

Abstract
Cardiac resynchronization therapy has been most typically achieved by biventricular stimulation. However, left ventricular (LV) free-wall pacing appears equally effective in acute and chronic clinical studies. Recent data suggest electrical synchrony measured epicardially is not required to yield effective mechanical synchronization, whereas endocardial mapping data suggest synchrony (fusion with intrinsic conduction) is important. To better understand this disparity, we simultaneously mapped both endocardial and epicardial electrical activation during LV free-wall pacing at varying atrioventricular delays (AV delay 0–150 ms) in six normal dogs with the use of a 64-electrode LV endocardial basket and a 128-electrode epicardial sock. The transition from dyssynchronous LV-paced activation to synchronous RA-paced activation was studied by constructing activation time maps for both endo- and epicardial surfaces as a function of increasing AV delay. The AV delay at the transition from dyssynchronous to synchronous activation was defined as the transition delay (AVt). AVt was variable among experiments, in the range of 44–93 ms on the epicardium and 47–105 ms on the endocardium. Differences in endo- and epicardial AVt were smaller (−17 to +12 ms) and not significant on average (−5.0 ± 5.2 ms). In no instance was the transition to synchrony complete on one surface without substantial concurrent transition on the other surface. We conclude that both epicardial and endocardial synchrony due to fusion of native with ventricular stimulation occur nearly concurrently. Assessment of electrical epicardial delay, as often used clinically during cardiac resynchronization therapy lead placement, should provide adequate assessment of stimulation delay for inner wall layers as well.
Keywords: resynchronization, mapping, heart failure
In the normal heart, the spread of ventricular electrical activation occurs within ∼40 ms via conduction through the Purkinje network, and contraction occurs with similar synchrony. In hearts with an intraventricular conduction delay of a left bundle branch (LBB) pattern, the electrical activation sequence is significantly altered, creating regions of early and late contraction separated by as much as 100 ms (10, 13, 14, 18). This occurs commonly in patients with dilated heart failure and those with right ventricular (RV) apex pacing and is an important contributor to ventricular dysfunction and worsening failure. Such dyssynchrony causes wasted work due to stretching of the unactivated region while the activated region contracts, resulting in a decline in cardiac output, increased end-systolic volume and wall stress, and delayed relaxation (4, 6, 12).
Cardiac resynchronization therapy (CRT) is a technique in which left ventricular (LV) or biventricular pacing is used to alter the sequence of electrical activation in the heart. CRT has been shown to improve LV efficiency and hemodynamic function in studies of HF with LBB block (LBBB) by increasing the synchrony of contraction (1, 2, 5, 9, 15). However, the relationship between changes in electrical synchrony and the resulting changes in the contractile or mechanical synchrony with CRT are poorly understood. Although intraventricular electrical delay is characterized by a wide QRS complex, clinical studies (7, 9, 11, 15) have shown a poor correlation between changes in QRS duration and hemodynamic improvement with CRT. Because the QRS complex is derived from potentials on the epicardium, this would indicate that epicardial electrical synchrony cannot predict functional improvement with CRT. Indeed, it has been shown in the canine model of HF with LBBB that improvements in hemodynamic function and mechanical synchrony with CRT are not correlated to epicardial electrical synchrony (10). Another animal study (16) of LBBB in nonfailing hearts, however, found that hemodynamic function was optimized with LV pacing only if endocar-dial electrical synchrony was also present. This was accomplished by selecting an atrioventricular (AV) delay that allowed fusion between the LV stimulus and intrinsic right bundle conduction. These results raise the hypothesis that endocardial and epicardial surface activation may exhibit different degrees of electrical fusion and thus synchrony with LV pacing such that hemodynamic improvement might be observed with increased endocardial electrical synchrony and presumably mechanical synchrony, without a clear correlation to epicardial electrical synchrony or QRS duration.
The present study tested this question by determining both epicardial and endocardial activation maps concomitantly in hearts paced from the LV free wall. We determined the extent of synchrony and the transition AV delay time required to yield fusion between the pacing activation and intrinsic conduction, and tested whether this is indeed different between endocardial versus epicardial surfaces.
MATERIALS AND METHODS
The study was conducted in accordance with the guidelines of the Animal Care and Use Committee of the National Heart, Lung, and Blood Institute.
Surgery
Six adult mongrel dogs weighing 15–20 kg were used. Anesthesia was induced with an initial intravenous injection of thiopental (25 mg/ml at 1 ml per 2 kg) and maintained after endotracheal intubation with isoflurane (0.8–2%). Ventilation was maintained with the use of a ventilator (model 900D, Siemens). Arterial blood pressure, arterial blood gases, and the ECG waveform were monitored throughout the study.
Epicardial electrode array
A median sternotomy was performed and the heart suspended in a pericardial cradle. A multielectrode sock was placed over the ventricular epicardium. The epicardial sock was composed of a nylon mesh with 128 copper electrodes sewn in an ordered fashion. In addition, six glass beads filled with iopamidol (18 μl, 80% by volume, Isovue-300, Bracco Diagnostic), 1% gadopentetate dimeglumine (Magnevist, Berlex Imaging), and water were attached to the sock as X-ray localization markers. Bipolar epicardial pacing electrodes were sutured onto the right atrial (RA) appendage and onto the free wall of the left ventricle. A ground reference electrode was sutured onto the fat pad at the root of the aorta. All sock and pacing wires were run directly out of the chest. An additional bipolar pair of pacing electrodes was sutured to the free wall of the right ventricle (RV) for studies not included in this report.
Endocardial electrode array
Heparin was administered (100 U/kg bolus, 50 U·kg−1 ·h−1). Under fluoroscopic guidance, a soft tip sheath (8.5-Fr, model 5660, Boston Scientific) was introduced into a femoral artery and advanced into the LV cavity over a standard guide wire. The guide wire was exchanged for a 64-electrode unipolar basket catheter (31 mm diameter, Constellation 8031, Boston Scientific), which was unsheathed in the LV apex. A 5-F pressure catheter (model SPC-350, Millar) was inserted via the right or left carotid artery into the LV chamber. In four animals, an endocardial pacing catheter was placed at the RV apex for studies not included in this report. Figure 1 shows a fluoroscopic image of the instrumented heart.
Fig. 1.
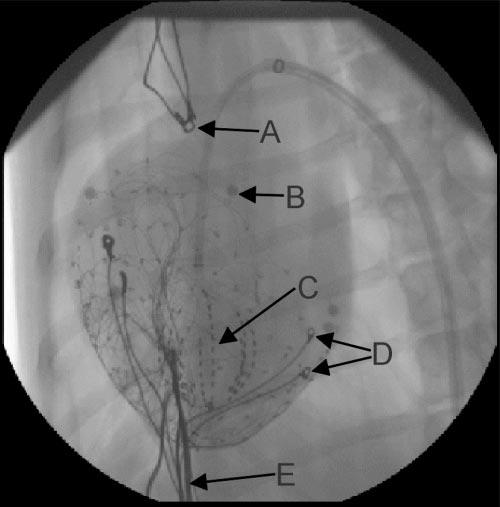
Fluoroscopic image of instrumented heart. A, right atrial (RA) pacing electrodes. B, localization marker. C, endocardial basket array. D, left ventricular (LV) pacing electrodes. E, epicardial sock wires.
Data Acquisition
Acquisition setup
Both endocardial and epicardial electrode arrays were connected to three 64-channel analog-to-digital (A/D) converter boards (model E1413C, Agilent). The pressure catheter signal was preamplified (model TCB-500, Millar) and connected to a fourth A/D board. The pacing leads were connected via photoelectric stimulus isolation units (model PSIU6, Grass Instrument) to time-synced stimulator outputs (models S11 and S88, Grass Instrument). The monitor output from one stimulator was connected directly to all four of the A/D boards to be used as a timing reference and as a verification of synchrony between the boards. All four A/D boards were connected to a 600-MHz computer and controlled by customized software (VEE version 5.0, Agilent). Unipolar electrograms from the sock and basket and LV pressure recordings were acquired in 10-s blocks encompassing ∼20 heartbeats.
Pacing protocol
The pacing protocol consisted of pacing the RA and LV with varying AV delays. The pacing rate was set at ∼25% above the native sinus rate for each animal, ranging from 120 to 150 min−1. Pacing was accomplished with a monophasic square wave current pulse with a duration of 2–5 ms and an amplitude of 20% above capture threshold for each pacing site. AV delay was incremented from 0 to 150 ms in major steps of 50 ms and minor steps of 10 ms (i.e., 0, 50, 100, 10, 60, 110, etc.). Pacing was stopped for ∼30 s between recordings.
Spatial digitization
After electrical recordings were obtained for all pacing conditions, the animal was euthanized with a bolus of potassium chloride. The heart was imaged in situ with an X-ray at several projection angles to locate the basket relative to the epicardial pacing electrodes and localization markers. The heart was then excised and filled with vinyl polysiloxane to maintain shape, and the sock electrode locations were digitized with the use of an articulated arm as previously described (8). The location of the localization markers and RV attachment were digitized at the same time.
Data Analysis
Activation times
For each AV delay, recordings from each endocardial (basket) and epicardial (sock) electrode were averaged over ∼20 heartbeats and differentiated with the use of a five-point finite difference estimate of the derivative. Electrical activation times, referenced to the ventricular pacing stimulus, were chosen as the point of the most negative derivative, indicating the time of local depolarization. Because of the pacing artifact, the first 10 ms after pacing were not considered in the picking of activation times. Activation time maps were then constructed for the ventricular epicardium with the use of the digitized points, and for the LV endocardium with the use of an 8 × 8 grid, for each AV delay.
Electrical activation delay magnitude
For both the epicardium and LV endocardium, an apex to base axis (long axis) was defined. Activation times were interpolated at evenly spaced points along the long axis and about that axis in the circumferential direction. For each AV delay and for both surfaces, the overall electrical activation delay magnitude (EADM), similar to that previously described for mechanical activation (17), was calculated with the use of the equation
| (1) |
where ϕc is the circumferential angle of the point, tact is its activation time, c and l are summation indexes ranging from 1 to C and 1 to L, respectively, and the summations are across evenly spaced circumferential (C) and longitudinal (L) indexes. EADM is large for a “bipolar” activation map with regions of early and late activation on opposite sides of the heart. EADM remains small despite a broad distribution of activation times without such spatial coherence. For the epicardial recordings, the most apical 20% of the activation times were not included in the analysis due to errors associated with assigning circumferential angles to apical points.
The curves of EADM by AV delay for each study, and for both epicardial and LV endocardial recordings, were parameterized by a least-square fit to an interpolation formula of the form
| (2) |
where P1–P5 are the fit parameters. The hyperbolic tangent was not based on a physiological model; it was chosen simply as an analytical interpolation function that fit the data very well. For comparison between curves, the EADM values were normalized with the use of the asymptotes of the interpolation fit. The top asymptote represents dyssynchronous EADM values associated with ventricular pacing, and the lower asymptote represents the more synchronous EADM associated with atrial or fused atrial and ventricular pacing, as shown in Fig. 2. The interpolation fit was also used to determine the transition AV delay (AVt), defined as the AV delay at which the EADM was midway between the asymptotic values, and the width of the transition zone, taken as 2 P3.
Fig. 2.
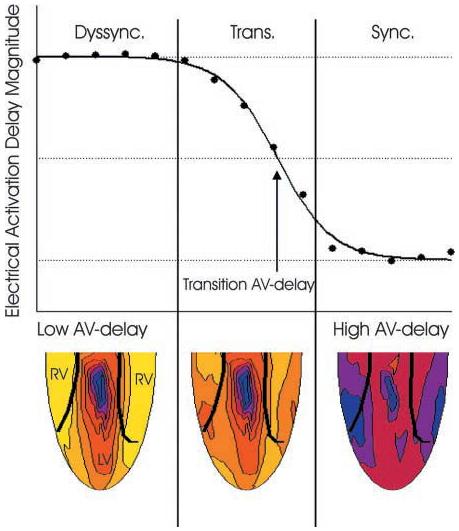
Example of electrical activation delay magnitude (EADM) plotted as a function of atrioventricular (AV) delay. At low AV delay values, corresponding to LV paced activation, EADM is at the top asymptote [highly dyssynchronous (dyssync) region]. At high AV delay values, corresponding to intrinsic activation from the RA pacing stimulus, electrical activation delay magnitude (EADM) is close to lower asymptote (synchronous region). The transition region represents fusion of the two activation patterns. The transition AV delay is defined as the AV delay at which the curve fit to the data crosses through the midpoint between the two asymptotes. Example epicardial electrical activation time maps are shown for each region, where blue is early and yellow is late. In the dyssynchronous region, maps demonstrate early activation near the LV pacing site and late activation of the RV. In the transition region, maps demonstrate fusion between the LV stimulus and Purkinje activation. In the synchronous region, the heart is activated synchronously and almost entirely through the Purkinje system.
Pressure
LV pressure recordings were averaged over ∼20 heartbeats and differentiated using a low-pass derivative filter to yield the rate of developed pressure over time (dP/dt) (3). The maximum dP/dt value (dP/dtmax) for each AV delay was calculated.
RESULTS
Electrical Activation Maps
Figure 3 displays example LV endocardial and epicardial electrical activation time maps for LV pacing with AV delay from 0 to 120 ms and for RA pacing. All ventricular pacing maps (Fig. 3, A-E) are plotted with the use of the LV pacing stimulus as the temporal reference while the RA pacing map (Fig. 3F) is plotted with the use of the RA pacing stimulus as the reference. In Fig. 3A, the AV delay is set to 0 ms, and the electrical activation on both the epicardium and endocardium proceeds from the LV free wall to the opposite side of the heart. With no AV delay, total dispersion in electrical activation time averaged 105.7 ± 11.2 ms on the epicardium and 43.9 ± 19.4 ms on the endocardium, with a transmural delay between the earliest epicardial activation and earliest endocardial activation of 30.5 ± 8.7 ms. In Fig. 3B (AV delay = 60 ms), activation patterns for both endocardial and epicardial maps are nearly identical to that of Fig. 3A, indicating that fusion between LV paced and intrinsic activation had not yet occurred. In Fig. 3C (AV delay = 80 ms), endocardial activation remained unchanged, yet on the epicardium, small regions of the RV were activated earlier than at a shorter AV delay, indicating that fusion is just beginning. In Fig. 3D (AV delay = 100 ms), the late-activated regions became more synchronous with early activated regions in both the endo- and epicardium because of fusion with the Purkinje system. This is most noticeable on the RV epicardium, which is activated in a pattern nearly identical to that seen with RA pacing (Fig. 3F). At longer AV delays, more of the heart was controlled by intrinsic activation. This is shown in Fig. 3E (AV delay = 120 ms), where most of the heart, with the exception of small endocardial and epicardial regions immediately below the pacing site, is activated in a pattern identical to that with RA pacing.
Fig. 3.
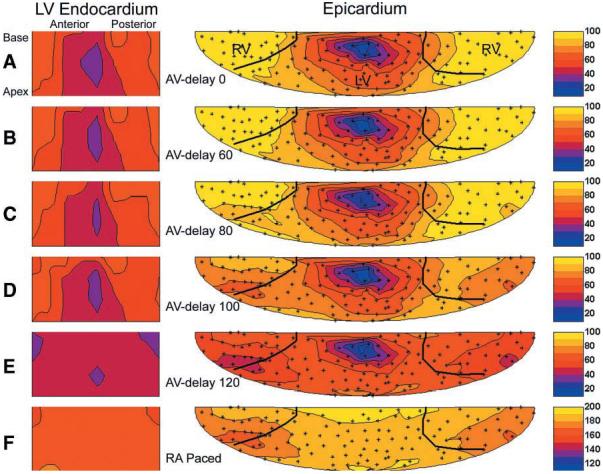
Endocardial and epicardial electrical activation maps for LV pacing at increasing AV delay and RA pacing. For both the epicardial and endocardial surface maps, the view is from the outside of the heart toward the inside. The broad black lines on the epicardial maps indicate the insertion of the RV as digitized. Endocardial and epicardial maps are centered about the earliest activation point for AV delay = 0. The small “+” symbols show the locations of the epicardial electrodes. Color transitions are in steps of 10 ms (see color bar on the right). Times are referenced to the LV pacing stimulus for LV pacing and to the RA pacing stimulus for RA pacing. A–F, different AV delays for left ventricular pacing in a single representative animal; F indicates right atrial pacing in same animal.
Electrical activation delay magnitude
To quantify the degree of electrical synchrony under different pacing conditions, the EADM index was constructed as described. Figure 4 shows endocardial and epicardial EADM as a function of AV delay for all animals. To compare endocardial synchrony to epicardial synchrony, EADM values were fit with the use of Eq. 2 and normalized by the top (dyssynchronous) and bottom (synchronous) asymptotes. The dyssynchronous asymptote represents LV-paced activation, whereas the synchronous asymptote represents activation through the intrinsic conduction system. The transition region represents activation patterns exhibiting fusion of the two excitation pathways. The AVt was defined as the delay corresponding to an EADM midway between the two asymptotes, as shown in Fig. 4. In one animal (animal D), the endocardial activation map for RA pacing was comparatively dyssynchronous; thus scaling to the asymptotes resulted in the more synchronous points of the transition falling below the range of the plot.
Fig. 4.
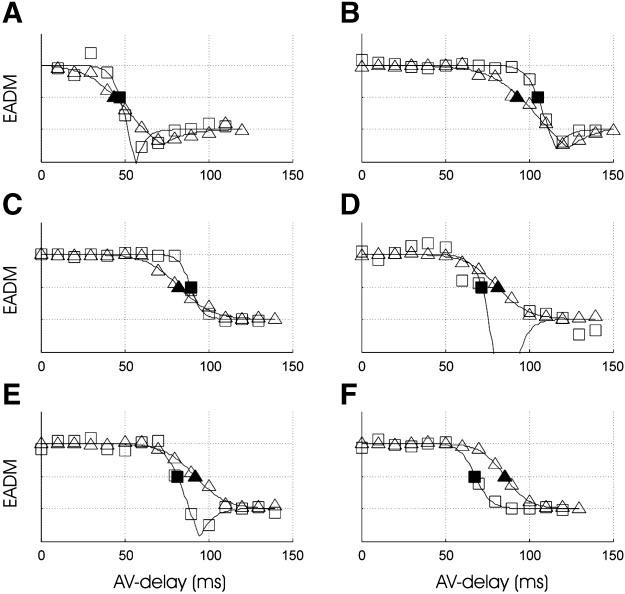
Electrical activation delay magnitude (EADM) normalized and plotted as a function of AV delay for the endocardium (□) and epicardium (●) for each animal (A–F). As AV delay is increased, endocardial and epicardial synchrony transition from the top (dyssynchronous) asymptote to the bottom (synchronous) asymptote. The endocardial (■) and epicardial (▲) transition AV delays (AVt) are shown.
EADM values for the dyssynchronous and synchronous asymptotes for each study are provided in Table 1. Over the six experiments, AVt was highly variable, ranging from 44 to 93 ms on the epicardium and 47–105 ms on the endocardium. Within a single experiment, the difference between endocardial and epicardial AVt delays ranged from −17 to 12 ms but was not significant on average (−5.0 ± 5.2 ms) and less than the width of the transition zone (19.2 ± 3.6 ms endocardial, 40.9 ± 9.6 ms epicardial). In no case was the transition to synchrony completed on one surface without substantial transition on the other surface.
Table 1.
Synchrony values
| Study |
||||||
|---|---|---|---|---|---|---|
| Parameter | A | B | C | D | E | F |
| EADM | ||||||
| Endocardial dyssynchronous | 18.1±7.6 | 19.4±1.6 | 24.5±0.2 | 18.8±4.6 | 25.9±2.0 | 24.9±0.3 |
| Endocardial synchronous | 5.4±7.6 | 4.3±1.6 | 4.9±0.2 | 12.3±4.6 | 14.9±2.0 | 11.7±0.3 |
| Epicardial dyssynchronous | 39.1±2.7 | 44.7±2.7 | 53.7±0.8 | 45.4±0.6 | 48.5±0.7 | 43.3±0.2 |
| Epicardial synchronous | 9.6±2.7 | 11.1±2.7 | 11.5±0.8 | 4.7±0.6 | 12.0±0.7 | 13.3±0.2 |
| Measured AVt | ||||||
| Endocardial | 47.2±3.5 | 105.4±1.0 | 89.5±0.4 | 71.8±3.2 | 81.5±2.1 | 68.0±1.0 |
| Epicardial | 43.9±3.0 | 93.4±2.8 | 82.6±1.1 | 81.3±0.9 | 92.1±1.0 | 85.5±0.3 |
| Difference (endo – epi) | 3.3±4.6 | 12.0±3.0 | 6.9±1.1 | −9.6±3.3 | −10.6±2.3 | −17.5±1.1 |
| Computed AVt | ||||||
| Endocardial | 48.3±0.1 | 108.6±0.4 | 101.0±0.2 | 75.4±1.2 | 82.2±0.7 | 74.9±0.3 |
| Epicardial | 43.5±1.1 | 97.2±1.4 | 92.2±0.5 | 84.4±0.5 | 89.7±0.5 | 85.4±0.6 |
| Difference (endo – epi) | 4.8±1.1 | 11.4±1.4 | 8.8±0.6 | −9.0±1.3 | −7.5±0.8 | −10.5±0.7 |
Values are means ± SE for 6 animals (A–F). The electrical activation delay magnitude (EADM) values are shown for the endocardial (endo) and epicardial (epi) synchronous and dyssynchronous asymptotes for each animal. Measured and computed values for the endocardial and epicardial transition AV delays (AVt) and their differences are shown.
LV pressures
LV pacing without fusion resulted in an 18.1 ± 10.5% decrease on average in dP/dtmax compared with synchronous activation (P < 0.01). There was no significant difference, however, between dP/dtmax evaluated without fusion and that evaluated at the AVt. The values of LV dP/dtmax at all three conditions for each experiment are shown in Fig. 5. LV dP/dtmax returned to synchronous activation baseline for AV delays >15 ms greater than the AVt, when the transition to endocardial and epicardial synchrony was nearly complete.
Fig. 5.
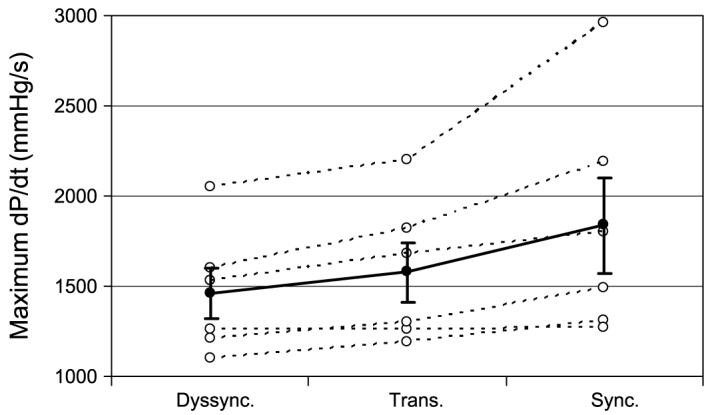
Maximum rate of developed pressure over time (dP/dt) shown for each experiment (○) evaluated under dyssynchronous electrical activation (Dyssync), pacing at the transition AV delay (Trans), and synchronous activation (Sync). For each condition, the average value (●) is shown ± SE.
EADM values computed from LV-paced and RA-paced activation maps
The curves of EADM versus AV delay for each study, and for both epicardial and LV endocardial recordings, were computed using only the activation pattern for RA pacing and a baseline activation pattern for LV pacing with 0-ms AV delay. For each value of AV delay, a computed LV pacing activation pattern was obtained by taking the minimum, for each electrode, of the RA pacing activation time and the baseline LV pacing activation time plus AV delay. At low AV delays, the computed LV pacing activation pattern was identical to the actual LV pacing activation pattern. At higher AV delays, the computed LV pacing activation pattern exhibited fusion with the RA pacing activation pattern. Figure 6 shows the computed EADM values from one experiment overlaid on the actual experimental data, demonstrating a strong agreement between the two curves. As shown in Table 1, AVt values from the computed curves predicted the actual AVt values with an average error of 4.5 ms on the endocardium and 3.2 ms on the epicardium and predicted the relative separation between the endocardial and epicardial curves with an average error of 2.4 ms. The activation time in any region of the heart could therefore be predicted by taking the minimum of the activation times obtained with either of the baseline patterns individually. This indicates that mechanical changes due to fusion did not have a measurable effect on activation patterns.
Fig. 6.
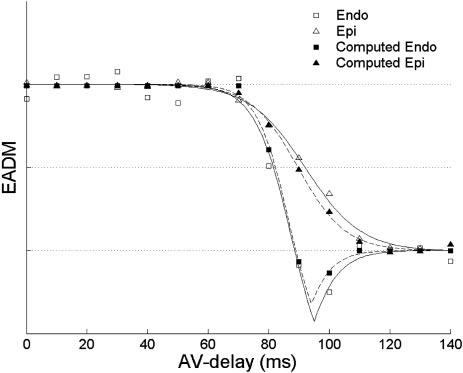
Example of computed EADM values for the endocardium and epicardium overlaid on the actual endocardial and epicardial EADM values for one animal.
DISCUSSION
In this study, simultaneous endocardial and epicardial activation time maps were obtained during LV pacing at incrementally increasing AV delay values. At low AV delays, activation patterns for both surfaces were dyssynchronous and originated from the LV pacing site. The degree of electrical dyssynchrony was consistent over a range of low AV delays, indicating that activation did not involve fusion with the atrial stimulus via the intrinsic conduction system. At long AV delays, EADM was smaller indicating more synchrony and ultimately also did not change as a function of AV delay, indicating that heart was primarily activated from the intrinsic conduction system rather than from the ventricular stimulus. Over a middle range of AV delays, endocardial and epicardial electrical synchrony increased with longer AV delay, indicating that an increasingly larger portion of the heart was being activated by intrinsic conduction. Importantly, we found the transition between these two stimulation patterns, indexed by AVt, was similar for both epi- and endocardial activation.
The AV delay at which electrical fusion was observed varied substantially among animals, but in each case the delay could be predicted from baseline maps. Whereas epicardial and LV endocardial surfaces exhibited slightly different transitions from dyssynchronous to synchronous activation, these differences were small compared with the width of the transition zone and the overall range of AV delays among the animals. For all experiments, the transition zones overlapped such that transition to synchrony on one surface was accompanied by similar transition on the other surface within a 20-ms AV delay. This point is demonstrated in Fig. 3, which shows endocardial and epicardial activation time maps at increasing AV delay. Whereas this particular heart demonstrated initial changes in epicardial electrical synchrony (AV delay = 80 ms) at a lower AV delay than that for endocardial changes (AV delay = 100 ms), both surfaces demonstrated a gradual progression from dyssynchronous to synchronous activation over the same range of AV delays (80–120 ms). This point is further illustrated in Fig. 4, which describes the progression from dyssynchronous to synchronous activation for all hearts. Although the endocardial transition AV delay could be greater or less than the epicardial transition AV delay, in no case was the transition to synchrony completed on one surface without substantial transition on the other surface.
The fact that the endocardium and epicardium demonstrate similar degrees of fusion of LV pacing with the intrinsic conduction system is clinically relevant to the study of CRT. Whereas CRT has been shown to improve contractile synchrony in the failing LBBB heart, there is a poor correlation between functional improvement and QRS width, a clinical measure of epicardial intraventricular dyssynchrony (7, 9, 11, 15). In an experimental HF animal model, Leclercq et al. (10), showed no clear correlation between epicardial electrical synchrony and functional improvement or mechanical synchrony. However, under CRT in the LBBB canine heart without failure, Verbeek et al. (16) found a correlation between endocardial synchrony and functional improvement. This discrepancy could be explained if, for some AV delay, LV pacing resulted in endocardial but not epicardial synchrony. From the results shown here in normal hearts, however, this does not appear to be the case. It appears likely that, under CRT, changes in endocardial synchrony are accompanied by corresponding changes in epicardial electrical synchrony. It remains possible that the similarity observed in the present study is somewhat different in cardiac failure with or without underlying interventricular conduction delay, and this will require further study.
Limitations
This study was conducted on normal rather than failing LBBB hearts, placing limits on its applicability to clinical cardiac resynchronization therapy. Whereas activation in the normal hearts remains synchronous at high AV delays due to conduction through the right and left bundles, in the LBBB model, a minimum EADM value would be achieved (maximum synchrony) at the AV delay corresponding to maximum fusion between the LV pacing stimulus and conduction through the right bundle. This study did not incorporate analysis of mechanical synchrony obtained from regional measures of the changes in myocardial strain, as was done previously with epicardial-only mapping in combination with MRI (8, 10). The methods to obtain mechanical measurements over a limited region of the heart must be developed to obtain local myocardial function at many AV delays.
REFERENCES
- 1.Auricchio A, Stellbrink C, Block M, Sack S, Vogt J, Bakker P, Klein H, Kramer A, Ding J, Salo R, Tockman B, Pochet T, Spinelli J. Effect of pacing chamber and atrioventricular delay on acute systolic function of paced patients with congestive heart failure. The Pacing Therapies for Congestive Heart Failure Study Group. The Guidant Congestive Heart Failure Research Group. Circulation. 1999;99:2993–3001. doi: 10.1161/01.cir.99.23.2993. [DOI] [PubMed] [Google Scholar]
- 2.Blanc JJ, Etienne Y, Gilard M, Mansourati J, Munier S, Boschat J, Benditt DG, Lurie KG. Evaluation of different ventricular pacing sites in patients with severe heart failure: results of an acute hemodynamic study. Circulation. 1997;96:3273–3277. doi: 10.1161/01.cir.96.10.3273. [DOI] [PubMed] [Google Scholar]
- 3.Bruce E. Biomedical Signal Processing and Signal Modeling. Wiley; New York: 2001. pp. 335–336. [Google Scholar]
- 4.Burkhoff D, Oikawa RY, Sagawa K. Influence of pacing site on canine left ventricular contraction. Am J Physiol Heart Circ Physiol. 1986;251:H428–H435. doi: 10.1152/ajpheart.1986.251.2.H428. [DOI] [PubMed] [Google Scholar]
- 5.Cazeau S, Leclercq C, Lavergne T, Walker S, Varma C, Linde C, Garrigue S, Kappenberger L, Haywood GA, Santini M, Bailleul C, Daubert JC. Effects of multisite biventricular pacing in patients with heart failure and intraventricular conduction delay. N Engl J Med. 2001;344:873–880. doi: 10.1056/NEJM200103223441202. [DOI] [PubMed] [Google Scholar]
- 6.Curry CW, Nelson GS, Wyman BT, Declerck J, Talbot M, Berger RD, McVeigh ER, Kass DA. Mechanical dyssynchrony in dilated cardiomyopathy with intraventricular conduction delay as depicted by 3D tagged magnetic resonance imaging. Circulation. 2000;101:E2. doi: 10.1161/01.cir.101.1.e2. [DOI] [PubMed] [Google Scholar]
- 7.Etienne Y, Mansourati J, Gilard M, Valls-Bertault V, Boschat J, Benditt DG, Lurie KG, Blanc JJ. Evaluation of left ventricular based pacing in patients with congestive heart failure and atrial fibrillation. Am J Cardiol. 1999;83:1138–1140. doi: 10.1016/s0002-9149(99)00031-4. A9. [DOI] [PubMed] [Google Scholar]
- 8.Faris O, Evans F, Ennis D, Helm P, Taylor J, Chesnick A, Guttman MA, Ozturk C, McVeigh E. A novel technique for cardiac electromechanical mapping with MRI tagging and an epicardial electrode sock. Ann Biomed Eng. 2003;31:430–440. doi: 10.1114/1.1560618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kass DA, Chen CH, Curry C, Talbot M, Berger R, Fetics B, Nevo E. Improved left ventricular mechanics from acute VDD pacing in patients with dilated cardiomyopathy and ventricular conduction delay. Circulation. 1999;99:1567–1573. doi: 10.1161/01.cir.99.12.1567. [DOI] [PubMed] [Google Scholar]
- 10.Leclercq C, Faris O, Tunin R, Johnson J, Kato R, Evans F, Spinelli J, Halperin H, McVeigh E, Kass DA. Systolic improvement and mechanical resynchronization does not require electrical synchrony in the dilated failing heart with left bundle-branch block. Circulation. 2002;106:1760–1763. doi: 10.1161/01.cir.0000035037.11968.5c. [DOI] [PubMed] [Google Scholar]
- 11.Nelson GS, Curry CW, Wyman BT, Kramer A, Declerck J, Talbot M, Douglas MR, Berger RD, McVeigh ER, Kass DA. Predictors of systolic augmentation from left ventricular preexcitation in patients with dilated cardiomyopathy and intraventricular conduction delay. Circulation. 2000;101:2703–2709. doi: 10.1161/01.cir.101.23.2703. [DOI] [PubMed] [Google Scholar]
- 12.Park RC, Little WC, O'Rourke RA. Effect of alteration of left ventricular activation sequence on the left ventricular endsystolic pressure-volume relation in closed-chest dogs. Circ Res. 1985;57:706–717. doi: 10.1161/01.res.57.5.706. [DOI] [PubMed] [Google Scholar]
- 13.Prinzen FW, Augustijn CH, Arts T, Allessie MA, Reneman RS. Redistribution of myocardial fiber strain and blood flow by asynchronous activation. Am J Physiol Heart Circ Physiol. 1990;259:H300–H308. doi: 10.1152/ajpheart.1990.259.2.H300. [DOI] [PubMed] [Google Scholar]
- 14.Prinzen FW, Hunter WC, Wyman BT, McVeigh ER. Mapping of regional myocardial strain and work during ventricular pacing: experimental study using magnetic resonance imaging tagging. J Am Coll Cardiol. 1999;33:1735–1742. doi: 10.1016/s0735-1097(99)00068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Touiza A, Etienne Y, Gilard M, Fatemi M, Mansourati J, Blanc JJ. Long-term left ventricular pacing: assessment and comparison with biventricular pacing in patients with severe congestive heart failure. J Am Coll Cardiol. 2001;38:1966–1970. doi: 10.1016/s0735-1097(01)01648-5. [DOI] [PubMed] [Google Scholar]
- 16.Verbeek XA, Vernooy K, Peschar M, Mutsaers D, van Hunnik A, Prinzen FW. Intraventricular but not interventricular resynchronization is required for optimal pacing therapy in hearts with left bundle branch block (Abstract) Circulation. 2002;106:II-380. [Google Scholar]
- 17.Wyman BT, Hunter WC, Prinzen FW, Faris OP, McVeigh ER. Effects of single- and biventricular pacing on temporal and spatial dynamics of ventricular contraction. Am J Physiol Heart Circ Physiol. 2002;282:H372–H379. doi: 10.1152/ajpheart.2002.282.1.H372. [DOI] [PubMed] [Google Scholar]
- 18.Wyman BT, Hunter WC, Prinzen FW, McVeigh ER. Mapping propagation of mechanical activation in the paced heart with MRI tagging. Am J Physiol Heart Circ Physiol. 1999;276:H881–H891. doi: 10.1152/ajpheart.1999.276.3.H881. [DOI] [PubMed] [Google Scholar]