity of this interaction is determined by the amino acids flanking the tyrosine residue. The SH2 domain of GRB2 prefers phosphotyrosine peptides with the consensus sequence pTyr-Ile/Val-Asn-X, in which the determining residues are hydrophobic amino acids in the +1 position and asparagine in the +2 position (Songyang et al, 1993; Songyang et al, 1994). Adapters are crucial in lymphocyte differentiation, proliferation and activation (Sosa, 1996; Pawson & Scott, 1997; Peterson et al, 1998; Rudd, 1998; Myung et al, 2000; Leo & Schraven, 2001; Kurosaki, 2002; Leo et al, 2002; Janssen & Zhang, 2003; Togni et al, 2004; Allam & Marshall, 2005) and many of them are important in antigen receptor-mediated signalling pathways. BLNK and LAB (Linker of Activated B cells) (Brdicka et al, 2002; Janssen et al, 2003) are central adaptors (Fu et al, 1998; Goitsuka et al, 1998; Ishiai et al, 1999; Minegishi et al, 1999; Pappu et al, 1999; Wollscheid et al, 1999; Kurosaki & Tsukada, 2000; Tan et al, 2001; Chiu et al, 2002; Taguchi et al, 2004) in the BCR signalling pathway. GCET2 is structurally similar to LAB and its T-cell functional homologue LAT in that they all have multiple tyrosine-based signalling motifs but lack any other defined domains or motifs. LAB and LAT have transmembrane domains and palmitoylation sites. GCET2 does not have a transmembrane domain, but it is constitutively localised in the cell membrane through myristoylation (¹MGN) and palmitoylation (43CFC). Lipid modifications, including myristylation, palmitoylation and farnesylation are important for the membrane localisation of signalling proteins, such as G proteins, RAS and SRC family PTKs. Myristylation and palmitoylation are also crucial for the membrane localisation and function of many members of the SRC family tyrosine kinases (Resh, 1994).
Pervanadate treatment of the human B cell lines, Daudi and DHL16, induced the phosphorylation of GCET2. Pervanadate is an extremely potent phosphotyrosine phosphatase (PTPs) inhibitor that works by irreversibly oxidising the catalytic cysteine of PTPs (Huyer et al, 1997) (Tsiani et al, 1998). The results indicate that GCET2 can be tyrosine-phosphorylated in B cells. To determine whether GCET2 can be phosphorylated by some of the major PTKs in lymphocyte signalling pathways, it was co-transfected with LCK, LYN, SYK, ZAP-70 or BTK into COS7 cells. GCET2 was phosphorylated in the presence of LCK, LYN or SYK, but not ZAP-70 or BTK. Three major PTK families, SRC, SYK and Tec, are important for the activation of B cells through the phosphorylation of their substrates. Upon BCR cross-linking, LYN undergoes auto-phosphorylation and the activated LYN phosphorylates the immunoreceptor tyrosine-based activation motif (ITAM) of Igα/β subunits of the BCR complex and sequentially phosphorylates SYK which is recruited to the phosphorylated tyrosines of the Igα/β subunits. Activated SYK then phosphorylates BTK, BLNK and LAB and BLNK serve as the central adaptor in BCR signalling (Fu et al, 1998). GCET2 is probably a substrate of LYN, SYK or a PTK downstream of these kinases. As BCR signalling is an important pathway that activates LYN and SYK and GCET2 is probably a substrate of these two PTKs, we tried to determine whether GCET2 functions in the BCR signalling pathway. We cross-linked the surface BCR of two human B-cell lines, Daudi and DHL16, but could not detect any phosphorylation of GCET2. Cross-linking of mouse B-cell surface BCR also failed to induce M17 phosphorylation (Data not shown). In addition, GCET2 was excluded from the rafts even after pervanadate treatment. These data indicate that GCET2 may not function in the BCR signalling pathway.
The GCET2 has five conserved possible tyrosine phosphorylation sites. In order to determine which of these tyrosines are phosphorylated, we constructed GCET2 mutants by mutating one or several of these five tyrosines, and we found that mutations in the first two tyrosines and the fifth tyrosine had no decrease in GCET2 phosphorylation. Single mutations in the third and the fourth tyrosines resulted in significantly decreased phosphorylation, and any combined mutations with either the third or the fourth tyrosine had a marked decrease in phosphorylation, indicating that the third and the fourth tyrosines are important for GCET2 phosphorylation. We could not detect any phosphorylation of mutants with only one intact tyrosine phosphorylation site (the W serial mutants) even after pervanadate treatment. The truncated GCET2 without the fifth tyrosine (T5) had no effect on GCET2 phosphorylation, but the truncated GCET2 without the last two tyrosines (T45) had no detectable phosphorylation when co-transfected with the PTKs. This indicates that the phosphorylation of a tyrosine residue depends on the presence of at least one other tyrosine residue and that the third and the fourth tyrosine residues are critical for phosphorylation to occur.
Our studies indicated that GCET2 is probably an adapter protein, which undergoes tyrosine phosphorylation after a cellular stimulation and recruits cytosolic proteins to the cell membrane to form a signalling complex. By determining the proteins recruited by GCET2 to the cell membrane, we may gain some insight on the pathways through which GCET2 functions. GCET2 very likely uses its phospho-tyrosines to bind to the SH2 domains of its partners. Indeed, the third tyrosine of GCET2 is within a sequence, YENV, which matches the consensus GRB2 binding site (YxN). Using immunoprecipitation assays, we found that GRB2 and GCET2 bind to each other after pervanadate treatment. The third conserved tyrosine of GCET2 is a consensus GRB2 binding site, and we determined that this tyrosine indeed is essential to GRB2 binding. GRB2 is a small cytoplasmic adaptor containing a central SH2 domain flanked by two SH3 domains (Suen et al, 1993; Fath et al, 1994). It is likely that GRB2 binds to GCET2 using its central SH2 domain and recruits additional proteins through its SH3 domains. GRB2 is involved in numerous signalling pathways and associates with many proteins, such as MEKK1, BLNK, VAV, ABL, SHP1/2 and SOS. Further studies on the SH3 domain binding proteins of GRB2 after association with GCET2 may give some information on the GRB2 signalling pathways mediated through GCET2.
The GCET2 is highly restricted to GC B cells and is likely to be important in the GC reaction. Recent double immunohistochemical staining on tonsil sections revealed that the majority of BCL6 and CD10-positive cells have GCET2 expression (Natkunam et al, 2005). GCET2 is an interleukin (IL)-4 inducible gene. IL-4 is a pleotropic cytokine regulating lymphocyte differentiation, proliferation and apoptosis (Nelms et al, 1998). IL-4 binds to its receptors on the cell surface, activates the JAK/STAT6 pathway and induces transcription of target genes. GC Bcells have high expression of IL-4 signalling pathway components (IL-4Rα, IRS, p110 subunit of phosphatidylinositol 3-kinase and PKC delta) and of IL-4 target genes, including BCL6 and GCET2. We propose a model of the activities of GCET2 in GCs. In GC B cells, GCET2 is upregulated by cytokines, such as IL-4 secreted by T-helper cell in the GCs. During the GC reaction, an extracellular signal induces the activation of LYN and SYK, and the active LYN and SYK then phosphorylate GCET2, which is constitutively localised in the cell membrane through myristoylation and palmitoylation. The phosphorylated GCET2 then recruits cytosolic proteins (e.g. GRB2) to the cell membrane to form a signalling complex. The recruited proteins then mediate downstream pathways, which are essential components of the GC reaction. Another interesting finding is that GCET2 is also highly expressed in the thymus (Lossos et al, 2003; Pan et al, 2003), very likely in the immature T cells, and this indicates that GCET2 may also play a role in T-cell maturation.
Acknowledgements
This study was supported by the Grant no. NIH U01-CA84967-05. The authors thank Dr Weiguo Zhang, Department of Immunology, Duke University School of Medicine, for helpful suggestions and BCR cross-linking of mouse A20 cell line.
References
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J, Jr, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Levy R, Wilson W, Grever MR, Byrd JC, Botstein D, Brown PO, Staudt LM. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Allam A, Marshall AJ. Role of the adaptor proteins Bam32, TAPP1 and TAPP2 in lymphocyte activation. Immunology Letters. 2005;97:7–17. doi: 10.1016/j.imlet.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Brdicka T, Imrich M, Angelisova P, Brdickova N, Horvath O, Spicka J, Hilgert I, Luskova P, Draber P, Novak P, Engels N, Wienands J, Simeoni L, Osterreicher J, Aguado E, Malissen M, Schraven B, Horejsi V. Non-T cell activation linker (NTAL): a transmembrane adaptor protein involved in immunoreceptor signaling. Journal of Experimental Medicine. 2002;196:1617–1626. doi: 10.1084/jem.20021405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavenagh MM, Whitney JA, Carroll K, Zhang C, Boman AL, Rosenwald AG, Mellman I, Kahn RA. Intracellular distribution of Arf proteins in mammalian cells. Arf6 is uniquely localized to the plasma membrane. Journal of Biological Chemistry. 1996;271:21767–21774. doi: 10.1074/jbc.271.36.21767. [DOI] [PubMed] [Google Scholar]
- Chiu CW, Dalton M, Ishiai M, Kurosaki T, Chan AC. BLNK: molecular scaffolding through ‘cis’-mediated organization of signaling proteins. EMBO Journal. 2002;21:6461–6472. doi: 10.1093/emboj/cdf658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoph T, Rickert R, Rajewsky K. M17: a novel gene expressed in germinal centers. International Immunology. 1994;6:1203–1211. doi: 10.1093/intimm/6.8.1203. [DOI] [PubMed] [Google Scholar]
- Dave SS, Wright G, Tan B, Rosenwald A, Gascoyne RD, Chan WC, Fisher RI, Braziel RM, Rimsza LM, Grogan TM, Miller TP, LeBlanc M, Greiner TC, Weisenburger DD, Lynch JC, Vose J, Armitage JO, Smeland EB, Kvaloy S, Holte H, Delabie J, Connors JM, Lansdorp PM, Ouyang Q, Lister TA, Davies AJ, Norton AJ, Muller-Hermelink HK, Ott G, Campo E, Montserrat E, Wilson WH, Jaffe ES, Simon R, Yang L, Powell J, Zhao H, Goldschmidt N, Chiorazzi M, Staudt LM. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. New England Journal of Medicine. 2004;351:2159–2169. doi: 10.1056/NEJMoa041869. [DOI] [PubMed] [Google Scholar]
- Epstein AL, Levy R, Kim H, Henle W, Henle G, Kaplan HS. Biology of the human malignant lymphomas. IV. Functional characterization of ten diffuse histiocytic lymphoma cell lines. Cancer. 1978;42:2379–2391. doi: 10.1002/1097-0142(197811)42:5<2379::aid-cncr2820420539>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Fath I, Schweighoffer F, Rey I, Multon MC, Boiziau J, Duchesne M, Tocque B. Cloning of a Grb2 isoform with apoptotic properties. Science. 1994;264:971–974. doi: 10.1126/science.8178156. [DOI] [PubMed] [Google Scholar]
- Fessart D, Simaan M, Laporte SA. c-Src regulates clathrin adapter protein 2 interaction with {beta}-arrestin and the angiotensin II type 1 receptor during clathrin-mediated internalization. Molecular Endocrinology. 2005;19:491–503. doi: 10.1210/me.2004-0246. [DOI] [PubMed] [Google Scholar]
- Fu C, Turck CW, Kurosaki T, Chan AC. BLNK: a central linker protein in B cell activation. Immunity. 1998;9:93–103. doi: 10.1016/s1074-7613(00)80591-9. [DOI] [PubMed] [Google Scholar]
- Gluzman Y. SV40-transformed simian cells support the replication of early SV40 mutants. Cell. 1981;23:175–182. doi: 10.1016/0092-8674(81)90282-8. [DOI] [PubMed] [Google Scholar]
- Goitsuka R, Fujimura Y, Mamada H, Umeda A, Morimura T, Uetsuka K, Doi K, Tsuji S, Kitamura D. BASH, a novel signaling molecule preferentially expressed in B cells of the bursa of Fabricius. Journal of Immunology. 1998;161:5804–5808. [PubMed] [Google Scholar]
- Husson H, Carideo EG, Neuberg D, Schultze J, Munoz O, Marks PW, Donovan JW, Chillemi AC, O’Connell P, Freedman AS. Gene expression profiling of follicular lymphoma and normal germinal center B cells using cDNA arrays. Blood. 2002;99:282–289. doi: 10.1182/blood.v99.1.282. [DOI] [PubMed] [Google Scholar]
- Huyer G, Liu S, Kelly J, Moffat J, Payette P, Kennedy B, Tsaprailis G, Gresser MJ, Ramachandran C. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. Journal of Biological Chemistry. 1997;272:843–851. doi: 10.1074/jbc.272.2.843. [DOI] [PubMed] [Google Scholar]
- Ishiai M, Kurosaki M, Pappu R, Okawa K, Ronko I, Fu C, Shibata M, Iwamatsu A, Chan AC, Kurosaki T. BLNK required for coupling Syk to PLC gamma 2 and Rac1-JNK in B cells. Immunity. 1999;10:117–125. doi: 10.1016/s1074-7613(00)80012-6. [DOI] [PubMed] [Google Scholar]
- Janssen E, Zhang W. Adaptor proteins in lymphocyte activation. Current Opinion in Immunology. 2003;15:269–276. doi: 10.1016/s0952-7915(03)00044-x. [DOI] [PubMed] [Google Scholar]
- Janssen E, Zhu M, Zhang W, Koonpaew S. LAB: a new membrane-associated adaptor molecule in B cell activation. Nature Immunology. 2003;4:117–123. doi: 10.1038/ni882. [DOI] [PubMed] [Google Scholar]
- Kelsoe G. The germinal center: a crucible for lymphocyte selection. Seminars in Immunology. 1996a;8:179–184. doi: 10.1006/smim.1996.0022. [DOI] [PubMed] [Google Scholar]
- Kelsoe G. Life and death in germinal centers (redux) Immunity. 1996b;4:107–111. doi: 10.1016/s1074-7613(00)80675-5. [DOI] [PubMed] [Google Scholar]
- Kim KJ, Kanellopoulos-Langevin C, Merwin RM, Sachs DH, Asofsky R. Establishment and characterization of BALB/c lymphoma lines with B cell properties. Journal of Immunology. 1979;122:549–554. [PubMed] [Google Scholar]
- Klein E, Klein G, Nadkarni JS, Nadkarni JJ, Wigzell H, Clifford P. Surface IgM-kappa specificity on a Burkitt lymphoma cell in vivo and in derived culture lines. Cancer Research. 1968;28:1300–1310. [PubMed] [Google Scholar]
- Koonpaew S, Janssen E, Zhu M, Zhang W. The importance of three membrane-distal tyrosines in the adaptor protein NTAL/LAB. Journal of Biological Chemistry. 2004;279:11229–11235. doi: 10.1074/jbc.M311394200. [DOI] [PubMed] [Google Scholar]
- Kurosaki T. Regulation of B-cell signal transduction by adaptor proteins. Nature Reviews Immunology. 2002;2:354–363. doi: 10.1038/nri801. [DOI] [PubMed] [Google Scholar]
- Kurosaki T, Tsukada S. BLNK: connecting Syk and Btk to calcium signals. Immunity. 2000;12:1–5. doi: 10.1016/s1074-7613(00)80153-3. [DOI] [PubMed] [Google Scholar]
- Leo A, Schraven B. Adapters in lymphocyte signalling. Current Opinion in Immunology. 2001;13:307–316. doi: 10.1016/s0952-7915(00)00220-x. [DOI] [PubMed] [Google Scholar]
- Leo A, Wienands J, Baier G, Horejsi V, Schraven B. Adapters in lymphocyte signaling. Journal of Clinical Investigation. 2002;109:301–309. doi: 10.1172/JCI14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossos IS, Alizadeh AA, Rajapaksa R, Tibshirani R, Levy R. HGAL is a novel interleukin-4-inducible gene that strongly predicts survival in diffuse large B-cell lymphoma. Blood. 2003;101:433–440. doi: 10.1182/blood-2002-06-1931. [DOI] [PubMed] [Google Scholar]
- Minegishi Y, Rohrer J, Coustan-Smith E, Lederman HM, Pappu R, Campana D, Chan AC, Conley ME. An essential role for BLNK in human B cell development. Science. 1999;286:1954–1957. doi: 10.1126/science.286.5446.1954. [DOI] [PubMed] [Google Scholar]
- Myung PS, Boerthe NJ, Koretzky GA. Adapter proteins in lymphocyte antigen-receptor signaling. Current Opinion in Immunology. 2000;12:256–266. doi: 10.1016/s0952-7915(00)00085-6. [DOI] [PubMed] [Google Scholar]
- Natkunam Y, Lossos IS, Taidi B, Zhao S, Lu X, Ding F, Hammer AS, Marafioti T, Byrne GE, Jr, Levy S, Warnke RA, Levy R. Expression of the human germinal center-associated lymphoma (HGAL) protein, a new marker of germinal center B cell derivation. Blood. 2005;105:3979–3986. doi: 10.1182/blood-2004-08-3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelms K, Huang H, Ryan J, Keegan A, Paul WE. Interleukin-4 receptor signalling mechanisms and their biological significance. Advances in Experimental Medicine and Biology. 1998;452:37–43. doi: 10.1007/978-1-4615-5355-7_5. [DOI] [PubMed] [Google Scholar]
- Pan Z, Shen Y, Du C, Zhou G, Rosenwald A, Staudt LM, Greiner TC, McKeithan TW, Chan WC. Two newly characterized germinal center B-cell-associated genes, GCET1 and GCET2, have differential expression in normal and neoplastic B cells. American Journal of Pathology. 2003;163:135–144. doi: 10.1016/S0002-9440(10)63637-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappu R, Cheng AM, Li B, Gong Q, Chiu C, Griffin N, White M, Sleckman BP, Chan AC. Requirement for B cell linker protein (BLNK) in B cell development. Science. 1999;286:1949–1954. doi: 10.1126/science.286.5446.1949. [DOI] [PubMed] [Google Scholar]
- Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- Peterson EJ, Clements JL, Fang N, Koretzky GA. Adaptor proteins in lymphocyte antigen-receptor signaling. Current Opinion in Immunology. 1998;10:337–344. doi: 10.1016/s0952-7915(98)80173-8. [DOI] [PubMed] [Google Scholar]
- Radha V, Rajanna A, Swarup G. Phosphorylated guanine nucleotide exchange factor C3G, induced by pervanadate and Src family kinases localizes to the Golgi and subcortical actin cytoskeleton. BMC Cell Biology. 2004;5:31. doi: 10.1186/1471-2121-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay M. Gene expression profiling to predict lymphoma outcome. Trends in Molecular Medicine. 2002;8:159. doi: 10.1016/s1471-4914(02)02296-7. [DOI] [PubMed] [Google Scholar]
- Resh MD. Myristylation and palmitylation of Src family members: the fats of the matter. Cell. 1994;76:411–413. doi: 10.1016/0092-8674(94)90104-x. [DOI] [PubMed] [Google Scholar]
- Rosenwald A, Staudt LM. Gene expression profiling of diffuse large B-cell lymphoma. Leukemia & Lymphoma. 2003;44 Suppl. 3:S41–S47. doi: 10.1080/10428190310001623775. [DOI] [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, Hurt EM, Zhao H, Averett L, Yang L, Wilson WH, Jaffe ES, Simon R, Klausner RD, Powell J, Duffey PL, Longo DL, Greiner TC, Weisenburger DD, Sanger WG, Dave BJ, Lynch JC, Vose J, Armitage JO, Montserrat E, Lopez-Guillermo A, Grogan TM, Miller TP, LeBlanc M, Ott G, Kvaloy S, Delabie J, Holte H, Krajci P, Stokke T, Staudt LM. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. New England Journal of Medicine. 2002;346:1937–1947. doi: 10.1056/NEJMoa012914. [DOI] [PubMed] [Google Scholar]
- Rudd CE. Lymphocyte signaling: adapting new adaptors. Current Biology. 1998;8:R805–R808. doi: 10.1016/s0960-9822(07)00505-2. [DOI] [PubMed] [Google Scholar]
- Secrist JP, Burns LA, Karnitz L, Koretzky GA, Abraham RT. Stimulatory effects of the protein tyrosine phosphatase inhibitor, pervanadate, on T-cell activation events. The Journal Of Biological Chemistry. 1993;268:5886–5893. [PubMed] [Google Scholar]
- Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ, Neel BG, Birge RB, Fajardo JE, Chou MM, Hanafusa H, Schaffhausen B, Cantley LC. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Shoelson SE, McGlade J, Olivier P, Pawson T, Bustelo XR, Barbacid M, Sabe H, Hanafusa H, Yi T, Ren R, Baltimore D, Ratnofsky S, Feldman RA, Cantley LC. Specific motifs recognized by the SH2 domains of Csk, 3BP2, fps/fes, GRB-2, HCP, SHC, Syk, and Vav. Molecular and Cellular Biology. 1994;14:2777–2785. doi: 10.1128/mcb.14.4.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa MA. The adaptor complexes: a bridge between the transmembrane proteins and clathrin lattices. Biocell. 1996;20:301–305. [PubMed] [Google Scholar]
- Suen KL, Bustelo XR, Pawson T, Barbacid M. Molecular cloning of the mouse grb2 gene: differential interaction of the Grb2 adaptor protein with epidermal growth factor and nerve growth factor receptors. Molecular and Cellular Biology. 1993;13:5500–5512. doi: 10.1128/mcb.13.9.5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi T, Kiyokawa N, Takenouch H, Matsui J, Tang WR, Nakajima H, Suzuki K, Shiozawa Y, Saito M, Katagiri YU, Takahashi T, Karasuyama H, Matsuo Y, Okita H, Fujimoto J. Deficiency of BLNK hampers PLC-gamma2 phosphorylation and Ca2+ influx induced by the pre-B-cell receptor in human pre-B cells. Immunology. 2004;112:575–582. doi: 10.1111/j.1365-2567.2004.01918.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JE, Wong SC, Gan SK, Xu S, Lam KP. The adaptor protein BLNK is required for b cell antigen receptor-induced activation of nuclear factor-kappa B and cell cycle entry and survival of B lymphocytes. Journal of Biological Chemistry. 2001;276:20055–20063. doi: 10.1074/jbc.M010800200. [DOI] [PubMed] [Google Scholar]
- Togni M, Lindquist J, Gerber A, Kolsch U, Hamm-Baarke A, Kliche S, Schraven B. The role of adaptor proteins in lymphocyte activation. Molecular Immunology. 2004;41:615–630. doi: 10.1016/j.molimm.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Tsiani E, Bogdanovic E, Sorisky A, Nagy L, Fantus IG. Tyrosine phosphatase inhibitors, vanadate and pervanadate, stimulate glucose transport and GLUT translocation in muscle cells by a mechanism independent of phosphatidylinositol 3-kinase and protein kinase C. Diabetes. 1998;47:1676–1686. doi: 10.2337/diabetes.47.11.1676. [DOI] [PubMed] [Google Scholar]
- Wienands J. Signal transduction elements of the B cell antigen receptor and their role in immunodeficiencies. Immunobiology. 2000;202:120–133. doi: 10.1016/S0171-2985(00)80059-5. [DOI] [PubMed] [Google Scholar]
- Wollscheid B, Wienands J, Reth M. The adaptor protein SLP-65/BLNK controls the calcium response in activated B cells. Current Topics in Microbiology and Immunology. 1999;246:283–289. doi: 10.1007/978-3-642-60162-0_35. discussion 288–289. [DOI] [PubMed] [Google Scholar]
- Zhang W, Trible RP, Samelson LE. LAT palmitoylation: its essential role in membrane microdomain targeting and tyrosine phosphorylation during T cell activation. Immunity. 1998;9:239–246. doi: 10.1016/s1074-7613(00)80606-8. [DOI] [PubMed] [Google Scholar]