

Abstract
The initial success of the first synthetic bcr-abl kinase inhibitor imatinib has been dampened by the emergence of imatinib-resistant disease in blast crisis CML. Here we report that the novel triterpenoid methyl-2-cyano-3,12 dioxoolean-1,9 diene-28-oate (CDDO-Me) potently induced cytotoxicity in imatinib-resistant KBM5 cells expressing the T315I mutation of bcr-abl (24 h EC50 = 540 nM). In long-term culture CDDO-Me abrogated the growth of human parental KBM5 and KBM5-STI cells with 96 h IC 50 values of 205 nM and 221 nM, respectively. In addition, CDDO-Me rapidly decreased the viability of murine lymphoid Ba/F3 cells expressing wild type p210, as well as the imatinib resistant E255K and T315I mutations of bcr-abl. The low dose effects of CDDO-Me are associated with inhibition of mitochondrial oxygen consumption while the cytotoxic effects appear to be mediated by a rapid and selective depletion of mitochondrial glutathione that accompanies the increased generation of reactive oxygen species and mitochondrial dysfunction. Interestingly, the mitochondriotoxic effects of CDDO-Me are followed by the rapid autophagocytosis of intracellular organelles or the externalization of phosphatidyl serine in different cell types. We conclude that alterations in mitochondrial function by CDDO-Me can result in autophagy or apoptosis of CML cells regardless of the mutational status of phosphatidyl CDDO-Me is in clinical trials and shows signs of clinical activity, with minimal side-effects and complete lack of cardiotoxicity. Studies in leukemias are in preparation.
Keywords: CDDO-Me, apoptosis, CML, mitochondria, glutathione
Introduction
Chronic myeloid leukemia is a clonal disease characterized by the accumulation of hematopoeitic progenitors carrying a (9;22) chromosomal translocation commonly known as the Philadelphia chromosome (Ph) that results in the expression of the oncogenic fusion kinase bcr-abl 1. Bcr-abl is a constitutively activated kinase that has been shown to activate MEK/ERK 2, PI3K 3, and JAK/STAT 4 signaling resulting in increased proliferation and resistance to chemotherapy 5. CML progresses from a chronic phase into a myeloid blast crisis phase accompanied by additional genetic and chromosomal abnormalities that cooperate with bcr-abl to drive disease progression. Treatment of CML with imatinib, a potent synthetic inhibitor of the bcr-abl kinase, produces high rates of hematologic and cytogenetic responses in the chronic phase of the disease making this agent a paradigm for molecularly targeted therapies 6,7. Unfortunately, imatinib induces only partial, short-lived responses in the blast crisis phase of the disease, and most patients develop resistance to this agent leading to disease recurrence 8. In fact, a recent long-term follow-up study of CML patients treated with imatinib reportedthat hematologic resistance to this agent occurred in 24% and 92% of patients in chronicand blast crisis, respectively 9. The decreased efficacy of imatinib in CML as a consequence of mutations within bcr-abl is best exemplified by the T315I mutation. Clinically, the T315I mutation is associated with a formidable therapeutic challenge because itmediates complete resistance to not only imatinib, but also to many of the next generation of ABL kinase inhibitors like dasatinib (Sprycel; Bristol-Myers Squibb) and the imatinib-related compound nilotinib (Tasigna; Novartis) 10. The emergence of T315I mutations in CML has given even greater urgency to develop more effective chemotherapeutics totreat this malignancy.
The novel triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) is effective in inducing apoptosis in a variety of tumor cell types including leukemia 11-14, multiple myeloma 15, breast, osteosarcoma 16, pancreatic 17, and skin 18. Furthermore, recent reports demonstrate that CDDO’s C28 methyl ester derivative, methyl-2-cyano-3,12-dioxooleana-1,9-dien-28-oate (CDDO-Me), is 5-fold more potent than CDDO as an antitumor agent in vitro 11,19. CDDO and CDDO-Me reportedly disrupted intracellular redox balance in U937 cells and multiple myeloma cells thereby activating the intrinsic apoptotic pathway 11,15, and CDDO-Me exhibited some selectivity in apoptosis induction between tumor and normal cells 19. Interestingly, recent evidence from our group indicates that CDDO induced the release of cytochrome c from isolated mitochondria via a cyclosporine A-independent permeability transition suggesting that this organelle may be a direct target of this agent 14,20.
Here we report that the CDDO derivative CDDO-Me is effective in abrogating the growth of imatinib resistant CML cells of human and mouse origin, and that the antiproliferative effects of this oleanic acid derivative appear to be initiated by rapid perturbations in mitochondrial function associated with increased oxidative stress. Interestingly, cytotoxic doses of CDDO-Me induced apoptotic or autophagic cell death in different cell types, and this is to our knowledge the first report demonstrating that the mitochondriotoxic effects of CDDO-Me can also activate autophagy. Autophagy, or programmed cell death II, is a pathway that recruits the endolysosomal system to digest intracellular components, presumably as a mode of survival during nutrient deprivation, but was more recently reported to be a form of cellular demise in cancer cells after a variety of chemotherapeutic insults 21. We hypothesize that CDDO-Me may be effective in treating CML, regardless of bcr-abl mutational status, by inducing programmed cell death (either apoptosis or authophagy) via the disruption of mitochondrial function.
Materials and Methods
Chemicals and Biochemicals
CDDO-Me was kindly provided by Dr. Edward Sausville (NCI) under the RAID program and by Dr. Michael Sporn (Dartmouth Medical College, Hanover NH). NAC was purchased from Sigma (St. Louis, MO). CMH2DCF-DA, CMXRos, and TMRM were all obtained from Molecular Probes (Eugene, OR). Z-VAD-fmk was purchased from Alexis Biochemicals (Axxora LLC, San Diego, CA). Phospho-p38 and p38 antibodies were purchased from Cell Signaling Technologies, Inc. (Beverly, MA). Hemeoxigenase-1 (HO-1) antibody was purchased from BD Biosciences (San Jose, CA) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody was purchased from Chemicon International (Temecula, CA). PARP1 antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and goat anti-mouse and anti-rabbit horseradish peroxidase-conjugated secondary antibodies were purchased from Bio-Rad (Hercules, CA). All other chemicals used were of the highest purity available.
Cell Lines
KBM5 cells were derived from a patient with myeloid blastic phase of CML; the cells contain multiple copies of the Philadelphia chromosome while lacking the normal ABL gene. KBM5 cells resistant to imatinib (KBM5-STI) were derived by Ricci et al. by chronic exposure of KBM5 cells to imatinib 22. KBM5-STI cells were able to grow in the presence of 2.0 μM STI571 and were maintained at this concentration. Cells were grown in RPMI 1640 medium supplemented with 10% fetal calf serum, 1% glutamine and 100 units/ml penicillin in a 37°C incubator containing 5% CO2. Interleukine-3 (IL-3)-dependent murine pro-B cell line BaF3 transfected with vector, wt-p210 (expressing p210bcr-abl), E255K or T315I were kindly provided by Dr. C. Sawyers and were cultivated in RPMI 1640 complemented with 10% fetal calf serum, 1% glutamine, 2ng/ml IL-3 (vector only), and 2μM puromycin 23. Viable cell numbers were quantitated in a Vi-Cell Cell Viability Analyzer (Beckman-Coulter, Fullerton, CA).
Human Subjects
Bone marrow or peripheral blood samples were obtained for in vitro studies from patients with chronic myeloid leukemia (CML); samples were collected during routine diagnostic procedures after informed consent was obtained in accordance with regulations and protocols approved by the Human Subjects Committee of the University of Texas M.D. Anderson Cancer Center (Houston, TX). Mononuclear cells were separated by Ficoll-Hypaque (SigmaChemical) density gradient centrifugation.
Measurement of mitochondrial membrane potential (ΔΨM)
After appropriate treatments, cells were washed twice in PBS and then resuspended in 100 μl of PBS containing 0.5 μg/ml MitoTracker CMXRos and 15 ng/ml MitoTracker Green, and incubated at 37°C for 45 min. Cells were then washed twice in PBS and analyzed by flow cytometry in a FACSCalibur flow cytometer using a 488 nm argon excitation laser. Alternatively, for confocal microscopy or short-timepoint measurements of ΔΨM cells were loaded with 50 nM of the potentiometric probe TMRM, treated as indicated, and analyzed by confocal microscopy or flow cytometry. Results presented are means +/- S.E. of three independent experiments.
Western Blot Analysis
Cells where harvested by centrifugation, washed twice in PBS, and resuspended in ice cold lysis buffer (1% Triton X-100, 45 mM KCl, 10mM Tris, pH 7.5), supplemented with proteaseand phosphatase inhibitors; then subjected to SDS-PAGE in 10% or 12% polyacrylamide gels followed by protein transfer to a Hybond-P membrane (Amersham PharmaciaBiotech, Little Chalfont, United Kingdom) and immunoblotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) blots were run in parallel as loading controls. Signals were detected by a PhosphorImager (Storm 860, version 4.0, Molecular Dynamics, Sunnyvale, CA).
Transmission electron microscopy
After appropriate treatments samples were fixed with a solution containing 3% glutaraldehyde plus 2% paraformaldehyde in 0.1 M cacodylate buffer, pH 7.3 for 1 hour. After fixation, the samples were washed and treated with 0.1% Millipore-filtered cacodylate buffered tannic acid, postfixed with 1% buffered osmium tetroxide for 30 min, and stained en bloc with 1% Millipore filtered uranyl acetate. The samples were dehydrated in increasing concentrations of ethanol, infiltrated, and embedded in Spurr’s low viscosity medium. The samples were polymerized in a 70°C oven for 2 days. Ultrathin sections were cut in a Leica Ultracut microtome (Leica, Deerfield, IL), stained with uranyl acetate and lead citrate in a Leica EM Stainer, and examined in a JEM 1010 transmission electron microscope (JEOL USA Inc., Peabody MA) at an accelerating voltage of 80 kV. Digital images were obtained using AMT Imaging System (Advanced Microscopy Techniques Corp, Danvers, MA).
Measurement of intracellular GSH by flow cytometry
Cells (3 × 105 cells/ml; 0.5 ml) were treated with CDDO-Me as indicated, or with 2 mM diethylmaleate (DEM) for 30 min. Cells were then collected by centrifugation, washed in PBS once, and resuspended in 0.2 ml PBS containing 400 nM Cell Tracker Green™ (CTG; Molecular Probes, Eugene OR) and incubated on ice protected from light for 10 min. Cells were then washed in PBS several times and CTG fluorescence was quantitated by flow cytometry. The mean CTG fluorescence from DEM treated samples was considered to be background and substracted accordingly. All experiments were done in duplicate and repeated at least thrice.
Measurement of oxygen consumption
Cells were resuspended in 1 mL fresh warm medium pre-equilibrated with 21% oxygen and placed in the sealed respiration chamber equipped with a thermostat control, a microstirring device, and a Clark-type oxygen electrode disc (Oxytherm, Hansatech Instrument, Cambridge, United Kingdom). The oxygen content in the cell suspension medium was constantly monitored and oxygen consumption rate was constantly monitored, and the signals were integrated using the software supplied by the manufacturer.
LC3B (isoform B of human microtubule-associated protein 1 light chain 3) immunohistochemistry
Anti-LC3B antibody against a synthetic peptide corresponding to the N-terminal 14 amino acids of isoform B of human LC3 and an additional cysteine (PSEKTFKQRRTFEQC) was prepared by immunization of a rabbit and then affinity purified on an immobilized peptide-Sepharose column (Covance, Denver, PA). After appropriate treatments, cells were harvested by centrifugation and fixed in 4% paraformaldehyde at room temperature for 10 minutes. Cells were then permeabilized by 0.1% Triton-X and washed extensively in PBS followed by incubation in 5% normal goat serum in PBS with 0.1% Tween-20 at 37°C for 1 hr. After incubation, LC3B antibody was added (1:100 dilution) and immune complexes were allowed to form at 4°C overnight. Cells were washed in PBS twice followed by incubation with an APC-conjugated anti-rabbit IgG for 30 minutes at room temperature. Cells were then mounted on DAPI containing medium (ProLong Gold; Invitrogen Carlsbad, CA) and analyzed by confocal microscopy on an Olympus IX71 inverted microscope.
Results
Submicromolar concentrations of CDDO-Me inhibit the growth of imatinib resistant CML cells in culture
Ricci et al. reported the development of an imatinb resistant CML cell line by chronicexposure of the previously described KBM5 CML cell line 24 to increasing concentrations of imatinib 22. This imatinib resistant cell line, KBM5-STI, was demonstrated to carry the T315I mutation in the ATP binding pocket of bcr-abl that has also been reported in a proportion of imatinib resistant patients 22,25. To investigate if CDDO-Me would be effective in preventing the proliferation of this clinically relevant imatinib resistance model, we cultured KBM5 and KBM5-STI cells in the presence of increasing concentrations of CDDO-Me (100 - 1000 nM) for 24, 48, 72, and 96 h. Under our experimental conditions, KBM5-STI cells were resistant to imatinib doses as high as 2 μM, whereas parental KBM5 cells underwent rapid apoptosis at this concentration of imatinib (not shown). Our results presented in Figure 1 A and B demonstrate that CDDO-Me decreased the numbers of viable cells of both KBM5 and KBM5-STI cells, with 96 h IC 50 values of 205 nM and 221 nM, respectively. To investigate if decreased cell cycle progression contributes to the antiproliferative action of CDDO-Me, we analyzed cell cycle distribution in cells treated with this agent, and found that at 72 h post-treatment CDDO-Me induced a similar increase (1.7 - 1.8 fold at 300 nM; data not shown) in the G1 phase of the cell cycle in both KBM5 and KBM5-STI cells. Finally, we investigated if imatinib resistance in different cell types would result in decreased sensitivity to CDDO-Me. For these experimentswe treated mouse lymphoid Ba/F3 cells expressing wild-type bcr-abl and the imatinib resistant E255K and T315I bcr-abl mutants with increasing concentrations of CDDO-Me. As illustrated in Figure 1 C, CDDO-Me rapidly decreased the viability of mouse Ba/F3 cells expressing wild-type bcr-abl (24 h IC50 = 250 nM), the E255K bcr-abl mutant (24 h IC50 = 94 nM), or the T315I bcr-abl mutant (24 h IC50 = 210 nM). Taken together, these data demonstrate that CDDO-Me effectively prevents the proliferation of imatinib resistant CML cells.
Figure 1.




Submicromolar concentrations of CDDO-Me abrogate the growth of imatinib resistant CML cells in culture. A) KBM5 cells were cultured with 100, 250, 500, and 1000 nM of CDDO-Me for the indicated times, and viable cells were quantitated as described in the methods B) KBM5-STI were cultured with CDDO-Me (100-1000 nM) for the indicated times and analyzed as above. C) Mouse lymphoid Ba/F3 cells expressing wild type bcr-abl, the T315I mutant of bcr-abl, the E255K mutant of bcr-abl, and a vector control were treated with increasing concentrations of CDDO-Me as indicated and viability was measured by trypan blue exclusion. D) Cells were cultured with 100 nM of CDDO-Me for 24 h and oxygen consumption was monitored in 1 × 107 cells as described in the materials and methods.
CDDO-Me decreases oxygen consumption in CML cell lines
We have previously reported that submicromolar concentrations of CDDO-Me inhibit oxygen consumption in AML cell lines, and that this effect precedes activation of the mitochondrial permeability transition 20. To investigate if CDDO-Me similarly affects mitochondrial metabolism in CML cell lines we monitored oxygen consumption in KBM5 and KBM5-STI cells after treatment with subcytotoxic doses of CDDO-Me. As shown in Figure 1 D, after 24 h treatment with 100 nM CDDO-Me oxygen consumption was decreased in KBM5 and KBM5-STI by 32 (from 16.9 to 11.5 nmoles/ml*min) and 28% (from 19.2 to 13.8 nmoles/ml*min), respectively (p<0.008). Interestingly, KBM5-STI cells had a 14% higher basal rate of oxygen consumption than parental KBM5 cells (p<0.002), suggesting the possibility that the imatinib resistance may be associated with differences in mitochondrial function.
CDDO-Me can induce apoptosis or autophagy in CML
To investigate if apoptosis contributes to the antiproliferative effects of CDDO-Me in CML cells, we examined mitochondrial membrane potential (ΔΨM) and phosphatidyl serine externalization in cells treated with increasing concentrations of CDDO-Me for 24 h. As illustrated inFigure 2 A, CDDO-Me induced a dose-dependent loss of ΔΨM in both KBM5 (24 h IC50 303 nM) and KBM5-STI cells (24 h IC50 540 nM). Surprisingly, in contrast to staurosporine, CDDO-Me induced significantly (p<0.007) less externalization of phosphatidyl serine in KBM5 cells at doses sufficient to induce marked loss of ΔΨM (Figure 2 B), suggesting the possibility that this agent is activating alternative modes of cell death. Similar findings were obtained in KBM5-STI cells after 24 and 96 h of exposure to CDDO-Me (not shown). Indeed, transmission electron microscopy revealed that both KBM5 and KBM5-STI cells formed extensive double membrane vesicles after exposure to cytotoxic concentrations of CDDO-Me, and this was accompanied by a decrease in cell size ruling out the possibility of oncosis (Figure 2 C). Furthermore, immunohistochemistry revealed an increased cytoplasmic staining of LC3B, a protein commonly associated with autophagosomes (Figure 2 D). In contrast, mouse Ba/F3 cells expressing wild-type bcr-abl, the E255K bcr-abl mutant, or the T315I bcr-abl mutant rapidly externalized PS in response to CDDO-Me (Figure 2 E), and similar observations were made in K562 cells (not shown) suggesting that autophagic cell death induced by this agent is not modulated by BCR-ABL per se.
Figure 2.





CDDO-Me induces programmed cell death that can be manifested by apoptosis or autophagy. A) KBM5 and KBM5-STI cells were treated with CDDO-Me as above for 24 h and ΔΨM was measured using CMXRos as described in the Materials and Methods. B) KBM5 cells were treated with CDDO-Me or staurosporine (STS) for 24 h and PS externalization and ΔΨM were analyzed by flow cytometry. * p<0.007. C) KBM5 and KBM5-STI cells were treated with 500 nM CDDO-Me for 24 h and processed for TEM as described in the Materials and Methods. Arrows indicate autophagosomes. D) The immunohistochemical expression of the autophagosomal protein LC3B was investigated in cells treated as above. E) Mouse lymphoid Ba/F3 cells expressing wild type bcr-abl, the T315I mutant of bcr-abl, the E255K mutant of bcr-abl, and a vector control were treated with increasing concentrations of CDDO-Me as indicated and PS externalization and ΔΨM were analyzed by flow cytometry.
CDDO-Me induces rapid generation of ROS that is associated with decreased mitochondrial membrane potential (ΔΨM) and precedes the loss of intracellular reduced GSX(GSH)
Because ROS is a component of oxidative stress-induced autophagy 26 we investigated if CDDO-Me induced the accumulation of ROS in KBM5 and KBM5-STI cells. Our results illustrate that 1 μM CDDO-Me induced a significant (p<0.02) 2.8-fold increase in the ROS-dependent CM-H2DCF fluorescence in KBM5 cells and a significant (p<0.008) 4.2-fold increase in KBM5-STI cells after a 3 h treatment (Figure 3 A) suggesting that ROS is indeed a component of the cytotoxicity of this agent in CML cells. Consistent with a mitochondriotoxic effect of ROS, the increase in ROS accompanied a decrease in ΔΨM of 50% (p<0.002) and 36% (p<0.002) in KBM5 and KBM5-STI cells, respectively (Figure 3 B). To investigate if oxidative stress in CML cells treated with CDDO-Me is associated with a decrease in the levels of reduced GSX (GSH) we measured the levels of intracellular GSH in KBM5 and KBM5-STI cells treated with 1 μM CDDO-Me. The results illustrated inFigure 3 C demonstrate that 1 μM CDDO-Me decreased the levels of GSH in a time-dependent manner in KBM5 cells (30% loss at 6 h; p<0.01) and KBM5-STI cells (80% loss at 6 h; p<0.001), albeit the latter cell type displayed a more rapid and pronounced decrease in the levels of GSH. These results are congruent with our observation that KBM5-STI cells generated more ROS in response to CDDO-Me treatment than KBM5 cells (Figure 3 A), and suggest that the increased generation of ROS induced by this agent contributes to oxidation of the GSX pool in CML cells. Our observations thus demonstrate that increased generation of ROS and oxidation of intracellular GSH are associated with CDDO-Me induced mitochondrial dysfunction and precede the onset of autophagy.
Figure 3.


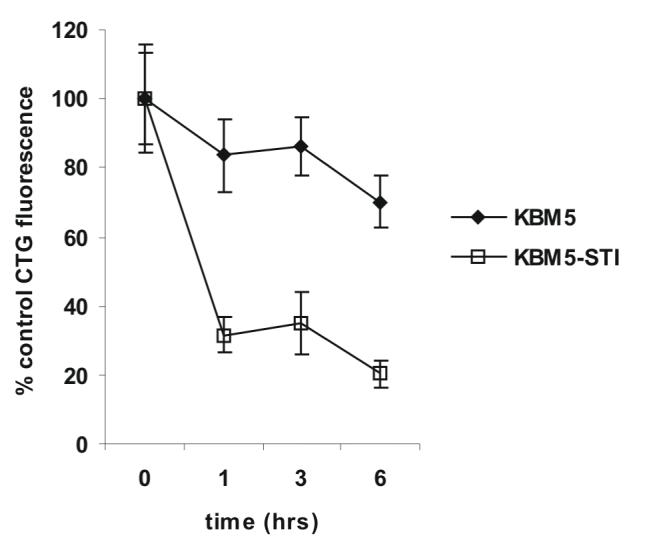
CDDO-Me induces rapid generation of ROS, mitochondrial dysfunction, and oxidation of intracellular GSH. A) Cells were treated with CDDO-Me as indicated and the generation of intracellular ROS was quantitated by flow cytometry as described in the Materials and Methods. B) Cells were loaded with the potentiometric probe TMRM, treated with CDDO-Me as indicated, and ΔΨM was quantitated by flow cytometry as described in the Materials and Methods. C) Cells were treated with CDDO-Me as indicated and loaded with the GSH specific probe Cell Tracker Green ™ followed by incubation in ice for 10 min. Cells were then extensively washed in PBS, and the GSH specific fluorescence of CTG was measured as described in the Materials and Methods.
CDDO-Me induces a rapid and selective depletion of mitochondrial GSX (GSXm) that leads to oxidation of cardiolipin
Cholesterol has been demonstrated to accumulate in the mitochondrial membrane and prevent the import of GSX from the cytosol leading to mitochondrial dysfunction 27,28. Since the triterpenoid structure of CDDO-Me shares some similarity to cholesterol we hypothesized that the rapid generation of ROS and decreased membrane potential induced by CDDO-Me may be mediated by perturbations in GSXm. We therefore quantitated biochemically the levels of GSXm and whole-cell GSX (GSXwc) in CML cells treated with CDDO-Me. The results presented in Figure 4 A demonstrate that 300 nM CDDO-Me selectively depleted GSXm by 69% (p<0.003) in KBM5 cells, and a similar effect was seen in KBM5-STI cells (65%; p<0.02) after 3 h treatment without any marked effect in GSXwc supporting the notion that the triterpenoid structure of CDDO-Me may mediate perturbations in GSXm flux. Cardiolipin is the most abundant phospholipid in the mitochondrial membrane, and has been shown to modulate the initiation of the intrinsic apoptotic pathway 29,30. Since GSXm is critical for the maintenance of cardiolipin 31-33, we examined the levels of cardiolipin in cells treated with CDDO-Me by staining with the cardiolipin-selective fluorochrome nonyl acrydine orange (NAO). NAO binds with high affinity and selectivity to reduced, but not oxidized cardiolipin making it a useful probe to study the redox status and quantity of this lipid in mitochondria 34. Notably, CDDO-Me potently increased the proportion of cells displaying decreased NAO staining (NAO(-)) in KBM5 cells (7.4-fold; p<0.00001), and to a lesser, but significant extent in KBM5-STI cells (2.7-fold; p<0.005 Figure 4 B). Since it has been previously demonstrated that depletion of GSXm results in increased expression of the oxidative stress responsive protein heme oxygenase 1 (HO-1) 35, we investigated if a similar response was elicited by CDDO-Me in CML cell lines. Western blot analysis of KBM5 and KBM5-STI cells treated with CDDO-Me indeed demonstrates that this agent markedly induces the expression of HO-1 in both cell lines (Figure 4 C). Taken together, the above data suggest that CDDO-Me-induced mitochondrial dysfunction in CML cells is mediated by the selective loss of GSXm that results in accumulation of ROS and the loss of cardiolipin in KBM5 and KBM5-STI cells.
Figure 4.



CDDO-Me induces early depletion of GSXm and oxidation of the mitochondrial phospholipids cardiolipin. A) KBM5 and KBM5-STI cells were treated with 300 nM of CDDO-Me for 3 h and the levels of GSX in whole-cell and mitochondrial extracts were quantitated biochemically as described in the Materials and Methods. B) Cells were treated as indicated and incubated with 10 nM of the cardiolipin specific dye nonyl acridine orange (NAO). Cells were then washed in PBS and NAO FL1 fluorescence was quantitated by flow cytomety. C) Cells were treated with 500 nM of CDDO-Me for 6 and 12 h and HO-1 expression was investigated by Western blot as described in the Materials and Methods.
CDDO-Me decreases viability, induces apoptosis, and provokes the loss of intracellular GSH in primary CML cells
To investigate if CDDO-Me would effectively decrease the viability of primary CML blast crisis cells in culture we exposed ex vivo leukemic cells derived from three patients in the blast crisis phase of CML. The results presented in Figure 5 A demonstrate that CDDO-Me decreases the viability of primary CML cells with a mean 24 h IC50 concentration of 599.7 ± 78.6 nM. Moreover, when the levels of intracellular GSH were measured after a short time exposure to CDDO-Me (3 h), we found that consistent with our observations in CML cell lines, CDDO-Me decreased the levels of GSH in all 3 patient samples with a mean EC50 concentration of 637 ± 27.3 nM (Figure 5 B). Flow cytometric analysis of these samples, as well as a T315I CML sample (#4), indicated that CDDO-Me induced loss of ΔΨM and externalization of phosphatidyl serine (Figure 5 C) suggesting that apoptosis and not autophagy is the preferential mode of cell death induced by CDDO-Me in primary CML samples regardless of the mutational status of bcr-abl. The doses required for cytotoxicity were not significantly different from the doses required to decrease intracellular GSH (p>0.1). Our results suggest that oxidative stress, represented by decreased GSH levels, is indeed associated with the loss of viability induced by CDDO-Me in primary CML cells.
Figure 5.



CDDO-Me induces GSH depletion and apoptosis in primary CML blast crisis cells. A) Cells obtained from patients in blast crisis CML were cultured ex vivo with increasing concentrations of CDDO-Me (0-1000 nM) for 24 h and viable cells were counted after trypan blue staining using a hemocytometer. B) Patient samples were treated with increasing concentrations of CDDO-Me (0-1000 nM) for 3 h and intracellular GSH was quantitated by flow cytometry as described in the Materials and Methods. C) Patient samples were treated as in A) and phosphatidyl serine externalization and ΔΨM were quantitated by flow cytometry as described in the Materials and Methods.
Discussion
The emergence of imatinib resistance in CML patients has been associated with the development of point mutations in the kinase domain of bcr-abl. Clinically, imatininb-resistance may be overcome by 1) novel bcr-abl inhibitors such as nilotinib and dasatanib which inhibit most clinically relevant bcr-abl mutants, except the T315I mutation, or 2) modulation of targets independent of bcr-abl, like farnesyl transferases or Aurora kinase 36-39. Albeit these approacheshave shown promising preliminary results, identifying additional targets for the treatment of CML is of utmost importance.
CDDO-Me has been reported to prevent the proliferation of AML cells, but the effects of this novel triterpenoid on CML cells in the context of imatinib resistance have not been investigated. Here we report that CDDO-Me effectively abrogates the growth of both parental, and imatinib resistant human KBM5 (KBM5-STI) CML cells that express the clinically relevant T315I mutation. This single base pair substitution (C1308T) was originally identified in 6 of 9 patients who displayed imatinib resistance 40, and subsequent work demonstrated that this mutation, as well as the E255K mutation previously identified in an imatinib resistant patient 41, resulted in a constitutively active kinase resistant to inhibition by imatinib in vitro 42. Notably, the T315I mutation also confers resistance to the novel bcr-abl inhibitor nilotinib and the src inhibitor dasatinib that have been reported to be 25-30 times more potent that imatinib against a variety of bcr-abl mutants 43. Albeit CDDO-Me at doses above 300 nM equally decreased the viability of both KBM5 and KBM5-STI cells, parental KBM5 cells appeared to be more sensitive to the growth inhibitory effects of low doses (100-300 nM) of CDDO-Me than KBM5-STI cells (57 vs 126 nM 120 h IC50), and this correlated with the increased sensitivity to cardiolipin oxidation induced by CDDO-Me (7.4 vs. 2.7-fold). In contrast, murine lymphoid Ba/F3 cells expressing wild type bcr-abl or the T315I mutant were equally sensitive to low doses of CDDO-Me, and in fact, Ba/F3 cells bearing the E255K bcr-abl mutant were more sensitive to the growth inhibitory effects of 500 nM of this agent (p<0.02). Taken together, our results indicate that molecular alterations associated with imatinib resistance, rather than imatinib resistance per se, modulate the cytotoxicity of CDDO-Me, but do not confer resistance to this agent.
Mechanistically, our data indicate that the antileukemia effects of CDDO-Me are mediated in large part by the induction of programmed cell death, which may manifest itself as apoptosis or autophagy. In addition, a moderate G1 to S cell cycle block may also contribute to the growth inhibitory effects of CDDO-Me in CML cells. In agreement with previous findings in AML, subcytotoxic doses of CDDO-Me induced a decrease in oxygen consumption in KBM5 and KBM5-STI cells, suggesting that a critical mitochondrial event is associated with the effects of this agent. At higher cytotoxic doses, CDDO-Me induced the rapid dissipation of ΔΨM, and this was accompanied by the increased generation of ROS that preceded a marked decrease in the levels of intracellular GSH. Our results suggest the possibility that a common mitochondrial target may mediate both apoptosis and autophagy, andthis is the first report to demonstrate that CDDO-Me can induce both types of programmed cell death.
The rapid dissipation of GSXm preceding the generation of ROS is in complete agreement with our previous observations in pancreatic cancer cell lines 17, and similar to our observation in AML cell lines, albeit in AML cell lines CDDO-Me did not generate ROS suggesting inherent mitochondrial differences in between AML and CML cells 20. In support of this notion, an earlier report demonstrated that bcr-abl conferred increased antoxidant capacity to CML cells 44,and Trachootham D et al. recently demonstrated that overexpression of bcr-abl in hematopoietic cells caused elevation of ROS levels rendering these cells susceptible to apoptosis induction by agents that inactivated antioxidant defenses 45. Taken together the above suggest that CDDO-Me may target bcr-abl expressing CML cells via its ability to decrease GSXm and GSH. Further studies are ongoing to investigate the mechanism of CDDO-Me induced GSXm depletion in CML cells, and how this effect contributes to apoptosis induction by this class of agents. Additionally, the role of mitochondrial respiration in modulating apoptosis or autophagy in the context of CDDO-Me is being investigated.
In summary, our findings indicate the potential clinical utility of CDDO-Me in CML, regardless of bcr-abl mutational status. Notably, Phase I clinical trials of this oral agent are currently ongoing at M. D. Anderson Cancer Center in patients with solid tumors, with first indications of clinical activity and notable lack of toxicity, in particular of any cardiotoxicity (46). The elucidation of the mechanism of CDDO-Me-induced cell killing will provide useful information to optimize anti-leukemic strategies targeting CML (especially among refractory cases) in upcoming clinical trials using this novel triterpenoid.
Acknowledgements
We would like to thank Rose Lauzon, Leslie Calvert, and Elena Vess for administrative assistance.
This research was supported by Leukemia and Lymphoma Society (M.K.) and the National Institutes of Health RO1 CA089346 (M.A.)
References List
- 1.Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004;103:4010–22. doi: 10.1182/blood-2003-12-4111. [DOI] [PubMed] [Google Scholar]
- 2.Mizuchi D, Kurosu T, Kida A, et al. BCR/ABL activates Rap1 and B-Raf to stimulate the MEK/Erk signaling pathway in hematopoietic cells. Biochem Biophys Res Commun. 2005;326:645–51. doi: 10.1016/j.bbrc.2004.11.086. [DOI] [PubMed] [Google Scholar]
- 3.Jain SK, Langdon WY, Varticovski L. Tyrosine phosphorylation of p120cbl in BCR/abl transformed hematopoietic cells mediates enhanced association with phosphatidylinositol 3-kinase. Oncogene. 1997;14:2217–28. doi: 10.1038/sj.onc.1201049. [DOI] [PubMed] [Google Scholar]
- 4.Danial NN, Rothman P. JAK-STAT signaling activated by Abl oncogenes. Oncogene. 2000;19:2523–31. doi: 10.1038/sj.onc.1203484. [DOI] [PubMed] [Google Scholar]
- 5.Steelman LS, Pohnert SC, Shelton JG, et al. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18:189–218. doi: 10.1038/sj.leu.2403241. [DOI] [PubMed] [Google Scholar]
- 6.Mauro MJ, Druker BJ. STI571: targeting BCR-ABL as therapy for CML. Oncologist. 2001;6:233–8. doi: 10.1634/theoncologist.6-3-233. [DOI] [PubMed] [Google Scholar]
- 7.Kantarjian H, O’Brien S, Cortes J, et al. Survival advantage with imatinib mesylate therapy in chronic-phase chronic myelogenous ;eukemia (CML-CP) after IFN-alpha failure and in late CML-CP, comparison with historical controls. Clin Cancer Res. 2004;10:68–75. doi: 10.1158/1078-0432.ccr-1035-3. [DOI] [PubMed] [Google Scholar]
- 8.Hochhaus A, Hughes T. Clinical resistance to imatinib: mechanisms and implications. Hematol Oncol Clin North Am. 2004;18:641–56. doi: 10.1016/j.hoc.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 9.Lahaye T, Riehm B, Berger U, et al. Response and resistance in 300 patients with BCR-ABL-positive leukemias treated with imatinib in a single center: a 4.5-year follow-up. Cancer. 2005;103:1659–69. doi: 10.1002/cncr.20922. [DOI] [PubMed] [Google Scholar]
- 10.Quintas-Cardama A, Kantarjian H, Cortes J. Flying under the radar: the new wave of BCR-ABL inhibitors. Nat Rev Drug Discov. 2007;6:834–48. doi: 10.1038/nrd2324. [DOI] [PubMed] [Google Scholar]
- 11.Ikeda T, Sporn M, Honda T, Gribble GW, Kufe D. The novel triterpenoid CDDO and its derivatives induce apoptosis by disruption of intracellular redox balance. Cancer Res. 2003;63:5551–58. [PubMed] [Google Scholar]
- 12.Inoue S, Snowden RT, Dyer MJ, Cohen GM. CDDO induces apoptosis via the intrinsic pathway in lymphoid cells. Leukemia. 2004;18:948–52. doi: 10.1038/sj.leu.2403328. [DOI] [PubMed] [Google Scholar]
- 13.Ito Y, Pandey P, Place A, et al. The novel triterpenoid 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid induces apoptosis of human myeloid leukemia cells by a caspase-8-dependent mechanism. Cell Growth Differ. 2000;11:261–7. [PubMed] [Google Scholar]
- 14.Konopleva M, Tsao T, Estrov Z, et al. The synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces caspase-dependent and -independent apoptosis in acute myelogenous leukemia. Cancer Res. 2004;64:7927–35. doi: 10.1158/0008-5472.CAN-03-2402. [DOI] [PubMed] [Google Scholar]
- 15.Ikeda T, Nakata Y, Kimura F, et al. Induction of redox imbalance and apoptosis in multiple myeloma cells by the novel triterpenoid 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid. Mol Cancer Ther. 2004;3:39–45. [PubMed] [Google Scholar]
- 16.Ito Y, Pandey P, Sporn MB, Datta R, Kharbanda S, Kufe D. The novel triterpenoid CDDO induces apoptosis and differentiation of human osteosarcoma cells by a caspase-8 dependent mechanism. Mol Pharmacol. 2001;59:1094–99. doi: 10.1124/mol.59.5.1094. [DOI] [PubMed] [Google Scholar]
- 17.Samudio I, Konopleva M, Hail N, Jr, et al. 2-Cyano-3,12-dioxooleana-1,9-dien-28-imidazolide (CDDO-Im) directly targets mitochondrial glutathione to induce apoptosis in pancreatic cancer. J Biol Chem. 2005;280:36273–82. doi: 10.1074/jbc.M507518200. [DOI] [PubMed] [Google Scholar]
- 18.Hail N, Jr, Konopleva M, Sporn M, Lotan R, Andreeff M. Evidence supporting a role for calcium in apoptosis induction by the synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) J Biol Chem. 2004;279:11179–87. doi: 10.1074/jbc.M312758200. [DOI] [PubMed] [Google Scholar]
- 19.Konopleva M, Tsao T, Ruvolo P, et al. Novel triterpenoid CDDO-Me is a potent inducer of apoptosis and differentiation in acute myelogenous leukemia. Blood. 2002;99:326–35. doi: 10.1182/blood.v99.1.326. [DOI] [PubMed] [Google Scholar]
- 20.Samudio I, Konopleva M, Pelicano H, et al. A novel mechanism of action of methyl-2-cyano-3,12 dioxoolean-1,9 diene-28-oate (CDDO-Me): direct permeabilization of the inner mitochondrial membrane to inhibit electron transport and induce apoptosis. Mol Pharmacol. 2006;69:1182–93. doi: 10.1124/mol.105.018051. [DOI] [PubMed] [Google Scholar]
- 21.Kondo Y, Kondo S. Autophagy and cancer therapy. Autophagy. 2006;2:85–90. doi: 10.4161/auto.2.2.2463. [DOI] [PubMed] [Google Scholar]
- 22.Ricci C, Scappini B, Divoky V, et al. Mutation in the ATP-binding pocket of the ABL kinase domain in an STI571-resistant BCR/ABL-positive cell line. Cancer Res. 2002;62:5995–8. [PubMed] [Google Scholar]
- 23.Gorre ME, EllwoodYen K, Chiosis G, Rosen N, Sawyers CL. BCR-ABL point mutants isolated from patients with imatinib mesylate-resistant chronic myeloid leukemia remain sensitive to inhibitors of the BCR-ABL chaperone heat shock protein 90. Blood. 2002;100:3041–4. doi: 10.1182/blood-2002-05-1361. [DOI] [PubMed] [Google Scholar]
- 24.Beran M, Pisa P, O’Brien S, et al. Biological properties and growth in SCID mice of a new myelogenous leukemia cell line (KBM-5) derived from chronic myelogenous leukemia cells in the blastic phase. Cancer Res. 1993;53:3603–10. [PubMed] [Google Scholar]
- 25.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–80. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 26.Yu L, Wan F, Dutta S, et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci USA. 2006;103:4952–7. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fernandez-Checa JC. Redox regulation and signaling lipids in mitochondrial apoptosis. Biochem Biophys Res Commun. 2003;304:471–9. doi: 10.1016/s0006-291x(03)00619-3. [DOI] [PubMed] [Google Scholar]
- 28.Colell A, Garcia-Ruiz C, Miranda M, et al. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology. 1998;115:1541–51. doi: 10.1016/s0016-5085(98)70034-4. [DOI] [PubMed] [Google Scholar]
- 29.Asumendi A, Morales MC, Alvarez A, Arechaga J, Perez-Yarza G. Implication of mitochondria-derived ROS and cardiolipin peroxidation in N-(4-hydroxyphenyl)retinamide-induced apoptosis. Br J Cancer. 2002;86:1951–6. doi: 10.1038/sj.bjc.6600356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalvez F, Bessoule JJ, Rocchiccioli F, Manon S, Petit PX. Role of cardiolipin on tBid and tBid/Bax synergistic effects on yeast mitochondria. Cell Death Differ. 2005;12:659–67. doi: 10.1038/sj.cdd.4401585. [DOI] [PubMed] [Google Scholar]
- 31.Nakagawa Y. Initiation of apoptotic signal by the peroxidation of cardiolipin of mitochondria. Ann NY Acad Sci. 2004;1011:177–84. doi: 10.1007/978-3-662-41088-2_18. [DOI] [PubMed] [Google Scholar]
- 32.Nakagawa Y. Role of mitochondrial phospholipid hydroperoxide glutathione peroxidase (PHGPx) as a antiapoptotic factor. Biol Pharm Bull. 2004;27:956–60. doi: 10.1248/bpb.27.956. [DOI] [PubMed] [Google Scholar]
- 33.Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem J. 2000;351:183–93. doi: 10.1042/0264-6021:3510183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Umansky V, Rocha M, Breitkreutz R, et al. Glutathione is a factor of resistance of Jurkat leukemia cells to nitric oxide-mediated apoptosis. J Cell Biochem. 2000;78:578–87. doi: 10.1002/1097-4644(20000915)78:4<578::aid-jcb7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 35.Rizzardini M, Lupi M, Bernasconi S, Mangolini A, Cantoni L. Mitochondrial dysfunction and death in motor neurons exposed to the glutathione-depleting agent ethacrynic acid. J Neurol Sci. 2003;207:51–8. doi: 10.1016/s0022-510x(02)00357-x. [DOI] [PubMed] [Google Scholar]
- 36.Giles FJ, Cortes J, Jones D, Bergstrom D, Kantarjian H, Freedman SJ. MK-0457, a novel kinase inhibitor, is active in patients with chronic myeloid leukemia or acute lymphocytic leukemia with the T315I BCR-ABL mutation. Blood. 2007;109:500–2. doi: 10.1182/blood-2006-05-025049. [DOI] [PubMed] [Google Scholar]
- 37.Kantarjian H, Giles F, Wunderle L, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354:2542–51. doi: 10.1056/NEJMoa055104. [DOI] [PubMed] [Google Scholar]
- 38.Talpaz M, Shah NP, Kantarjian H, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354:2531–41. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 39.Kantarjian HM, Cortes J. New strategies in chronic myeloid leukemia. Int J Hematol. 2006;83:289–93. doi: 10.1532/IJH97.06024. [DOI] [PubMed] [Google Scholar]
- 40.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–80. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 41.Barthe C, Gharbi MJ, Lagarde V, et al. Mutation in the ATP-binding site of BCR-ABL in a patient with chronic myeloid leukaemia with increasing resistance to STI571. Br J Haematol. 2002;119:109–111. doi: 10.1046/j.1365-2141.2002.03708.x. [DOI] [PubMed] [Google Scholar]
- 42.Yamamoto M, Kurosu T, Kakihana K, Mizuchi D, Miura O. The two major imatinib resistance mutations E255K and T315I enhance the activity of BCR/ABL fusion kinase. Biochem Biophys Res Commun. 2004;319:1272–5. doi: 10.1016/j.bbrc.2004.05.113. [DOI] [PubMed] [Google Scholar]
- 43.O’Hare T, Walters DK, Stoffregen EP, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65:4500–05. doi: 10.1158/0008-5472.CAN-05-0259. [DOI] [PubMed] [Google Scholar]
- 44.Mayerhofer M, Florian S, Krauth MT, et al. Identification of heme oxygenase-1 as a novel BCR/ABL-dependent survival factor in chronic myeloid leukemia. Cancer Res. 2004;64:3148–54. doi: 10.1158/0008-5472.can-03-1200. [DOI] [PubMed] [Google Scholar]
- 45.Trachootham D, Zhou Y, Zhang H, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–52. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 46.Hong D, Kurzrock R, Supko JG, et al. Phase I trial with a novel orally administered synthetic triterpenoid RTA 402 (CDDO-Me) in patients with solid tumors and lymphoid malignancies; Proceedings of the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics; San Francisco, CA. 2007 October 22-26; p. 188. Abstract B82. [Google Scholar]