

Abstract
The epidermal growth factor (EGF)/EGF-receptor (ErbB1-4) family is involved in the biology of multiple myeloma (MM). In particular, ErbB-specific inhibitors induce strong apoptosis of myeloma cells (MMC) in vitro. To delineate the contribution of the 10 EGF-family ligands to the pathogenesis of MM, we have assessed their expression and biological activity. Comparing Affymetrix DNA-microarray-expression-profiles of CD138-purified plasma-cells from 65 MM-patients and 7 normal individuals to those of plasmablasts and B-cells, we found 5/10 EGF-family genes to be expressed in MMC. Neuregulin-2 and neuregulin-3 were expressed by MMC only, while neuregulin-1, amphiregulin and TGF-α were expressed by both MMC and normal plasma-cells. Using real-time PCR, we found HB-EGF, amphiregulin, neuregulin-1 and epiregulin to be expressed by cells from the BM-environment. Only the EGF-members able to bind heparan-sulphate proteoglycans (HSPGs) – neuregulin-1, amphiregulin, HB-EGF – promote the growth of MMC. Those ligands strongly bind MMC through HSPGs. The binding and the MMC growth activity was abrogated by heparitinase, heparin or deletion of the HS-binding domain. The number of HS-binding EGF ligand molecules bound to MMC was higher than 105 molecules/cell and paralleled that of syndecan-1. Syndecan-1, the main HSPG present on MM cells, likely concentrates high levels of HS-binding-EGF-ligands at the cell membrane and facilitates ErbB-activation. Altogether, our data further identify EGF-signalling as promising target for MM-therapy.
Keywords: B-Lymphocytes; metabolism; Cell Proliferation; Epidermal Growth Factor; genetics; metabolism; Flow Cytometry; Gene Expression; Gene Expression Profiling; Hematopoietic Stem Cells; metabolism; Heparan Sulfate Proteoglycans; metabolism; Humans; Ligands; Middle Aged; Multiple Myeloma; metabolism; Oligonucleotide Array Sequence Analysis; Plasma Cells; metabolism; RNA, Messenger; analysis; Receptor; Epidermal Growth Factor; genetics; metabolism; Reverse Transcriptase Polymerase Chain Reaction; Signal Transduction; physiology; Syndecan-1; metabolism
Introduction
Multiple myeloma (MM) is a hematological malignancy characterized by the accumulation of malignant plasma cells in the bone marrow (BM). Despite recent therapeutic advances, the disease remains incurable with a median survival of approximately 3–4 years (Kumar et al., 2003). MM cells (MMC) are dependent on several growth factors and cytokines, produced by the MMC themselves or by the BM microenvironment. Besides the well known MMC factors, interleukin-6 (IL-6) (Kawano et al., 1988; Klein et al., 1989) and insulin-like growth factor-1 (IGF-1) (Georgii-Hemming et al., 1996; Jelinek et al., 1997; Ferlin et al., 2000), an increasing number of additional factors are being identified, providing novel therapeutic targets for myeloma (Klein et al., 2003).
The epidermal growth factor (EGF) receptor family comprises 4 members – ErbB1 (EGFR), ErbB2, ErbB3 and ErbB4 – that are involved in the development of numerous types of human cancers. Expression and/or activation of ErbB receptors are altered in many epithelial tumors and are involved in tumor progression (Holbro et al., 2003). This has lead to the development of ErbB-specific inhibitors that are now at various stages of clinical development (Hynes & Lane, 2005). In multiple myeloma, we have previously demonstrated that MMC express ErbB receptors, and that their activation is required for in vitro survival of MMC in a majority of patients. Indeed, a pan-ErbB inhibitor induced strong apoptosis of MMC cultured for five days with their BM environment in 71% of the patients (Mahtouk et al., 2004). When the ErbB-specific inhibitor was combined with dexamethasone or anti-IL-6 antibody, apoptosis was increased leading to an almost complete elimination of viable MMC while non-MMC were unaffected (Mahtouk et al., 2004). For two of the 10 EGF-ligands – HB-EGF and AREG – we have shown that they support the growth of MMC in cooperation with IL-6 (Mahtouk et al., 2005; Mahtouk et al., 2004). However, the significance of other EGF-family ligands in MM has yet not been elucidated.
Ten ligands have been described for ErbB receptors. They can be subdivided in 3 groups according to their specificity: the first one includes EGF, amphiregulin (AREG), and transforming growth factor-α (TGF-α) which bind to ErbB1/EGFR exclusively. The second one includes heparin-binding EGF-like growth factor (HB-EGF), betacellulin (BTC) and epiregulin (EPR) which bind to both ErbB1/EGFR and ErbB4. The last one includes the 4 neuregulins (NRG1, NRG2, NRG3 and NRG4) which bind to ErbB3 and/or ErbB4 (Harris et al., 2003). ErbB2 has no ligand but is the preferred heterodimerization partner for other ErbBs (Citri et al., 2003). A common feature to 4 out of the 10 EGF-family members (HB-EGF, AREG, NRG1 and NRG2) is their ability to bind heparin and heparan sulfate proteoglycans (HSPGs) (Higashiyama et al., 1993; Johnson & Wong, 1994; Paria et al., 1999). NRG1 and NRG2 bind HSPGs through an immunoglobulin (Ig)-like domain, located terminally to the EGF-like domain (Carraway et al., 1997; Loeb & Fischbach, 1995).
A hallmark of plasma cell differentiation is the expression of the syndecan-1 HSPG at a high density. Syndecan-1 is expressed on normal and malignant plasma cells (PC) (Wijdenes et al., 1996) and is now widely used to identify and purify PC (Costes et al., 1999; Sun et al., 1997). Syndecan-1 is found predominantly on epithelial cells. It is involved in several cellular processes, which rely on interactions with extra-cellular matrix proteins, growth factors, chemokines and adhesion molecules (Couchman, 2003; Rapraeger, 2000). In myeloma, syndecan-1 has been shown to colocalize with growth factors in the uropods of MMC (Borset et al., 2000), and to promote hepatocyte growth factor (HGF) activity on MMC (Derksen et al., 2002).
To delineate the contribution of all 10 EGF-family members to the pathogenesis of MM, we studied here their expression and biological activity in MMC. We found 5/10 EGF-family genes to be expressed in MMC compared to normal BMPC, plasmablastic cells (PPC) and B cells, 5/10 genes to be expressed in some human myeloma cell lines (HMCLs) and 4/10 genes to be expressed by cells from the BM-environment. Among all EGF-ligands, only those able to bind HSPGs can promote the growth of MMC. In particular, we show for the first time that NRG1 is a growth factor for MMC. We show evidence that HSPGs, mainly syndecan-1, concentrate high levels of HS-binding-EGF-ligands at the MMC membrane, in the proximity of ErbB-receptors. This HSPG-dependant accumulation of EGF-family members is required for their MMC growth activity.
Results
Gene expression profile of EGF-family members in purified myeloma cells
Gene expression profile of the 10 EGF-family members was evaluated with U133A+B Affymetrix microarrays. Two genes (NRG2 and NRG3) were overexpressed in MMC compared to B cells, PPC, and BMPC (Figure 1A). The median NRG2 expression in myeloma cells was 3-, 6.5-, and 10-fold higher than that in BMPC (P = .031), PPC (P = .002), or B cells (P < 10−4), respectively (Figure 1A). The median NRG3 expression in myeloma cells was 5-, 40-, and 80-fold higher than that in BMPC (P = .016), PPC (P < 10−4), or B cells (P < 10−4), respectively (Figure 1A). Furthermore, NRG2 and NRG3 had an Affymetrix “present call” in MMC exclusively. The call (“present” or “absent”) is determined by the GCOS-software and indicates whether a gene is reliably expressed or not (Liu et al., 2002). NRG2 was “present” in 26% (16/65) of the myeloma samples and NRG3 in 58% (39/65) of them, and they were both “absent” in all B cell, BMPC and PPC (Figure 1A). Two other genes (NRG1 and TGF-α) were overexpressed in MMC and BMPC compared to B cells and PPC (Figure 1A). Although the median NRG1 expression in MMC was not statistically significantly different to that found in B cells, PPC or BMPC, NRG1 was present in 28% (18/65) of the myeloma samples and 43% (3/7) of the BMPC samples but was “absent” in all B cells and PPC. NRG1 expression in MMC + BMPC was statistically significantly higher than that found in B cells + PPC (p = .05). TGF-α was detected in 72% of myeloma samples, and was statistically significantly overexpressed in MMC (median value = 67) compared to B cells (P = .007) and PPC (P < 10−4). TGF-α was also detected in 6/7 BMPC samples with a median value similar to that of MMC. Regarding HMCLs (n=20), the median expression levels of NRG2, NRG3, NRG1 and TGF-α were lower than those found in MMC (Figure 1A). As for the MMC, their expression was heterogeneous: NRG2, NRG3, NRG1 and TGF-α had a “present” call in 15%, 15%, 10% and 60% of the HMCLs, respectively (Figure 1A). In a previous study (Mahtouk et al., 2005), we also found that AREG was overexpressed in MMC compared to B cells, PPC and BMPC. AREG was expressed in BMPC but at a significant lower level than in MMC and was not expressed in HMCLs (data not shown). Of note, 65/65 patients expressed at least one of the five EGF-family ligands – AREG, NRG1, NRG2, NRG3 and TGF-α – in MMC. 5/65 patients expressed only one of the 5 ligands, 16/65 two of them, 32/65 three of them, 10/65 four of them, and 2/65 all the 5 ligands. There was no significant difference in NRG1, NRG2, NRG3 or TGF-α expression between MMC of patients with stage I, II or III MM (Figure 1A). The expression of NRG1, NRG2, NRG3 and TGF-α was validated by real-time RT-PCR on 4 B cell samples, 7 PPC samples, 3 BMPC samples, 7 MMC samples and the 20 HMCLs (Figure 1B).
Figure 1. Overexpression of NRG1, NRG2, NRG3 and TGF-α in multiple myeloma.
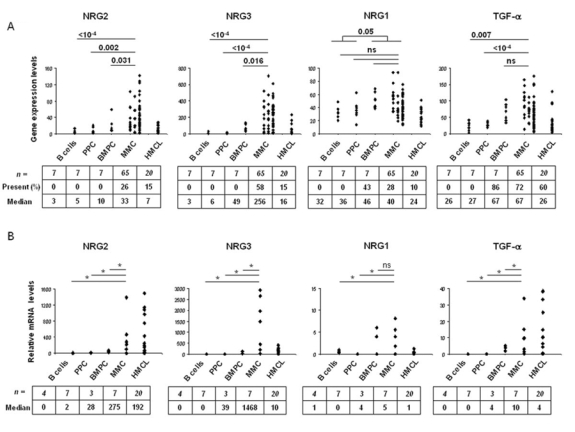
A. Gene expression profiles were determined in B cells, PPC, BMPC, MMC of 65 patients and 20 human myeloma cell lines (HMCLs). Data from MM patients are presented with 3 columns. The left, middle and right columns correspond to MMC from patients at stage I, II or III, respectively. Statistical comparisons were made with a Mann-Whitney test. p values are indicated, “ns” indicates that differences between samples are “not significant” (P >.05). Percentages of presence and median values are indicated under each graphic. B. Real-time RT-PCR analysis was completed as decribed in material and methods. * indicates that differences between samples are significant (P ≤ .05). Median values are indicated under each graphic.
Other 5 EGF family members – HB-EGF, EGF, EPR, BTC, and NRG4 – displayed a very weak expression level associated with an “absent” Affymetrix call in almost all primary MMC samples included in the study (percentage of presence < 10%) (Figure 2A). Again, real-time RT-PCR confirmed the Affymetrix data (Figure 2B). According to our previous data (Mahtouk et al., 2004), the HB-EGF gene showed a different expression pattern in HMCLs compared to MMC and was highly expressed in 8/20 HMCLs (Figure 2B). Of note, 19/20 HMCLs expressed at least one of the 10 EGF-family ligands, 9/20 HMCLs expressed one of the 10 ligands, 5/20 2 of them, 3/20 3 of them and 2/20 4 of them.
Figure 2. Gene expression profile of EGF family members.
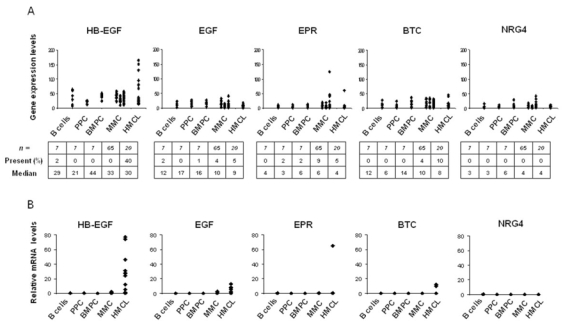
A. Gene expression profiles were determined in B cells, PPC, BMPC, MMC of 65 patients and 20 HMCLs. Data from MM patients are presented with 3 columns. The left, middle and right columns correspond to MMC from patients at stage I, II or III, respectively. Statistical comparisons were made with a Mann-Whitney test. Percentages of presence and median values calculated over all samples are indicated under each graphic. B. Real-time RT-PCR analysis was completed as described in material and methods.
Expression of EGF family members by the bone-marrow environment compared to myeloma cells
We investigated whether – besides MMC themselves – the BM microenvironment could be a source of EGF family members, thus delivering a paracrine growth signal to MMC. Using real-time RT-PCR, we looked for their relative expression in 7 samples of BM environment cells depleted from MMC (< 1% MMC, see “Material and Methods” section) compared to purified MMC. Among the 5 EGF-family members overexpressed in MMC, AREG and NRG1 were also expressed by the BM environment with a median value of 21 and 8, respectively (Figure 3A). NRG2, NRG3 and TGF-α were weakly expressed by the BM environment compared to MMC. Regarding the other 5 EGF family members, EPR and HB-EGF were highly expressed by the BM environment compared to MMC (Figure 3B). EGF, BTC and NRG4 were weakly or not expressed both in the BM environment and in MMC (Figure 3C).
Figure 3. EGF-family members mRNA quantification in the tumor environment compared to myeloma cells.

Real-time RT-PCR was performed on RNA samples isolated from primary MMC and BM mononuclear cells depleted of myeloma cells (BM) from 7 patients. Real-time RT-PCR analysis was made as described in Materials and Methods. Bars indicate median values.
Only HS-binding EGF-family members have a myeloma cell growth activity
To clarify the involvement of EGF family members in myeloma, we looked for their effect on MMC growth. We used 2 IL-6 dependant HMCLs (XG-7, XG-12) that express all 4 ErbB receptors, and the XG-5 HMCL that lacks ErbB4 (Mahtouk et al., 2005). HMCLs were cultured at a low cell density to limit the biological activity of autocrine EGF-ligands produced by the HMCLs themselves. This is the case for XG-7 cells that express HB-EGF, and whose spontaneous proliferation when cultured at a concentration of 5×105 cells/ml is inhibited by a pan-ErbB inhibitor (data not shown). Regarding ErbB1-specific ligands, AREG increased the growth of the 3 HMCLs (P < .05), which confirms our previous data (Mahtouk et al., 2005). No stimulatory effect was found with EGF or TGF-α, in spite of the expression of their specific receptor (Figure 4A). Regarding ErbB1/4-specific ligands, HB-EGF was also a potent myeloma cell growth factor (P < .05) in agreement with our previous observations (Mahtouk et al., 2004). In contrast, neither BTC nor EPR could stimulate myeloma cell growth (Figure 4A). Regarding ErbB3/4-specific ligands, NRG1 increased the growth of the XG-5, XG-7 and XG-12 HMCLs 18-, 5.2- and 4.7-fold, respectively (P < .05). The effect of NRG2-4 could not be investigated because the recombinant growth factors are not available commercially. AREG, HB-EGF and NRG1 also protected myeloma cells from apoptosis induced by IL-6 deprivation (Figure 4B). Again, no effect was found with EGF (Figure 4B), BTC, EPR and TGF-α (data not shown). Finally, AREG, HB-EGF and NRG1 dramatically promoted the proliferation of myeloma cells, 26-, 28-, and 22-fold, respectively (Figure 4C). As expected, a pan-ErbB inhibitor (PD169540) reversed the HS-binding EGF-ligands-induced proliferation of XG-7 cells (Figure 4C). The inhibitor was used at 1-μM concentration, which is known to inhibit specifically ErbB1, ErbB2, and ErbB4 kinase activities, without affecting a large panel of other kinases (Mahtouk et al., 2004). EGF (Figure 4C), BTC, EPR and TGF-α (data not shown) had no effect on myeloma cell proliferation. These data indicate that the ability to promote myeloma cell growth is not a common feature of all members of the EGF family. Only HS-binding EGF-ligands can enhance survival and proliferation of IL-6-dependent HMCLs.
Figure 4. Only heparan-sulfate binding EGF-family members have a myeloma cell growth factor activity.
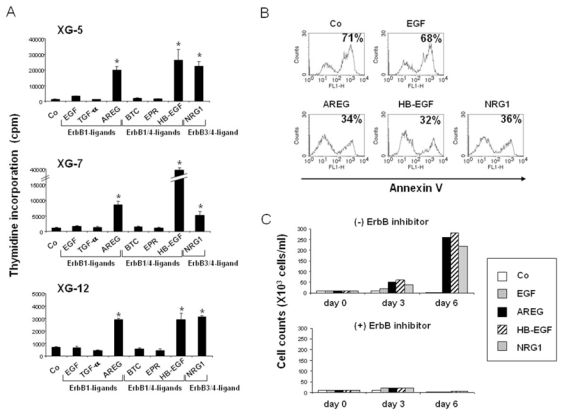
A. XG-5, XG-7 and XG-12 cells were IL-6-starved for 3 h and cultured in RPMI1640 culture medium and 5% FCS with 5 pg/ml IL-6, with or without 1 μg/ml of recombinant EGF, TGF-α, AREG, BTC, EPR, HB-EGF or NRG1. Cells were cultured for 6 days and pulsed for 12 h with tritiated thymidine at the end of the culture. Data are means ± SD of the tritiated thymidine incorporation determined on sixplicate culture wells and are those of one experiment representative of three. * Indicates that the mean value is statistically significantly different from that obtained without EGF family proteins (Co), using a Student t-test (P ≤ .05). B. XG-7 cells were IL-6-starved for 3 h and cultured in RPMI1640 culture medium and 5% FCS with 5 pg/ml IL-6, with or without 1 μg/ml of recombinant EGF, AREG, HB-EGF or NRG1. Cells were cultured for 6 days and stained with FITC-annexin V to determine the percentage of apoptotic cells. Results are of one experiment representative of three. C. Cells were cultured for 6 days, with or without a pan-ErbB inhibitor (PD169540), and were counted at day 3 and day 6 of the culture. Data are of one experiment representative of three.
Myeloma cells bind high levels of HS-binding EGF-family members through membrane HSPG
HB-EGF, AREG and NRG1 have a heparan sulphate (HS)-binding domain in contrast to EGF, TGF-α, BTC and EPR. This suggests that interaction of EGF-family members with cell surface HSPGs might be required to promote their MMC growth activity. To explore this hypothesis, we looked for the ability of MMC to bind EGF-ligands. XG-7 cells were preincubated with saturating concentrations of recombinant AREG, HB-EGF, NRG1 or EGF, washed, and stained with corresponding specific antibodies. As shown in figure 5A, a strong labelling of MMC was found with anti-AREG, anti-HB-EGF or anti-NRG1 antibodies when MMC were preincubated with either AREG or HB-EGF or NRG1 — mean fluorescence intensities (MFI) of respectively 1530, 1840 and 560 —, suggesting that MMC could bind high amounts of these EGF-family ligands. No staining of MMC was detected with an anti-EGF antibody when MMC were preincubated with EGF (data not shown). AREG, HB-EGF, or NRG1 binding to MMC was dependant on HS chains since it was abrogated by pre-treatment with heparitinase, an enzyme that cleaves heparan-sulfate chains of HSPGs (Figure 5A). Heparitinase did not affect syndecan-1 protein expression (Figure 5B). The binding of the three HS-binding EGF-family ligands to MMC was also abrogated by heparin (Figure 5C). These data indicate that a HSPG present on MMC membrane can bind large amounts of HS-binding EGF family ligands.
Figure 5. Myeloma cells bind HS-binding EGF-ligands through heparan-sulfate chains.
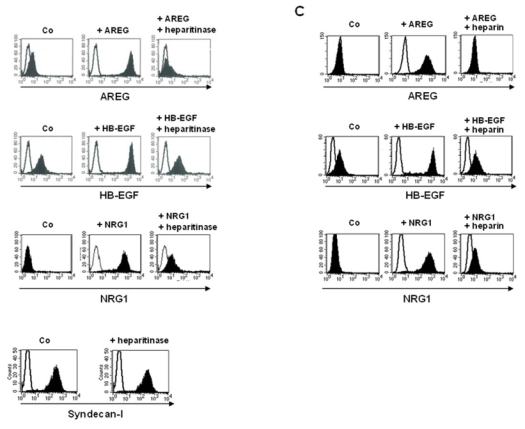
A. Cells were incubated with or without (Co) 1 μg/ml of AREG or HB-EGF or NRG1, washed, and stained with corresponding antibodies (black histogram) or isotype control (open histogram), followed by PE-conjugated secondary antibody. When indicated, cells were pretreated with 10 mU/ml of heparitinase for 1 hour. The fluorescence was determined using a FACSCalibur cytometer. B. Surface expression of syndecan-1 was determined on XG-7 cells with a PE-conjugated anti-CD138 antibody (black histogram) and a PE-isotype control (open histogram), before and after treatment with 10 mU/ml heparitinase for 1 hour. C. Cells were incubated with or without (Co) 1 μg/ml of AREG or HB-EGF or NRG1, pre-incubated with 4IU/ml heparin for one hour when indicated, washed, and stained with corresponding antibodies (black histogram) or isotype control (open histogram), followed by PE-conjugated secondary antibody. All results are those of one experiment representative of five.
Syndecan-1 is the main membrane HSPG on MMC able to bind HS-binding EGF members
Ten membrane HSPGs have been identified up to now: 4 members of the syndecan family and 6 members of the glypican one. We assessed their gene expression profile using Affymetrix U133A+B microarrays. The data are summarized in Table 1. Syndecan-1 was highly expressed and displayed a “present” Affymetrix call in all MMC and BMPC samples and in 19/20 (95%) HMCLs but was “absent” in PPC and B cells, according to our previous data (Tarte et al., 2002). Other HSPG genes, including syndecan-2, -3 and -4 and the six glypican genes, were not expressed (“absent” call) by most myeloma cell samples, and displayed a very weak signal compared to syndecan-1. This lack of expression cannot be attributed to a failed detection by a non-functional Affymetrix probeset, as a high expression of the 10 HSPG genes could be detected on various normal tissues according to the data available (Su et al., 2004) (data not shown). A further HSPG, the variant v3 of CD44, was very weakly expressed on HMCLs (MFI ranging from 5 to 15, data not shown), in agreement with previous reports (Derksen et al., 2002; Van Driel et al., 2002). Thus, syndecan-1 is the major HSPG present on the surface of plasma cells.
Table 1.
Gene expression profile of heparan-sulfate proteoglycans.
| B cells (n=7) | PPCs (n=7) | BMPCs (n=7) | MMC (n=65) | HMCLs (n=20) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Median | P (%) | Median | P (%) | Median | P (%) | Median | P (%) | Median | P (%) |
| Syndecan-1 | 19 | 0 | 29 | 0 | 1126 | 100 | 1286 | 100 | 347 | 95 |
| Syndecan-2 | 19 | 0 | 4 | 0 | 4 | 0 | 22 | 0 | 2 | 1 |
| Syndecan-3 | 3 | 0 | 3 | 0 | 3 | 0 | 10 | 0 | 3 | 0 |
| Syndecan-4 | 69 | 29 | 54 | 14 | 54 | 3 | 55 | 14 | 39 | 2 |
| Glypican-1 | 10 | 0 | 13 | 0 | 18 | 0 | 17 | 0 | 11 | 0 |
| Glypican-2 | 36 | 0 | 32 | 0 | 32 | 0 | 67 | 0 | 28 | 4 |
| Glypican-3 | 30 | 0 | 22 | 0 | 22 | 8 | 37 | 0 | 16 | 0 |
| Glypican-4 | 11 | 0 | 14 | 0 | 14 | 2 | 26 | 0 | 15 | 2 |
| Glypican-5 | 2 | 0 | 4 | 0 | 4 | 0 | 3 | 0 | 2 | 0 |
| Glypican-6 | 7 | 0 | 4 | 0 | 4 | 9 | 7 | 0 | 7 | 3 |
Gene expression profile of syndecan-1-4 and glypican-1-6 was determined with Affymetrix U133A+B DNA microarrays in seven B cell samples, seven PPC samples, seven BMPC samples, purified malignant plasma cells of 65 patients with MM (MMC) and 20 HMCLs. The median expression level and the percentage of samples that display a “present” call (P (%)) according to the Affymetrix definition are indicated for each gene.
To further investigate the role of syndecan-1 for the binding of EGF members, we quantified the number of recombinant AREG molecules that could be bound by MMC. Data are shown in Table 2. As expected from the high MFI found in Figure 5A, very large numbers of AREG molecules (≥ 105 molecules/cell) were bound to the membrane of the 3 HMCLs. In addition, the numbers of bound AREG molecules paralleled those of syndecan-1 present on the surface of MMC. In agreement with the data presented in Figure 5B, binding was abrogated by pre-treatment with heparin. As we failed to down-regulate syndecan-1 expression and the binding of EGF-ligands to MMC using syndecan-1 specific siRNA (this can be explained by the very high syndecan-1 expression level), we used XG-10, a HMCL that does not express syndecan-1, as a control (see table 1 and Figure 6A). As shown in Figure 6B and Table 2, XG-10 cells were unable to bind large levels of AREG, HB-EGF or NRG1. In addition, XG-10 cells were not stimulated by HB-EGF or NRG1 although they expressed ErbB4 (Mahtouk et al., 2005) (results not shown). Altogether, these data indicate that syndecan-1 is the main membrane HSPG on MMC able to bind HS-binding EGF members.
Table 2.
Number of syndecan-1 molecules expressed and AREG molecules bound on myeloma cells.
| Syndecan-1a (molecules/cell) | AREGb (molecules/cell) | AREG + heparinb (molecules/cell) | |
|---|---|---|---|
| XG-5 | 158097 (+/− 9660) | 213902 (+/−13679) | 76 (+/− 50) |
| XG-7 | 168479 (+/− 17713) | 172241 (+/− 20814) | 229 (+/− 224) |
| XG-12 | 592758 (+/− 13505) | 431487(+/−30418) | 511 (+/− 338) |
| XG-10 | 100(+/−29) | 94(+/−16) | 74(+/−21) |
Cells were labelled with the MI15 anti-syndecan-1 MoAb.
Cells were incubated with 1 μg/ml of AREG (pre-incubated without or with 4IU/ml heparin), washed and labelled with an AREG-specific mouse MoAb. The quantification of molecule numbers bound to cell membrane was done with QIFIKIT (DakoCytomation), using a set of calibrating beads precoated with an average of 0, 3,400, 15,000, 69,000, 206,000, and 591,000 mouse IgG molecules/bead. Data are means (+/− SD) of the number of molecules determined on three independent experiments.
Figure 6. Syndecan-1 negative myeloma cells are not able to bind EGF-family members.
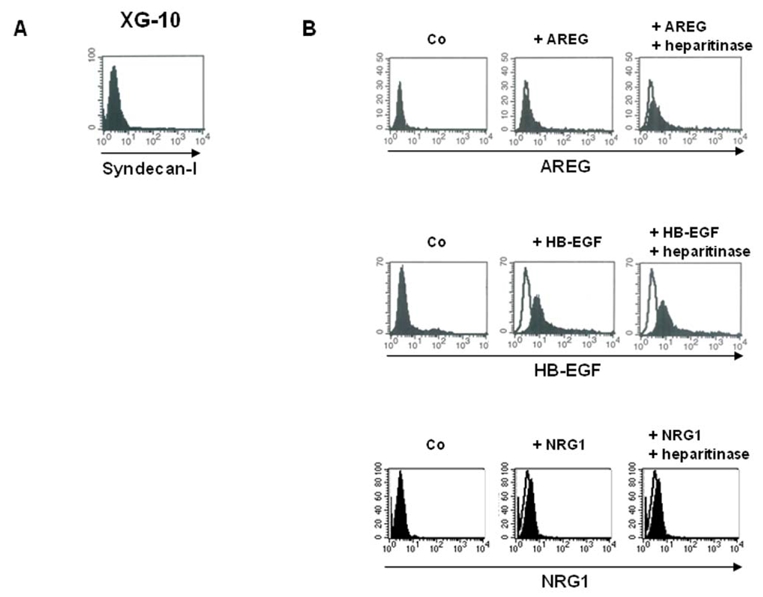
A. Surface expression of syndecan-1 was determined on the XG-10 myeloma cell line with a PE-conjugated anti-CD138 antibody (black histogram) and a PE-isotype control (open histogram). B. XG-10 cells were incubated with or without (Co) 1 μg/ml of AREG or HB-EGF or NRG1, washed, and stained with the corresponding antibodies (black histogram) or isotype control (open histogram), followed by PEconjugated secondary antibody. When indicated, cells were pretreated with 10 mU/ml of heparitinase for 1 hour. The fluorescence was determined using a FACSCalibur cytometer. Results are those of one experiment representative of five.
The binding of EGF-family members to myeloma cell HSPG is required for their myeloma cell growth activity
To demonstrate that interaction of HS-binding EGF-family members with HSPGs is a prerequisite for their myeloma cell growth activity, we used two proof elements. First, heparin, used as a competitor of membrane syndecan-1, completely abolished the MMC growth promoted by AREG, HB-EGF or NRG (Figure 7A), as well as it abolished the binding of the growth factor to the MMC membrane (Figure 5C). The inhibitory effect of heparin was dose-dependant. A significant 40% inhibition was found with 0.1 IU/ml (P ≤ .05), a 88% inhibition with 1 IU/ml, and a complete inhibition (99% inhibition) was obtained with 10–100 IU/ml (Figure 7B). Secondly, we used a feature of NRG1 that distinguishes it from HB-EGF and AREG. The NRG1 HS-binding domain (Ig-like domain) is separated from the ErbB binding domain (EGF-like domain) by a spacer region (Loeb & Fischbach, 1995). This makes it possible to obtain 2 forms of recombinant NRG1, one including the Ig-like domain (NRG1), and one without it (Ig−-NRG1). Of interest, Ig−-NRG1 had no effect on XG-5, XG-7, or XG-12 HMCL contrary to NRG1 (Figure 7C). As expected, no binding of recombinant NRG1 devoid of the HS-binding Ig domain (Ig−-NRG1) could be found on XG-7 cells, whereas a strong labelling was obtained with the HS-binding NRG1 (Figure 7D).
Figure 7. Binding of EGF-family members to HSPG is required for their myeloma cell growth factor activity.
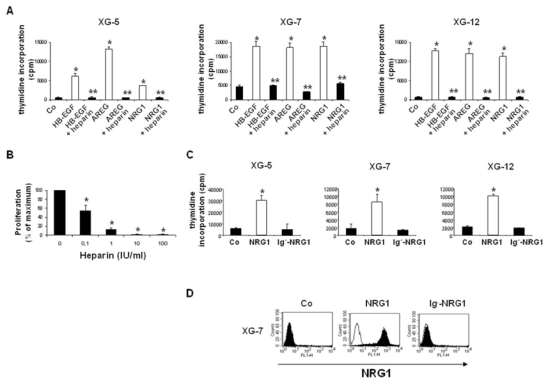
A. HMCLs were cultured with 5 pg/ml IL-6, with or without 1 μg of recombinant HB-EGF, AREG or NRG1. When indicated, growth factors were pre-incubated for one hour with 4IU/ml heparin at 4°C. * Indicates that the mean value is statistically significantly different from that obtained without growth factor, using a Student t-test (P ≤ .05). ** Indicates that the mean value is statistically significantly different from that obtained without heparin, using a Student t-test (P ≤ .05). B. XG-7 cells were cultured with 5 pg/ml IL-6 and 1 μg of recombinant HB-EGF, pre-incubated for one hour at 4°C with different concentrations of heparin. * Indicates that the mean value is statistically significantly different from that obtained without adding heparin, using a Student t-test (P ≤ .05). C. HMCLs cultured with 5 pg/ml IL-6, with or without 1 μg of recombinant NRG-1 or Ig--NRG1. * Indicates that the mean value is statistically significantly different from that obtained without adding NRG1 or Ig--NRG1, using a Student t-test (P ≤ .05). In all experiments, data are means ± SD of the tritiated thymidine incorporation determined on sixplicate culture wells and are those of one experiment representative of three. D. XG-7 cells were incubated with or without (Co) 1 μg/ml of recombinant NRG1 or Ig−-NRG1, washed, and stained with anti-NRG-1 antibody (black histogram), or isotype control (open histogram), followed by FITC-conjugated secondary antibody, and analyzed by FACS. Results are those of one experiment representative of three.
Discussion
We have previously shown that EGF-family receptors are frequently expressed on MMC (Mahtouk et al., 2005) and that their activation is required for survival of MMC cultured in vitro with their BM environment (Mahtouk et al., 2004). We have shown that 2 members of the EGF family - AREG and HB-EGF - can support the growth of myeloma cells (Mahtouk et al., 2005; Mahtouk et al., 2004). The aim of the current study was to examine the global significance of all EGF-family members in MM. Using Affymetrix microarrays and real-time RT-PCR validations, we first provide a global picture of the expression of the 10 EGF-family members in MMC and throughout PC differentiation. Data are summarized in figure 8. We show here that 7 of 10 EGF-family members are expressed by MMC and/or by cells from the BM environment. Five genes - AREG, TGF- α, NRG1, NRG2 and NRG3 - are expressed by primary MMC. Among them, two – NRG2 and NRG3 – are “myeloma genes”, i.e they are significantly overexpressed in MMC compared to B cells, PPC and BMPC, and display an Affymetrix “present call” in MMC exclusively. Three other genes – AREG, TGF- α and NRG1 – are “plasma cell genes”, i.e they are expressed both in normal and malignant PC but not in B cells and PPC. Thus they are induced during late plasma cell differentiation, and their expression on MMC is – at least in part – the reflection of their normal counterpart. In addition, 4 EGF-family member genes (HB-EGF, AREG, NRG1 and EPR) are expressed by the BM environment of MM patients. HB-EGF expression by the BM environment confirms our previously published data (Mahtouk et al., 2004). Five of 10 EGF-family members – TGF- α, NRG1, NRG2, NRG3 and HB-EGF – are expressed by a large panel of 20 HMCLs, indicating that autocrine activation loops may also be present in myeloma cell lines. 4 of those 5 genes – TGF-α, NRG1, NRG2 and NRG3 – are common to MMC and HMCLs (see Figure 1). Regarding AREG and HB-EGF, there is an opposite pattern between HMCLs and MMC. None of the HMCLs express AREG contrary to MMC (Mahtouk et al., 2005). HB-EGF is not expressed by MMC but is expressed by the BM microenvironment and by some HMCLs. Thus, HMCLs have acquired the ability to express growth factors, initially produced by the BM environment, and to use them as growth factors.
Figure 8. Syndecan-1, ErbB receptors and EGF-ligands are induced during plasma cell differentiation and malignant transformation.
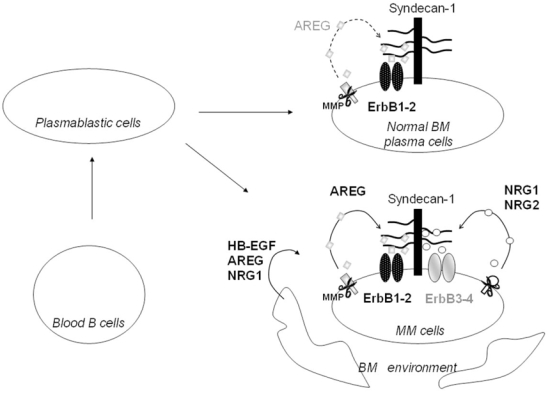
Blood B cells and plasmablastic cells neither express syndecan-1, nor ErbB receptors, nor EGF-ligands. Late plasma cell differentiation is associated with the development of a possible functional autocrine loop involving ErbB1 and ErbB2, their specific ligand AREG, and its co-receptor syndecan-1. Malignant transformation is associated with the development of an additional possible autocrine loop involving ErbB3 and ErbB4, their specific ligands NRG1-2, and their co-receptor syndecan-1. NRG1, AREG and HB-EGF are also produced by cells from the myeloma BM environment. Only the 4 ligands with a HS-binding domain, i.e that are able to promote myeloma cell growth, are represented here. MMP = metalloproteinase.
The high expression of several EGF-family members by MMC and/or the microenvironment provided the rationale to investigate their involvement in myeloma biology. We first observed that the ability to stimulate MMC growth was not a common feature to all EGF-ligands. EGF, BTC, EPER, TGF-α had no effect on MMC proliferation in spite of the expression of their specific receptors, ErbB1-4. Only the EGF family members able to bind HS chains - AREG, HB-EGF and NRG1 - could stimulate the growth of MMC, which suggested a major contribution of HSPGs for the growth factors activity of EGF-family ligands. In agreement with our previous data (Mahtouk et al., 2004), this effect was abrogated by an pan-ErbB inhibitor, indicating that the myeloma cell growth activity of HS-binding EGF-members is mediated by ErbB receptors. In the current experiments, cells were cultured at a low concentration to limit the biological activity of autocrine loops involving EGF-ligands. Indeed, we have previously shown that autocrine HB-EGF contributes to the IL-6-induced growth of some IL-6 dependant HMCLs cultured at a high cell density (De Vos et al., 2001; Wang et al., 2002; Mahtouk et al., 2004). Compiling our previous data about ErbB expression in HMCLs with those of EGF-ligand expression, we provide here a comprehensive description of autocrine loops in HMCLs. Those data are summarized in Table 3 and indicate that functional autocrine loops can be found in 10/20 HMCLs. In those 10 HMCls, at least one HS-binding EGF-ligand (i.e AREG, HB-EGF, NRG1-3) and its corresponding specific receptor are both expressed. We could not examine the function of NRG2 and NRG3 since those 2 growth factors have been little studied and recombinant proteins are not commercially available. NRG2 and NRG3 are myeloma genes and the high expression of NRG3 in MMC of some patients is intriguing. One can anticipate NRG2 to be a myeloma cell growth factor because NRG1 and NRG2 are closely related proteins that have the same specificity for ErbB receptors and have a HS-binding (Ig-like) domain (Carraway et al., 1997). NRG3 does not have this HS-binding Ig-like domain (Zhang et al., 1997) but we cannot exclude that it can also bind HSPGs. The role of NRG2 and NRG3 will deserve further studies.
Table 3.
Autocrine loops involving EGF-members and their receptors in HMCLs.
| HMCLs | ErbB1 | ErbB2/ErbB3 | ErbB4 |
|---|---|---|---|
| XG1 | NRG1 | HB-EGF/NRG1 | |
| XG2 | NRG2 | ||
| XG3 | |||
| XG4 | HB-EGF | HB-EGF | |
| XG5 | |||
| XG6 | |||
| XG7 | HB-EGF | HB-EGF | |
| XG10 | |||
| XG11 | |||
| XG12 | NRG1 | NRG1 | |
| XG13 | HB-EGF | ||
| XG14 | |||
| XG16 | NRG3 | ||
| XG19 | |||
| XG20 | HB-EGF | NRG2 | HB-EGF/NRG2/NRG3 |
| LP1 | NRG2 | NRG2 | |
| OPM2 | HB-EGF | NRG3/HB-EGF | |
| RPMI | |||
| SKMM | |||
| U266 |
Gene expression profile of EGF-ligands was determined with Affymetrix U133A+B DNA microarrays in 20 HMCLs. ErbB receptor expression was determined by real-time PCR (Mahtouk et al., 2005). Only HS-binding EGF-ligands are indicated in the table. They are also indicated only when the corresponding specific receptor (i.e ErbB1 for AREG, ErbB1 and ErbB4 for HB-EGF, ErbB2/ErbB3 and ErbB4 for NRG1 and NRG2 and ErbB4 for NRG3) is also expressed in the HMCL. ErbB3 has no transducing activity and is active only as a heterodimer with ErbB2.
The restriction of the MMC growth activity to HS-binding EGF-ligands can be explained by the binding of large amounts of these factors by MMC. The binding was abrogated when MMC were pretreated with heparitinase, that cleaves HS chains, or when growth factor were pre-incubated with heparin, demonstrating the involvement of HS chains. We demonstrate that in MMC, the binding to a membrane HSPG is absolutely required for the MMC growth activity of EGF-family members. Indeed, heparin abrogated the stimulatory effect of AREG, HB-EGF and NRG1 on MMC proliferation. In addition, a truncated form of NRG1 corresponding to the EGF domain without the HS-binding domain was unable to bind to MMC and to promote myeloma cell growth, contrary to the complete form of NRG1. This NRG1 truncated form was previously reported to be biologically active on MCF-7 cells that expressed ErbB4 (Karey & Sirbasku, 1988). Using real-time RT-PCR, we have previously shown that expression of ErbB receptors is low on MMC. However, their activation is critical for MMC biology as a pan-ErbB inhibitor (which blocks the kinase activity of all 3 ErbB receptors with transducing activity) induced dramatic apoptosis of patient’s myeloma cells cultured in vitro together with their environment and without adding exogenous growth factors (Mahtouk et al., 2004). The binding of very high numbers of HS-binding EGF-family molecules to the MMC membrane makes it possible to understand why we found a potent growth factor activity of those ligands although their specific receptors are weakly expressed. HSPG probably increase the effective concentration at the cell membrane and then facilitate ErbB activation. An additional explanation might be that HSPGs induce a conformational change in EGF-ligands which would stabilize them in an active conformation, as it was shown for the chemokine IL-8 (Goger et al., 2002). As the non HS-binding EGF-ligands have no effect on myeloma cells, one can speculate that they are unable to activate their receptors. However, as previously mentioned (Mahtouk et al., 2004), we could not directly study the phosphorylation of ErbB receptors because their low density on the MMC surface makes it difficult to immunoprecipitate them.
Several arguments suggest that syndecan-1 is the major membrane HSPG involved in the binding of EGF members: (i) among the 11 known membrane HSPG (Belting, 2003), syndecan-1 was the main one to be expressed by normal and malignant plasma cells. (ii) The number of HS-binding EGF family members bound on MMC was very large (1.7 to 4.4 × 105 molecules/cell) and was in the same range than the number of membrane syndecan-1 molecules. (iii) XG-10 myeloma cells, which express virtually no syndecan-1 molecules on the surface (around 100 molecules/cell), could not bind HB-EGF or AREG and were not stimulated by HB-EGF, in spite of ErbB4 expression (data not shown). It is noteworthy that syndecan-1, ErbB receptors and HS-binding EGF-family ligands are concomitantly expressed during normal plasma cell differentiation at the stage of plasma cells (Figure 8).
The function of syndecan-1 has been largely described in normal and tumor epithelial cells where it plays major roles, partly due to its ability to bind growth factors and chemokines (Bernfield et al., 1999; Sanderson et al., 2004). On MMC, the function of syndecan-1 is not fully elucidated yet. The soluble form of syndecan-1 is detected in the serum of patients with MM (Dhodapkar et al., 1997) and is an indicator of poor prognosis (Klein et al., 1999; Seidel et al., 2000b). Soluble syndecan-1 also accumulates within the tumor BM environment of MM patients (Bayer-Garner et al., 2001; Seidel et al., 2000a). As this soluble form of syndecan-1 is biologically active (Dhodapkar et al., 1997), one can speculate that MMC are likely “bathed” in high concentrations of EGF-family ligands (produced by MMC themselves and/or by the BM environment) that are either bound to MMC via membrane syndecan-1 or sequestrated within the extracellular matrix via soluble syndecan-1, in the proximity to the MMC. Derksen at al. reported that syndecan-1 act as a coreceptor for hepatocyte growth factor (HGF) to promote HGF/met signalling in MM cells (Derksen et al., 2002). Together with our findings, this suggests that interaction of HS-binding growth factors with syndecan-1 might be a general mechanism to promote MMC growth. Whether our present data can be extended to other myeloma growth factors is under active investigation in the laboratory. Two recent studies demonstrated that APRIL, a growth factor for myeloma cells (Moreaux et al., 2004), can bind HSPG (Hendriks et al., 2005; Ingold et al., 2005). One study reported that IL-6, the major known MMC growth factor (Klein et al., 1989), is a heparin-binding cytokine and that a heparin mimetic inhibits the biological activity of IL-6 and the binding to its receptors (Mummery & Rider, 2000). However, we failed to find a large binding of IL-6 molecules to MMC, which can be detected with anti-IL-6 MoAbs and FACS analysis (unpublished data).
The failing of MMC to bind HB-growth factors in the presence of heparin, and the resulting reduction of EGF-ligand-induced proliferation of MMC suggest that heparin – or its non-anti-coagulant derivatives (Kragh & Loechel, 2005 ) – could be of interest for MM treatment. Of note, the HB-EGF-induced proliferation of myeloma cells was inhibited by 46% with 0.1 IU/ml of heparin, which is in the range of therapeutic concentrations. One recent study has reported that a short-term treatment with heparin could improve the survival of patients with metastatic epithelial cancers (Klerk et al., 2005). One possible mechanism might be that heparin competes with HS for the binding of growth factors preventing their stimulatory effect on tumor cells. Taken together with our previous data showing that a pan-ErbB inhibitor induces strong apoptosis of primary MMC, the present study emphasize that EGF-signalling should be considered as a potential therapeutic target in multiple myeloma. ErbB-specific inhibitors, heparin, or a combinantion of those molecules could be of major therapeutic benefit for the treatment of MM patients.
Materials and Methods
Cell samples
MMC were purified from 65 consecutive patients with MM at diagnosis (median age, 59 years) after informed consent was given. According to Durie-Salmon classification, 12 patients were in stage IA, 12 in stage IIA, 38 in stage IIIA, and three in stage IIIB. 11 patients had IgAκ MM, 7 IgAλ MM, 26 IgGκ MM, 9 IgGλ MM, 7 Bence-Jones κ MM, 3 Bence-Jones λ MM, and 2 non-secreting MM. BMPCs were obtained from healthy donors after informed consent was given. Plasma cells were purified with anti-CD138 MACS microbeads (Miltenyi-Biotec, Paris, France). PPC were generated from purified CD19+ peripheral blood B cells in vitro as previously described (Tarte et al., 2000). BM mononuclear cells from seven patients were obtained by removing MMC with CD138 Milteny microbeads (< 1% plasma cells). Human IL-6 dependant myeloma cell lines (HMCLs) were obtained in our laboratory (Rebouissou et al., 1998; Zhang et al., 1994). They were routinely maintained in RPMI1640, 10% fetal calf serum, and 2 ng/ml of IL-6.
Reagents
Recombinant IL-6 was purchased from Abcys SA (Paris, France) and EGF-family growth factors were purchased from R&D Systems (Minneapolis, MN). The antibodies used were PE-conjugated anti-CD138 (Beckman-Coulter, Marseilles, France) and anti-EGF-family growth factors all from R&D Systems.
Microarray hybridization
RNA was extracted with the RNeasy Kit (Quiagen, Valencia, CA). Biotinylated complementary RNA (cRNA) was amplified with a double in-vitro transcription, and hybridized to the human U133 A and B GeneChip microarrays according to manufacturer’s instructions (Affymetrix). Fluorescence intensities were quantified and analyzed using the GECOS software (Affymetrix).
Real-time reverse transcriptase-polymerase chain reaction
For real-time RT-PCR, we used the assay-on-demand primers and the TaqMan Universal Master Mix from Applied Biosystems (Courtaboeuf, France) according to the manufacturer’s instructions. Real-time RT-PCR was performed using the ABI prism 7000 Sequence Detection system. Data were analyzed as previously described (Mahtouk et al., 2005) and were normalized to GAPDH for each sample. MCF-7 cells (for NRG3 and NRG4) or A431 cells (for all other genes) were used as control cell lines and were assigned the arbitrary value of 100.
Myeloma cell proliferation assay
Cells were IL-6 starved for 3 hours and cultured for 6 days in 96-well flat-bottomed microtiter plates at 104 cells/well in 200 μl of RPMI 1640 culture medium and 5% FCS, with a low concentration of IL-6 (5 pg/ml). Growth factors (1 μg) were added at the beginning of the culture in six culture wells per group. At the end of the culture, cells were pulsed with tritiated thymidine (Amersham Pharmacia Biotech, Orsay, France) for 12 h, harvested and counted as reported previously (De Vos et al., 2001).
Detection of apoptotic cells and cell counts
Myeloma cells were cultured for 3 days in 24-well flat-bottomed microtiter plates at 104 cells/well in 1 ml of RPMI 1640 medium with 5% FCS and various growth factors. Cells were counted at day 3 and 6 of the culture. At day 6, they were washed once in PBS and suspended in Annexin-V FITC solution (Boehringer, Mannheim, Germany). Fluorescence was analyzed with a FACScan flow cytometer.
Detection of growth factor binding by FACS
Cells were incubated with 1 μg/mL of HB-EGF, AREG, NRG1 or Ig−-NRG1 for one hour at 4°C and washed twice in PBS before incubation with the corresponding antibodies. The fluorescence was determined using a FACSCalibur flow-activated cytometer (Becton Dickinson). To cleave the HS chains, myeloma cells were pretreated with 10 mU/ml heparitinase (EC 4.2.2.8, Sigma, St Louis, MO, USA) for one hour at 37°C. When indicated, added recombinant EGF-ligands were pre-incubated with 4 IU/ml of heparin (Heparin Choay®, 25000 UI/5 ml) for one hour at 4°C.
Quantitative analysis of immunofluorescence staining
The quantification of syndecan-1 or AREG molecules bound to the membrane of MMC was done using the DAKO QIFIKIT (DakoCytomation, Glostrup, Denmark) according to the manufacturer’s instructions.
Statistical analysis
Gene Expression Profiles were analyzed with our bioinformatics platform (RAGE, remote analysis of microarray gene expression, http://rage.montp.inserm.fr) designed by T. Reme (INSERM U475, Montpellier, France). Statistical comparisons were made with the non-parametric Mann-Whitney test or the Student t-test.
Acknowledgments
This work was supported by grants from the Ligue Nationale Contre le Cancer (équipe labellisée), Paris, France, and from the Association Guillaume Espoir, St Genis Laval, France.
References
- Bayer-Garner IB, Sanderson RD, Dhodapkar MV, Owens RB, Wilson CS. Mod Pathol. 2001;14:1052–8. doi: 10.1038/modpathol.3880435. [DOI] [PubMed] [Google Scholar]
- Belting M. Trends Biochem Sci. 2003;28:145–51. doi: 10.1016/S0968-0004(03)00031-8. [DOI] [PubMed] [Google Scholar]
- Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, et al. Annu Rev Biochem. 1999;68:729–77. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- Borset M, Hjertner O, Yaccoby S, Epstein J, Sanderson RD. Blood. 2000;96:2528–36. [PubMed] [Google Scholar]
- Carraway KL, 3rd, Weber JL, Unger MJ, Ledesma J, Yu N, Gassmann M, et al. Nature. 1997;387:512–6. doi: 10.1038/387512a0. [DOI] [PubMed] [Google Scholar]
- Citri A, Skaria KB, Yarden Y. Exp Cell Res. 2003;284:54–65. doi: 10.1016/s0014-4827(02)00101-5. [DOI] [PubMed] [Google Scholar]
- Costes V, Magen V, Legouffe E, Durand L, Baldet P, Rossi JF, et al. Hum Pathol. 1999;30:1405–11. doi: 10.1016/s0046-8177(99)90160-0. [DOI] [PubMed] [Google Scholar]
- Couchman JR. Nat Rev Mol Cell Biol. 2003;4:926–37. doi: 10.1038/nrm1257. [DOI] [PubMed] [Google Scholar]
- De Vos J, Couderc G, Tarte K, Jourdan M, Requirand G, Delteil MC, et al. Blood. 2001;98:771–780. doi: 10.1182/blood.v98.3.771. [DOI] [PubMed] [Google Scholar]
- Derksen PW, Keehnen RM, Evers LM, van Oers MH, Spaargaren M, Pals ST. Blood. 2002;99:1405–10. doi: 10.1182/blood.v99.4.1405. [DOI] [PubMed] [Google Scholar]
- Dhodapkar MV, Kelly T, Theus A, Athota AB, Barlogie B, Sanderson RD. Br J Haematol. 1997;99:368–71. doi: 10.1046/j.1365-2141.1997.3893203.x. [DOI] [PubMed] [Google Scholar]
- Ferlin M, Noraz N, Hertogh C, Brochier J, Taylor N, Klein B. British Journal of Haematology. 2000;111:626–634. doi: 10.1046/j.1365-2141.2000.02364.x. [DOI] [PubMed] [Google Scholar]
- Georgii-Hemming P, Wiklund HJ, Ljunggren O, Nilsson K. Blood. 1996;88:2250–2258. [PubMed] [Google Scholar]
- Goger B, Halden Y, Rek A, Mosl R, Pye D, Gallagher J, et al. Biochemistry. 2002;41:1640–6. doi: 10.1021/bi011944j. [DOI] [PubMed] [Google Scholar]
- Harris RC, Chung E, Coffey RJ. Exp Cell Res. 2003;284:2–13. doi: 10.1016/s0014-4827(02)00105-2. [DOI] [PubMed] [Google Scholar]
- Hendriks J, Planelles L, de Jong-Odding J, Hardenberg G, Pals ST, Hahne M, et al. Cell Death Differ. 2005;12:637–48. doi: 10.1038/sj.cdd.4401647. [DOI] [PubMed] [Google Scholar]
- Higashiyama S, Abraham JA, Klagsbrun M. J Cell Biol. 1993;122:933–40. doi: 10.1083/jcb.122.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbro T, Civenni G, Hynes NE. Exp Cell Res. 2003;284:99–110. doi: 10.1016/s0014-4827(02)00099-x. [DOI] [PubMed] [Google Scholar]
- Hynes NE, Lane HA. Nat Rev Cancer. 2005;5:341–54. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- Ingold K, Zumsteg A, Tardivel A, Huard B, Steiner QG, Cachero TG, et al. J Exp Med. 2005;201:1375–83. doi: 10.1084/jem.20042309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinek DF, Witzig TE, Arendt BK. J Immunol. 1997;159:487–96. [PubMed] [Google Scholar]
- Johnson GR, Wong L. J Biol Chem. 1994;269:27149–54. [PubMed] [Google Scholar]
- Karey KP, Sirbasku DA. Cancer Res. 1988;48:4083–92. [PubMed] [Google Scholar]
- Kawano M, Hirano T, Matsuda T, Taga T, Horii Y, Iwato K, et al. 1988;332:83–85. doi: 10.1038/332083a0. [DOI] [PubMed] [Google Scholar]
- Klein B, Li XY, Lu ZY, Jourdan M, Tarte K, Brochier J, et al. Curr Top Microbiol Immunol. 1999;246:335–41. doi: 10.1007/978-3-642-60162-0_41. [DOI] [PubMed] [Google Scholar]
- Klein B, Tarte K, Jourdan M, Mathouk K, Moreaux J, Jourdan E, et al. Int J Hematol. 2003;78:106–13. doi: 10.1007/BF02983377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein B, Zhang XG, Jourdan M, Content J, Houssiau F, Aarden L, et al. Blood. 1989;73:517–526. [PubMed] [Google Scholar]
- Klerk CP, Smorenburg SM, Otten HM, Lensing AW, Prins MH, Piovella F, et al. J Clin Oncol. 2005;23:2130–5. doi: 10.1200/JCO.2005.03.134. [DOI] [PubMed] [Google Scholar]
- Kragh M, Loechel F. Int J Oncol. 2005;27:1159–67. [PubMed] [Google Scholar]
- Kumar A, Loughran T, Alsina M, Durie BG, Djulbegovic B. Lancet Oncol. 2003;4:293–304. doi: 10.1016/s1470-2045(03)01077-5. [DOI] [PubMed] [Google Scholar]
- Liu WM, Mei R, Di X, Ryder TB, Hubbell E, Dee S, et al. Bioinformatics. 2002;18:1593–9. doi: 10.1093/bioinformatics/18.12.1593. [DOI] [PubMed] [Google Scholar]
- Loeb JA, Fischbach GD. J Cell Biol. 1995;130:127–35. doi: 10.1083/jcb.130.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahtouk K, Hose D, Reme T, De Vos J, Jourdan M, Moreaux J, et al. Oncogene. 2005 doi: 10.1038/sj.onc.1209699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahtouk K, Jourdan M, De Vos J, Hertogh C, Fiol G, Jourdan E, et al. Blood. 2004;103:1829–37. doi: 10.1182/blood-2003-05-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreaux J, Legouffe E, Jourdan E, Quittet P, Reme T, Lugagne C, et al. Blood. 2004;103:3148–57. doi: 10.1182/blood-2003-06-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummery RS, Rider CC. J Immunol. 2000;165:5671–9. doi: 10.4049/jimmunol.165.10.5671. [DOI] [PubMed] [Google Scholar]
- Paria BC, Elenius K, Klagsbrun M, Dey SK. Development. 1999;126:1997–2005. doi: 10.1242/dev.126.9.1997. [DOI] [PubMed] [Google Scholar]
- Rapraeger AC. J Cell Biol. 2000;149:995–8. doi: 10.1083/jcb.149.5.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebouissou C, Wijdenes J, Autissier P, Tarte K, Costes V, Liautard J, et al. Blood. 1998;91:4727–4737. [PubMed] [Google Scholar]
- Sanderson RD, Yang Y, Suva LJ, Kelly T. Matrix Biol. 2004;23:341–52. doi: 10.1016/j.matbio.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Seidel C, Borset M, Hjertner O, Cao D, Abildgaard N, Hjorth-Hansen H, et al. Blood. 2000a;96:3139–46. [PubMed] [Google Scholar]
- Seidel C, Sundan A, Hjorth M, Turesson I, Dahl IM, Abildgaard N, et al. Blood. 2000b;95:388–92. [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, et al. Proc Natl Acad Sci U S A. 2004;101:6062–7. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun RX, Lu ZY, Wijdenes J, Brochier J, Hertog C, Rossi JF, et al. Journal of Immunological Methods. 1997;205:73–79. doi: 10.1016/s0022-1759(97)00056-2. [DOI] [PubMed] [Google Scholar]
- Tarte K, De Vos J, Thykjaer T, Zhan F, Fiol G, Costes V, et al. Blood. 2002;100:1113–22. [PubMed] [Google Scholar]
- Tarte K, Fiol G, Legouffe E, Rossi JF, Klein B. Blood. 2000;96:514a. [PubMed] [Google Scholar]
- Van Driel M, Gunthert U, van Kessel AC, Joling P, Stauder R, Lokhorst HM, et al. Leukemia. 2002;16:135–43. doi: 10.1038/sj.leu.2402336. [DOI] [PubMed] [Google Scholar]
- Wang YD, De Vos J, Jourdan M, Couderc G, Lu ZY, Rossi JF, et al. Oncogene. 2002;21:2584–92. doi: 10.1038/sj.onc.1205355. [DOI] [PubMed] [Google Scholar]
- Wijdenes J, Vooijs WC, Clement C, Post J, Morard F, Vita N, et al. Br J Haematol. 1996;94:318–23. doi: 10.1046/j.1365-2141.1996.d01-1811.x. [DOI] [PubMed] [Google Scholar]
- Zhang D, Sliwkowski MX, Mark M, Frantz G, Akita R, Sun Y, et al. Proc Natl Acad Sci U S A. 1997;94:9562–7. doi: 10.1073/pnas.94.18.9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XG, Gaillard JP, Robillard N, Lu ZY, Gu ZJ, Jourdan M, et al. Blood. 1994;83:3654–3663. [PubMed] [Google Scholar]