

Abstract
Trinucleotide repeats frequently expand and contract in humans and model organisms. Protein factors that modulate this process have been found by candidate gene approaches or mutant screens for increased expansion rates. To extend this effort, Saccharomyces cerevisiae mutants with higher CAG CTG repeat contraction rates were sought using a disruption library. This screen identified Mrc1, the homolog of human Claspin, which mediates the replication and DNA damage checkpoints, and also couples the replicative helicase and polymerase. Genetic analysis showed that Mrc1, along with Tof1 and Csm3, inhibits instability in two distinct ways. Contraction rates of (CAG)20 tracts are elevated by loss of Mrc1, Tof1 or Csm3, but not by defects in most replication checkpoint or DNA damage checkpoint proteins. The three proteins likely inhibit contractions primarily through their coupling activity, which would prevent accumulation of single-strand template DNA prior to the formation of aberrant secondary structure. In contrast, expansion rates of (CTG)13 are elevated in strains defective for Mrc1, Tof1, Csm3, Mec1, Ddc2, Rad24, Ddc1, Mec3, Rad17, Rad9, Rad53 or Chk1, suggesting that the DNA damage checkpoint inhibits expansions after formation of repeat-dependent structures. Together, these results indicate that at least two Mrc1-dependent mechanisms function to reduce CAG•CTG repeat instability.
Keywords: trinucleotide repeat, expansion, checkpoint, DNA damage response, replicational coupling
1. Introduction
Trinucleotide repeats (TNRs) are unstable repetitive DNA elements found in both coding and non-coding regions of numerous human genes. Expansions in specific TNRs cause at least 15 heritable neurodegenerative human diseases, including Huntington’s disease and fragile X syndrome [1, 2]. Expansion patterns follow non-Mendelian inheritance patterns in afflicted families [3], indicating that complex and unique molecular mechanisms underlie the propensity of triplet repeats to expand and contract [1, 2]. Disease-causing TNRs almost exclusively have the sequence (CNG)n, and single-stranded DNA containing these repeats readily forms secondary structures in vitro that correlate strongly with the genetic instability of these sequences in vivo [4, 5]. Furthermore, DNA polymerases in vitro [6] and replication forks in E. coli [7] and yeast [8] have difficulty synthesizing G-C rich TNRs. These and other observations led to well supported replication-based models for TNR instability in proliferating cells that are all founded on the premise that aberrant replication of the lagging strand is linked to secondary structure formation in single stranded DNA (ssDNA) [1, 2, 9]. Generation of ssDNA on the nascent strand of the Okazaki fragment may trigger hairpin formation, allowing formation of this crucial structured intermediate that ultimately yields an expansion. Similarly, generation of excess ssDNA on the template strand is thought to permit collapse into a hairpin, and aberrant synthesis past this hairpin would result in contraction on one strand. Thus, for both expansions and contractions, the availability of ssDNA at TNRs is a critical factor determining the likelihood of hairpin formation and subsequent genetic instability.
Expansions and contractions in somatic cells can exhibit differing levels of instability in various tissues [10–13], suggesting that tissue-specific trans-acting factors modulate TNR instability. In accordance with this idea, several pathways in yeast modulate TNR mutagenesis, including Okazaki fragment maturation [14–16] and post-replication repair [17, 18]. To identify novel trans factors, we performed a blind screen for S. cerevisiae mutants that increase rates of TNR contractions. This screen revealed an mrc1 mutation, suggesting that Mrc1 protein normally prevents contractions in wild-type cells. We focused on Mrc1 because of recent findings implicating it and associated proteins in limiting accumulation of ssDNA, discussed below, and also in prevention of chromosome fragility and instability in yeast with a long, disease-length (CAG)85 tract [19, 20].
Mrc1 was initially identified as a mediator of the replication checkpoint [21], which responds to stalled replication forks arising from treatment with hydroxyurea (HU). In the presence of a stalled fork, ssDNA coated with RPA stimulates the recruitment of Mec1/Ddc2 (in yeast) or ATR/ATRIP (in humans) to the replication fork [22, 23]. Mec1 phosphorylates and activates Mrc1, which recruits and facilitates the activation of the effector kinase Rad53 (Chk2 in humans). Activated Rad53 then phosphorylates a variety of downstream targets, resulting in the inhibition of late origin firing and the upregulation of genes involved in DNA repair [24]. The loss of Rad53, combined with HU treatment, leads to excess ssDNA formation at the replication fork that is detectable by electron microscopy [25]. Mrc1 is also involved in a second checkpoint, the intra-S phase DNA damage response. A number of proteins in the DNA damage response overlap with those in the checkpoint response, including Mrc1, Mec1/Ddc2, Rad53 and others [26]. This overlap may be due to damage sensing through promotion of single-strand gaps. However the DNA damage response also requires additional factors, such as the alternative clamp loader Rad24 and the alternative clamp Rad17/Mec3/Ddc1 (9-1-1 in humans) [26]. Thus phenotypes associated with defects in Rad24, Rad17, Mec3 or Ddc1 distinguish the DNA damage response from the replication checkpoint. In addition to signaling, Mrc1 also has a structural role at the replication fork that is central to normal replisome function [27]. Mrc1 functions with Tof1 and Csm3 to form the replication pausing complex, which maintains fork stability and prevents the uncoupling of helicase and polymerase activities under conditions of replication stress [28, 29]. In cells lacking Mrc1, Tof1 or Csm3, helicase activity occurs without polymerization, and leads to accumulation of excess ssDNA [30]. Thus Mrc1 is involved both in preventing accumulation of ssDNA through its structural role, and response to ssDNA via the replication checkpoint and the DNA damage response [29].
The evidence summarized above shows that genetic instability at TNRs is potentially suppressed by Mrc1 either through replicational coupling to avoid ssDNA and secondary structure formation, or to checkpoint response(s) after structure formation to reduce the likelihood of completing the mutagenic process. Previous work showed that long CAG•CTG tracts, which are disease-causing in humans, can be further destabilized by defects in the DNA damage response [19] or Mrc1 [20]. Our independent discovery of an mrc1 mutant that also destabilized shorter CAG•CTG runs, more similar to those seen in normal humans, suggested that checkpoint activities help prevent triplet repeat mutations between genetically stable, subthreshold alleles and the longer, unstable tracts that can give rise to further mutation and disease in humans. Furthermore we found that Mrc1, Tof1 and Csm3 are highly selective in protecting TNRs from instability, and that they use two distinct mechanisms to help avoid CAG•CTG repeat expansions and contractions. Together these results significantly extend what is known about Mrc1, Tof1 and Csm3 and their action at TNRs.
2. Materials and methods
2.1. Saccharomyces cerevisiae strains
Most strains used in this study were derived from BY4741 (MAT-a his3Δ1 leu2Δ0 met15Δ0 ura3Δ0), a derivative of Saccharomyces cerevisiae strain S288C (Open Biosystems). Mutants used in this study were created by targeted deletion and confirmed by PCR, and when possible, by phenotypic traits such as UV or hydroxyurea sensitivity. TNR-containing plasmids were digested and integrated into the yeast genome; single integrants were confirmed as described previously [31].
2.2. Plasmids
The pBL94 vector was used to construct all TNR-containing plasmids as described previously [32]. The dinucleotide repeat-containing plasmid, pSH44 [33], was a gift from Tom Petes, Duke University. The CEN/ARS based recovery plasmid pMRC1 and the pmrc1AQ mutant plasmid [34] were gifts from Stephen Elledge, Harvard University.
2.3. Genetic assays and molecular analysis of mutated TNR alleles
Expansion and contraction rates were measured by fluctuation analysis as described previously [31, 32] and as shown in Fig. 1. Mutation rates were calculated by the method of the median [35]. Single-colony PCR analysis of expansions and contractions was performed as previously described [32, 36], and rates were corrected by multiplying the percent bona fide expansions/contractions by the apparent mutation rates obtained by fluctuation analysis [31]. Dinucleotide mutation rates were measured as described previously [33]. Forward mutation rates for the CAN1 gene were determined by fluctuation analysis using selection for canavanine resistance. At least two independent clones were tested for all the above assays to ensure reproducibility. Statistical analyses for data shown in Table 2 were performed using the t-test (two-tailed distribution and two-sample equal variance) and P values of less than 0.05 were considered statistically significant. Any outliers were determined using the Q-test. For contraction and expansion experiments in Table 1, mutant strains were directly compared to wild-type in each experiment, and the results are expressed as fold change in rate compared to wild-type. Statistical analyses were performed using the Wilcoxon Mann Whitney test. P values of less than 0.05 were considered statistically significant.
Figure 1.
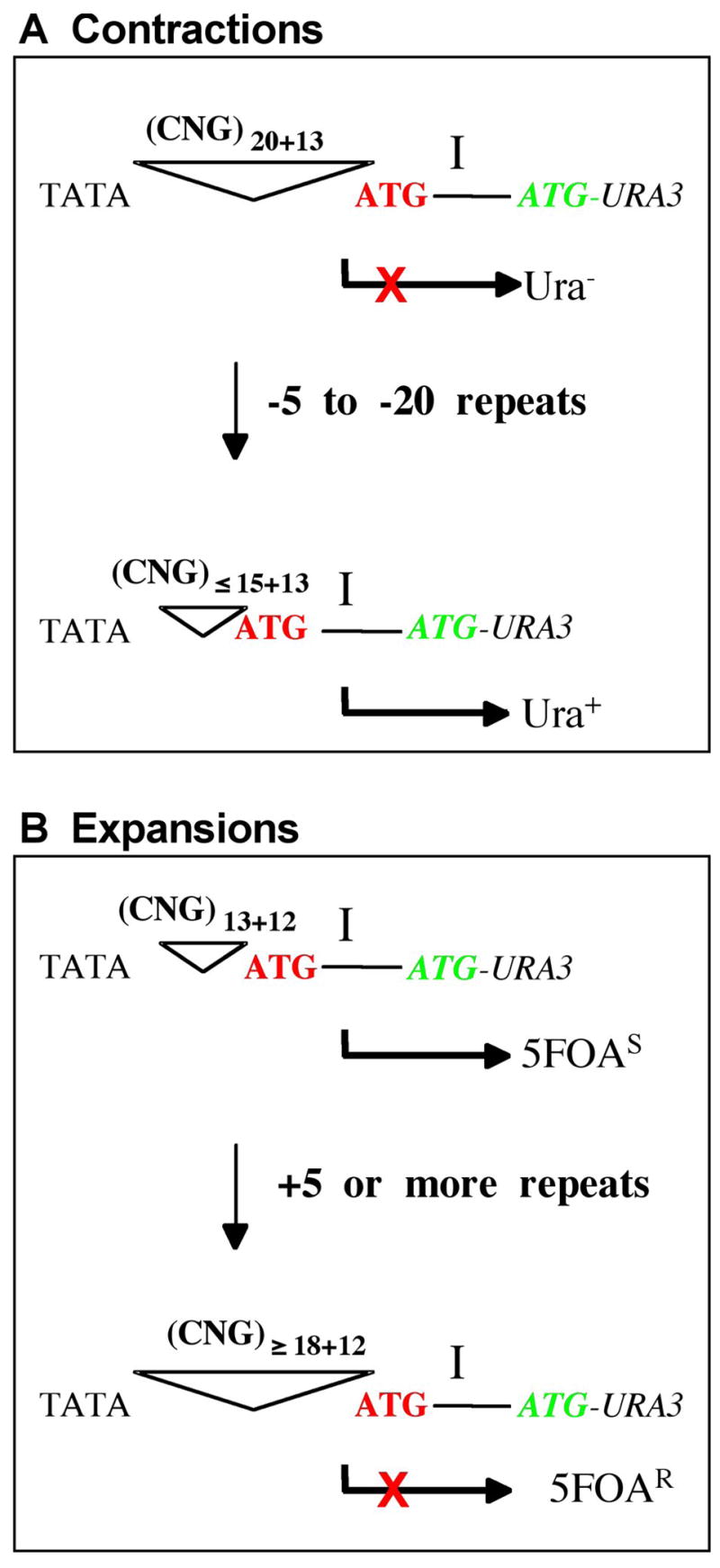
A genetic assay to monitor trinucleotide repeat (TNR) contractions and expansions in yeast. The regulatory region controlling expression of the reporter gene URA3 is shown. Important features include the TATA box, the trinucleotide region, an out-of-frame initiator codon (in red), the preferred transcription initiation site “I”, and the start of the URA3 gene with initiator ATG codon in green. Anticipated transcription is shown as the right-angle arrow. For both panels, the top strand (i.e., the sense strand of URA3) is the lagging strand template. (A) Yeast cells that have undergone a TNR contraction can be selected by their ability to grow in the absence of uracil. The starting strain is Ura− due to the inserted triplet repeat sequence, (CNG)20+13. (This nomenclature refers to 20 repeats of the trinucleotide CNG, where N=any nucleotide, plus 39 bp of randomized, genetically inert sequence [39]. The total DNA length is therefore equivalent to 33 repeats.) Insertion of this many nucleotides between the TATA box and the preferred transcription initiation site places “I” too far from the TATA box, such that transcription is predicted to begin upstream. This incorporates an out-of-frame ATG (red), resulting in translational incompetence (indicated by X) and leading to a non-functional URA3 product. Cells have a Ura− phenotype. If a contraction occurs, losing 5 to 20 repeats, initiation will occur at the proper site “I,” leading to expression of URA3 and attainment of the Ura+ phenotype. (B) Yeast that have undergone an expansion can be identified by growth in the presence of 5FOA. (CNG)13+12 refers to 13 repeats of the trinucleotide CNG plus 36 bp of randomized sequence [32]. The total DNA length is equivalent to 25 repeats. Proper initiation at “I” results in functional expression of URA3, which confers sensitivity to the drug 5-fluoroorotic acid (5FOA). If the TNR expands, gaining 5 or more repeats, upstream transcription initiation will include the red out-of-frame ATG, resulting in translational incompetence and resistance to 5FOA.
TABLE 2.
Spontaneous Mutation Rates
| Mutation rate and genotype | Mean no. of mutations/cell generation (±SD), 10−n | Ratio (fold over wild type) |
|---|---|---|
| CAN1 forward mutations (×10−7) | ||
| Wild type | 3.0 ± 0.0 | 1.0 |
| mrc1 | 1.8 ± 0.9 | 0.6 |
| tof1 | 3.0 ± 1.8 | 1.0 |
| rad27 | 23 ± 6.6 | 7.5 |
| Dinucleotide repeat mutations (×10−6) | ||
| Wild type | 1.7 ± 0.1 | 1.0 |
| mrc1 | 3.7 ± 1.4 | 2.2 |
| tof1 | 3.6 ± 0.9 | 2.1 |
| Expansions of (CTA)25 (×10−8) | ||
| Wild type | < 4.9 | 1.0 |
| mrc1 | < 3.1 | 0.6 |
| tof1 | < 3.1 | 0.6 |
TABLE 1.
CAG•CTG Repeat Mutation Rates
| Ratio (fold over wild type)
|
||
|---|---|---|
| Genotype | Contractions of (CAG)20 | Expansions of (CTG)13 |
| mrc1 | 7.1* | 4.0* |
| mrc1 + pMRC1 | 1.6 | 1.7 |
| mrc1 + pmrc1AQ | 2.0 | 3.7* |
| tof1 | 8.6* | 7.4* |
| csm3 | 6.5* | 6.8* |
| sml1 | 0.7 | 1.3 |
| sml1 mec1 | 1.6 | 6.0* |
| sml1 ddc2 | 3.7* | 4.9* |
| rad17 | 1.4 | 3.7* |
| rad24 | 1.0 | 6.4* |
| ddc1 | 1.4 | 4.5* |
| mec3 | 0.8 | 3.5* |
| rad9 | 2.2 | 8.2* |
| chk1 | 0.8 | 7.7* |
| sml1 rad53 | 3.7* | 3.0* |
Significantly different from wild type (P < 0.05). The wild type rate for contractions is 1.4 × 10−7 per cell generation, and for expansions is 1.8 × 10−6 per cell generation.
2.4. Genetic screen for modifiers of TNR stability
To identify novel proteins involved in preventing TNR contractions, a random screen of yeast insertion mutants was performed. A plasmid disruption library [37] was used to disrupt random genes in yeast strain BY4741 containing a (CAG)20 contraction reporter. A high throughput replica plating method was used to identify mutants with increased rates of instability, based on excess number of papillae when transferred to media lacking uracil. Mutants with increased rates of contractions as compared to wild type were further analyzed by fluctuation analysis. Several mutants showed increased rates of TNR contractions; mutant genes were identified by Vectorette PCR as described in [38].
3. Results
3.1. CAG repeat contractions are inhibited by Mrc1, Tof1 and Csm3
To find trans-acting factors that help prevent TNR instability, a yeast gene disruption library was used to screen for mutants with increased rates of TNR contractions. Screens have not previously been focused on mutants affecting contractions; therefore this approach complements previous screens that identified mutants with elevated expansion rates [17, 38]. Also, contractions in wild type yeast show a threshold-like effect, unique to TNRs, near 22 repeats [39]. Therefore a (CAG)20-URA3 reporter was chosen, based on the hypothesis that mutants that elevate contraction rates near the threshold might be highly selective for TNR instability. The strain is initially Ura−, but contractions removing 5 repeats or more generate an Ura+ phenotype (Fig. 1A) [32]. This strain was transformed with a library of LEU2-marked disruption cassettes [37] and approximately 15,000 Leu+ isolates were screened for increased rates of CAG repeat contractions. The initial positives were screened twice more with increasing stringency. Using Vectorette PCR (described in [38]) we identified the disrupted MRC1 gene in a mutant with 3.6-fold increased rates of contractions. These preliminary findings were confirmed when another mrc1 strain, acquired commercially, displayed a 7.1 fold increase in contractions (Table 1). Furthermore, the contraction rate phenotype could be rescued in both strain backgrounds by add-back of the wild type gene on the low copy pMRC1 plasmid (Table 1 and data not shown). Strains with tof1 or csm3 mutations had elevated contraction rates of 8.6- and 6.5-fold over wild type, similar in magnitude to the mrc1 strain, and consistent with the functional interdependence of Mrc1, Tof1 and Csm3 [30]. Analysis of the contraction size spectra showed that mrc1, tof1 and csm3 mutants all yielded −16 to −20 repeat changes, overlapping the range seen in wild type cells [39]. We conclude that the lack of Mrc1, Tof1 or Csm3 leads to a higher rate of contractions, rather than the appearance of a new size class of contractions.
3.2. Mrc1, Tof1 and Csm3 are selective for preventing TNR instability
It is of particular interest to know if Mrc1, Tof1 and Csm3 selectively protect triplet repeats from instability or whether these proteins have a general effect to repress mutation rates throughout the genome. To assess the mutator selectivity of mrc1 and tof1 mutants, three additional spontaneous mutator assays were performed (Table 2). Forward mutations in the CAN1 gene, resulting in resistance to canavanine, detect many types of inactivating alterations. There was no detectable increase in the rate of canavanine resistance, compared to wild type, in strains lacking either Mrc1 or Tof1 (Table 2). In contrast, a rad27 control strain lacking flap endonuclease 1 had an elevated rate, consistent with a previous report [40]. The second assay measures frameshift mutations of a dinucleotide repeat tract, (GT)16.5, which most often occur as changes of ±1–2 repeats [33] rather than the larger mutations associated with TNRs. The rate of dinucleotide repeat changes was not statistically different from wild type for both mrc1 and tof1 (Table 2), whereas the rate in an msh2 mismatch repair mutant is elevated several hundred fold [17]. In the third assay, we tested whether the TNR phenotype of mrc1 and tof1 mutants was dependent on the ability of the TNR to adopt stable secondary structure, which is generally believed to be very important in the mechanism of instability [1, 2]. Expansions were measured for a CTA repeat, which has poor structure forming ability in vitro [4] and is genetically stable in yeast as a (CTA)25 allele [32]. There was no detectable increase in CTA expansion rates in cells lacking Mrc1 or Tof1 (Table 2), suggesting the phenotype of increased contractions occurs by a mechanism that requires the ability of the TNR to form secondary structures. In summary, these data support the idea that cells lacking Mrc1 or Tof1 (or, by inference, Csm3) are selectively defective in preventing TNR mutagenesis.
3.3. The replication stalling complex, Mrc1/Tof1/Csm3, reduces CAG repeat contraction rates independently of replication checkpoint and DNA damage checkpoint factors
In addition to its replication coupling role, Mrc1 also acts as a mediator of the replication checkpoint and the DNA damage checkpoint. To determine whether CAG repeat contractions are inhibited by either checkpoint, contraction rates were measured in additional mutants. We took advantage of a specific signaling deficient mutant form of Mrc1 called mrc1AQ. This mutant has its Mec1 kinase target phosphorylation sites modified so that it is unable to mediate signaling but is still capable of performing its coupling role [34]. When pmrc1AQ was introduced into an mrc1 knockout strain, the contraction phenotype was nearly normal (Table 1), suggesting that checkpoint signaling is dispensable for the prevention of contractions. Similarly, loss of the upstream kinase Mec1 showed no significant increase in contraction rates (Table 1), although there was a marginally significant increases in contraction rates in the strains lacking the Mec1-associated protein Ddc2 and the downstream kinase Rad53. Complementation by pmrc1AQ and the lack of a mec1 phenotype suggests that, for the most part, the checkpoint signaling function of Mrc1 is dispensable for the prevention of TNR contractions. To test the influence of the replication checkpoint and the DNA damage checkpoint, we investigated (CAG)20 contraction rates for strains lacking the RFC-like Rad24 protein, components of the yeast 9-1-1 complex (Rad17, Mec3 and Ddc1), the signaling mediator Rad9 or the effector kinase Chk1. There was no significant contraction phenotype in strains lacking any of these factors (Table 1). Together, the lack of a contraction phenotype in most replication checkpoint or DNA damage checkpoint mutants suggests that the mechanism for preventing contractions may be different for the near-threshold tracts of 20 repeats used here versus the longer, 85 repeat alleles reported previously [19].
Efficient coupling of the replicative helicase and polymerase helps minimize single-stranded template DNA [25, 30]. We considered the possibility that the absence of coupling in mrc1, tof1 or csm3 strains would lead to more single-stranded template that could fold into a stable secondary structure and lead to a higher rate of contractions. If so, treatment of cells with hydroxyurea to increase ssDNA levels might give an elevated contraction phenotype. Wild type cells were grown continuously in the presence of 50 mM HU and then assayed for (CAG)20 contractions. Modestly higher contraction rates were observed in HU treated cells compared to untreated controls (2.0-fold, P = 0.037). In contrast, there was no significant increase (<1.4-fold) in contraction rates when mrc1 cells were treated with HU. Together with the absence of a checkpoint effect on contractions, the HU result supports the coupling mechanism for preventing contractions. We conclude that the replication coupling activity of Mrc1, Tof1 and Csm3, not their checkpoint activities, is most important for inhibiting contractions of CAG repeats near the apparent threshold length
3.4. Expansions are inhibited by the DNA damage checkpoints
Are expansions inhibited by Mrc1, Tof1 and Csm3, and, if so, do these proteins act through their coupling activity or in conjunction with checkpoint activation? Expansions were assessed using a (CTG)13 repeat reporter (Fig. 1B) because this allele length in our system lies near the apparent threshold for expansions [32]. Similar to the increases seen for contraction rates in cells lacking Mrc1, Tof1 or Csm3 proteins, expansion rates of a (CTG)13 repeat tract were elevated 4.0- to 7.4-fold in mrc1, tof1 and csm3 strains (Table 1). To test a longer repeat tract, the expanion rate of (CAG)25 in the mrc1 mutant was found to be 8-fold increased above wild type (mrc1 9.0 (±4.8) × 10−7 per cell generation; wild type 1.2 (±1.3) × 10−7 per cell generation; P < 0.05). These data show that Mrc1 helps prevent expansions of CAG•CTG tracts of 13–25 repeats, in accordance with a previous report for an 85 repeat allele [20].
However, contrary to what is seen for contractions, the checkpoint activity of Mrc1, Tof1 and Csm3 seems to be key for blocking expansions. First, when Mrc1-deficient cells were supplemented with the signaling deficient mrc1AQ mutant, the mutant phenotype on (CTG)13 was not suppressed (3.7-fold, compared to 4.0-fold for the mrc1 strain). Second, there was significant increase in expansion rates for both mec1 (6.0-fold) and ddc2 (4.9-fold) mutants compared to the sml1 (1.3-fold) parental strain. Together with the mrc1AQ result, this suggests the replication and/or DNA damage checkpoint is an important inhibitor of expansions for CTG tracts near the apparent threshold. Third, increased expansion rates, 3.5- to 6.4-fold above wild type, were seen in strains specifically lacking DNA damage checkpoint components, such as the alternate clamp loader component Rad24, or any member of the PCNA-like Rad17, Mec3, and Ddc1. Fourth, strains lacking the downstream mediator Rad9 or the effector kinases Chk1 or Rad53 also exhibited expansion rates that were 3.5- to 8.2-fold higher than wild type. All of the mutants tested for expansions gave phenotypes of similar magnitude (3.5- to 8.2-fold), consistent with a DNA damage checkpoint response to help block CTG expansions. The range of expansion sizes in all mutants (+5 to +10 repeats) was similar to wild type [17] suggesting that the higher rates are due to more expansions of the same size rather than appearance of a new size category. Clearly, expansions and contractions are inhibited by different Mrc1-dependent mechanisms for the repeat lengths tested here.
There was a formal possibility that the drug used to select cells with an expansion, 5-fluoroorotic acid (5FOA; Fig. 1B), might trigger a DNA damage response in wild type cells and slow their growth relative to the checkpoint mutants. Thus the higher expansion rates in the mutants might be an artifact of faster growth than wild type cells on 5FOA. This was tested in growth experiments on media containing 5FOA compared to rich media (YPD; yeast extract/peptone/dextrose). To mimic an expanded allele, cells were transformed with the URA3 reporter in the unexpressed configuration, due to the presence of 33 CAG repeats. Comparison of the growth of the wild type strain and representative mutants (mrc1, mrc1 + pMRC1, sml1, and sml1 ddc2) revealed no consistent differences between the strains. We deem it unlikely that the expansion phenotype observed for checkpoint mutants is due to differences in growth rate on 5FOA media used here.
4. Discussion
This study used an unbiased mutant screen and found that disruption of MRC1 leads to consistently elevated expansion and contraction rates of CAG•CTG repeat tracts. Subsequent analysis showed that Tof1 and Csm3 are also necessary to inhibit both classes of TNR mutations. In the absence of any of these proteins, triplet repeats expand and contract significantly more often than normal. The starting lengths for the expansion and contraction reporters were specifically chosen to monitor length changes near the threshold. These length changes thereby span the range between genetically stable, subthreshold alleles and the longer, unstable tracts that can give rise to further mutation and disease in humans. The demonstration that Mrc1, Tof1 and Csm3 inhibit instability for subthreshold alleles confirms and extends previous studies showing that long, expanded alleles of 85-155 CAG repeats are also prone to fragility and contraction in the absence of the DNA damage checkpoint pathway [19], and to fragility, contraction and expansion in the absence of Mrc1 [20]. Our work provides two additional important insights to the role of Mrc1, Tof1 and Csm3. First, mrc1 and tof1 mutants have a highly selective mutator phenotype where only structure-forming CAG•CTG tracts are destabilized. Other sequences, including the CAN1 gene, poly(GT) tracts and unstructured CTA repeats are not affected by mrc1 or tof1 mutations. These findings are entirely consistent with the structure forming requirement of pathogenic TNRs as a major part of accepted models of instability [1, 2]. Furthermore this selectivity strongly supports the idea, as described below for expansions, that the DNA damage checkpoint responds to TNR-mediated secondary structure or to subsequently processed forms [19, 20]. The second important finding was that expansions and contractions are differentially sensitive to defects in DNA damage checkpoint factors, indicating that at least two mechanisms are at play in preventing triplet repeat instability through Mrc1, Tof1 and Csm3.
Two lines of evidence suggest that checkpoint signaling is not important to prevent contractions of (CAG)20 in our system. First, the high rate of contractions in the mrc1 strain was complemented by a mutant allele, mrc1AQ, which is selectively defective in checkpoint responses [34]. Second, contraction rates are also elevated by loss of Tof1 or Csm3, but not by defects in most replication checkpoint or DNA damage checkpoint proteins (Mec1, Rad17, Rad24, Ddc1, Mec3, Rad9 or Chk1). There was a small contraction phenotype in sml1 ddc2 and sml1 rad53 strains, although the reasons for this remain unclear. Nonetheless, these data suggest that Mrc1, Tof1 and Csm3 inhibit contractions primarily through their coupling activity on the replicative helicase and polymerase. In mrc1, tof1 or csm3 mutants, the helicase becomes uncoupled from the polymerase, leading to accumulation of single-stranded template [25, 30] and spontaneous secondary structure formation at single-stranded CAG or CTG repeats [1, 2, 9]. DNA synthesis at the shortened template would lead to a contraction. The major role of Mrc1, Tof1 and Csm3 in this model is to prevent contractions by minimizing single-strand template DNA. More complex models envisage repeat-mediated breakage, leading to recombinational repair, single-strand annealing or inability to restart a stalled replication fork at the TNR. These break repair activities typically require recombinational proteins such as Rad52 but previous data in our system showed no contraction phenotype in a rad52 mutant [41]. We favor the simpler model where Mrc1, Tof1 and Csm3 help prevent contractions by limiting the accumulation of single-strand template DNA prior to the formation of aberrant secondary structure.
Our data suggest that expansions of subthreshold CTG tracts are prevented by DNA damage checkpoint response to TNR-mediated secondary structure, such as a hairpin, or to subsequently processed forms. The major difference from contractions is that expansions are inhibited by the DNA damage checkpoint after structure formation. Defects in either checkpoint fail to respond to aberrant secondary structure, which persists and leads to expansions. This model is supported by the mutator specificity of mrc1 and tof1 mutants, and by the fact that the mrc1AQ mutant was defective in blocking expansions. Similarly high rates of expansions occurred in mutants defective in both replication and DNA damage checkpoints (mec1, ddc2, mrc1, tof1, csm3 or rad53) or in strains selectively deficient in the DNA damage checkpoint (rad24, ddc1, mec3, rad17, rad9 or chk1). It is possible that both checkpoints are capable of responding to the damage intermediate, but epistasis experiments could not be performed due to inviability of double mutants. It is unlikely that recombination plays a role in promoting or preventing expansions in our system since no increase in expansion rates was observed in rad52 or mrc1 rad52 mutants (data not shown) and because previous studies showed no rad51 or rad52 phenotype on expansions [17, 36]. Mec1 dependent signaling through Mrc1 specifically responds to replication stalling at DNA lesions [34]. In the case of triplet repeats, our data suggest that repeat-mediated secondary structure formation adds to or possibly replaces replication stalling as the signal. Alternatively, the aberrant secondary structure could be cleaved or otherwise altered enzymatically to produce the signal. Finally, unreplicated single stranded DNA, resulting from downstream priming, may serve as the signal for checkpoint activation. Although we currently lack the molecular tools to examine what is occurring as the replication fork encounters a triplet repeat in vivo, it is likely that the absence of the DNA damage checkpoint creates an opportunity for mutagenic replication or repair leading to an expansion.
The results of this study are largely in agreement with previous reports of DNA damage response to expanded CAG tracts of 85-115 repeats [19, 20]. However, there are some interesting differences. One key difference is that we found an expansion phenotype for mutations in many genes that inactivate the DNA damage checkpoint. In contrast, the earlier work found a significant expansion phenotype only for mrc1 [19, 20]. This distinction could be due to differences in assay sensitivity; our genetic assay can distinguish changes in expansion rate over several orders of magnitude [32] whereas the bulk PCR-based method has a more limited range. Also, longer tracts show instability in wild type cells at relatively high frequencies in the ~1–3% range, so there is less sensitivity available to distinguish an elevated mutant phenotype. Another difference was that contractions of long CAG tracts are sensitive to the absence of Mec1, Ddc2, Rad17, Rad24 or Rad53 [19] whereas we found no significant change in contraction rates for these mutations (Table 1). Perhaps the fragility associated with long tracts stimulates a checkpoint response that helps avoid contractions, whereas the shorter tracts examined in our study do not break often enough to generate a checkpoint response. In summary, it is now clear from previous work [19, 20] and from this study that the DNA damage checkpoint helps avoid instability of CAG•CTG repeat tracts. We show here that triplet repeat sequences are selectively protected by Mrc1, Tof1 and Csm3, and that they act in two distinct ways to inhibit contractions through replicational coupling activity and expansions via the DNA damage checkpoint.
Acknowledgments
The authors gratefully acknowledge Tony Mertz for technical assistance. This work was supported by National Institutes of Health grant GM61961 and by Science Foundation of Ireland grant 06/IN.1/B73 (to R.S.L.), by National Cancer Institute training grant T32 CA09476 and by a graduate fellowship from the University of Nebraska Medical Center (both to D.F.R.) and by National Cancer Institute Cancer Center Support Grant P30 CA36727 (to the Eppley Institute).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pearson CE, Edamura KN, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6:729–742. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- 2.Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 3.Richards RI, Sutherland GR. Dynamic mutations: a new class of mutations causing human disease. Cell. 1992;70:709–712. doi: 10.1016/0092-8674(92)90302-s. [DOI] [PubMed] [Google Scholar]
- 4.Gacy AM, Goellner G, Juranic N, Macura S, McMurray CT. Trinucleotide repeats that expand in human disease form hairpin structure in vitro. Cell. 1995;81:533–540. doi: 10.1016/0092-8674(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 5.Pearson CE, Sinden RR. Alternative structures in duplex DNA formed within the trinucleotide repeats of the myotonic dystrophy and fragile X loci. Biochemistry. 1996;35:5041–5053. doi: 10.1021/bi9601013. [DOI] [PubMed] [Google Scholar]
- 6.Petruska J, Hartenstine MJ, Goodman MF. Analysis of strand slippage in DNA polymerase expansions of CAG/CTG triplet repeats associated with neurodegenerative diseases. J Biol Chem. 1998;273:5204–5210. doi: 10.1074/jbc.273.9.5204. [DOI] [PubMed] [Google Scholar]
- 7.Samadashwily GM, Raca G, Mirkin SM. Trinucleotide repeats affect DNA replication in vivo. Nature Genet. 1997;17:298–304. doi: 10.1038/ng1197-298. [DOI] [PubMed] [Google Scholar]
- 8.Pelletier R, Krasilnikova AS, Samadishwily GM, Lahue RS, Mirkin SM. Replication and expansion of trinucleotide repeats in yeast. Mol Cell Biol. 2003;23:1349–1357. doi: 10.1128/MCB.23.4.1349-1357.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang S, Jaworski A, Ohshima K, Wells RD. Expansion and deletion of CTG repeats from human disease genes are determined by the direction of replication in E. coli. Nat Genet. 1995;10:213–218. doi: 10.1038/ng0695-213. [DOI] [PubMed] [Google Scholar]
- 10.Monckton DG, Wong LJ, Ashizawa T, Caskey CT. Somatic mosaicism, germline expansions, germline reversions and intergenerational reductions in myotonic dystrophy males: small pool PCR analyses. Hum Mol Genet. 1995;4:1–8. doi: 10.1093/hmg/4.1.1. [DOI] [PubMed] [Google Scholar]
- 11.Fortune MT, Vassilopoulos C, Coolbaugh MI, Siciliano MJ, Monckton DG. Dramatic, expansion-biased, age-dependent, tissue-specific somatic mosaicism in a transgenic mouse model of triplet repeat instability. Hum Mol Genet. 2000;9:439–445. doi: 10.1093/hmg/9.3.439. [DOI] [PubMed] [Google Scholar]
- 12.Seznec H, Lia-Baldini AS, Duros C, Fouquet C, Lacroix C, Hofmann-Radvanyi H, Junien C, Gourdon G. Transgenic mice carrying large human genomic sequences with expanded CTG repeat mimic closely the DM CTG repeat intergenerational and somatic instability. Hum Mol Genet. 2000;9:1185–1194. doi: 10.1093/hmg/9.8.1185. [DOI] [PubMed] [Google Scholar]
- 13.van den Broek WJAA, Nelen MR, Wansink DG, Coerwinkel MM, te Riele H, Groenen PJTA, Wieringa B. Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum Mol Genet. 2002;11:191–198. doi: 10.1093/hmg/11.2.191. [DOI] [PubMed] [Google Scholar]
- 14.Schweitzer JK, Livingston DM. Expansions of CAG repeat tracts are frequent in a yeast mutant defective in Okazaki fragment maturation. Hum Mol Genet. 1998;7:69–74. doi: 10.1093/hmg/7.1.69. [DOI] [PubMed] [Google Scholar]
- 15.Freudenreich CH, Kantrow SM, Zakian VA. Expansion and length-dependent fragility of CTG repeats in yeast. Science. 1998;279:853–856. doi: 10.1126/science.279.5352.853. [DOI] [PubMed] [Google Scholar]
- 16.Spiro C, Pelletier R, Rolfsmeier ML, Dixon MJ, Lahue RS, Gupta G, Park MS, Chen X, Mariappan SVS, McMurray CT. Inhibition of FEN-1 processing by DNA secondary structure at trinucleotide repeats. Mol Cell. 1999;4:1079–1085. doi: 10.1016/s1097-2765(00)80236-1. [DOI] [PubMed] [Google Scholar]
- 17.Bhattacharyya S, Lahue RS. Yeast Srs2 DNA helicase selectively blocks expansions of trinucleotide repeats. Mol Cell Biol. 2004;24:7324–7330. doi: 10.1128/MCB.24.17.7324-7330.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daee DL, Mertz T, Lahue RS. Postreplication repair inhibits CAG•CTG repeat expansions in Saccharomyces cerevisiae. Mol Cell Biol. 2007;27:102–110. doi: 10.1128/MCB.01167-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lahiri M, Gustafson TL, Majors ER, Freudenreich CH. Expanded CAG repeats activate the DNA damage checkpoint pathway. Mol Cell. 2004;15:287–293. doi: 10.1016/j.molcel.2004.06.034. [DOI] [PubMed] [Google Scholar]
- 20.Freudenreich CH, Lahiri M. Structure-forming CAG/CTG repeat sequences are sensitive to breakage in the absence of Mrc1 checkpoint function and S-phase checkpoint signaling: implications for trinucleotide repeat expansion diseases. Cell Cycle. 2004;3:1370–1374. doi: 10.4161/cc.3.11.1246. [DOI] [PubMed] [Google Scholar]
- 21.Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJH, Bousset K, Furuya K, Diffley JFX, Carr AM, Elledge SJ. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Biol. 2001;3:958–965. doi: 10.1038/ncb1101-958. [DOI] [PubMed] [Google Scholar]
- 22.Zou L, Liu D, Elledge SJ. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci U S A. 2003;100:13827–13832. doi: 10.1073/pnas.2336100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Majka J, Niedziela-Majka A, Burgers PM. The checkpoint clamp activates Mec1 kinase during initiation of the DNA damage checkpoint. Mol Cell. 2006;24:891–901. doi: 10.1016/j.molcel.2006.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Branzei D, Foiani M. The Rad53 signal transduction pathway: replication fork stabilization, DNA repair, and adaptation. Exp Cell Res. 2006;312:2654–2659. doi: 10.1016/j.yexcr.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 25.Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- 26.Longhese MP, Clerici M, Lucchini G. The S-phase checkpoint and its regulation in Saccharomyces cerevisiae. Mutat Res. 2003;532:41–58. doi: 10.1016/j.mrfmmm.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 27.Szyjka SJ, Viggiani CJ, Aparicio OM. Mrc1 is required for normal progression of replication forks throughout chromatin in S. cerevisiae. Mol Cell. 2005;19:691–697. doi: 10.1016/j.molcel.2005.06.037. [DOI] [PubMed] [Google Scholar]
- 28.Zegerman P, Diffley JFX. Lessons in how to hold a fork. Nat Struct Biol. 2003;10:778. doi: 10.1038/nsb1003-778. [DOI] [PubMed] [Google Scholar]
- 29.Tourrière H, Pasero P. Maintenance of fork integrity at damaged DNA and natural pause sites. DNA Repair. 2007;6:900–913. doi: 10.1016/j.dnarep.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 30.Nedelcheva MN, Roguev A, Dolapchiev LB, Shevchenko A, Taskov HB, Shevchenko A, Steward AF, Stoynov SS. Uncoupling of unwinding from DNA synthesis implies regulation of MCM helicase by Tof1/Mrc1/Csm3 checkpoint complex. J Mol Biol. 2005;347:509–521. doi: 10.1016/j.jmb.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 31.Dixon MJ, Bhattacharyya S, Lahue RS. Genetic assays for triplet repeat instability in yeast. Methods Mol Biol. 2004;277:29–45. doi: 10.1385/1-59259-804-8:029. [DOI] [PubMed] [Google Scholar]
- 32.Rolfsmeier ML, Dixon MJ, Pessoa-Brandao L, Pelletier R, Miret JJ, Lahue RS. Cis-elements governing trinucleotide repeat instability in Saccharomyces cerevisiae. Genetics. 2001;157:1569–1579. doi: 10.1093/genetics/157.4.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henderson ST, Petes TD. Instability of simple sequence DNA in Saccharomyces cerevisiae. Mol Cell Biol. 1992;12:2749–2757. doi: 10.1128/mcb.12.6.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Osborn AJ, Elledge SJ. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes & Dev. 2003;17:1755–1767. doi: 10.1101/gad.1098303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lea DE, Coulson CA. The distribution of the number of mutants in bacterial populations. J Genet. 1948;49:264–284. doi: 10.1007/BF02986080. [DOI] [PubMed] [Google Scholar]
- 36.Miret JJ, Pessoa-Brandao L, Lahue RS. Orientation-dependent and sequence-specific expansions of CTG/CAG trinucleotide repeats in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1998;95:12438–12443. doi: 10.1073/pnas.95.21.12438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burns N, Grimwade B, Ross-Macdonald PB, Choi EY, Finberg K, Roeder GS, Snyder M. Large-scale analysis of gene expression, protein localization, and gene disruption in Saccharomyces cerevisiae. Genes Dev. 1994;8:1087–1105. doi: 10.1101/gad.8.9.1087. [DOI] [PubMed] [Google Scholar]
- 38.Bhattacharyya S, Rolfsmeier ML, Dixon MJ, Wagoner K, Lahue RS. Identification of RTG2 as a modifier gene for CAG•CTG repeat instability in Saccharomyces cerevisiae. Genetics. 2002;162:579–589. doi: 10.1093/genetics/162.2.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dixon MJ, Lahue RS. DNA elements important for CAG•CTG repeat thresholds in Saccharomyces cerevisiae. Nucleic Acids Res. 2004;32:1289–1297. doi: 10.1093/nar/gkh292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tishkoff DX, Filosi N, Gaida GM, Kolodner RD. A novel mutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinct from DNA mismatch repair. Cell. 1997;88:253–263. doi: 10.1016/s0092-8674(00)81846-2. [DOI] [PubMed] [Google Scholar]
- 41.Miret JJ, Pessoa-Brandao L, Lahue RS. Instability of CAG and CTG trinucleotide repeats in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:3382–3387. doi: 10.1128/mcb.17.6.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]