

Abstract
The final step in heme biosynthesis, insertion of ferrous iron into protoporphyrin IX, is catalyzed by protoporphyrin IX ferrochelatase (E.C. 4.99.1.1). It is demonstrated that the pre-steady state human ferrochelatase (R115L) shows a stoichiometric burst of product formation and substrate consumption, consistent with a rate determining step following metal-ion chelation. Detailed analysis shows that chelation requires at least two steps, rapid binding followed by a slower (k ca. 1 s−1) irreversible step, provisionally assigned to metal ion chelation. Comparison with steady-state data reveals that the rate-determining step in the overall reaction, converting free porphyrin to free metalloporphyrin, occurs after chelation and is most probably product release. We have measured rate constants for significant steps on the enzyme and demonstrate that metal-ion chelation, with a rate constant of 0.96 s−1, is around 10 times faster than the rate determining step in the steady-state (kcat 0.1 s−1). The effect of an additional E343D mutation is apparent at multiple stages in the reaction cycle with a seven fold drop in kcat and three fold drop in kchel. This conservative mutation primarily affects events occurring after metal ion chelation. Further evaluation of structure-function data on site-directed mutants will therefore require both steady state and pre-steady state approaches.
Keywords: porphyrin biosynthesis, ferrochelatase, transient kinetics
The final step in heme biosynthesis, insertion of ferrous iron into protoporphyrin IX, is catalyzed by protoporphyrin IX ferrochelatase (protoheme ferro-lyase, E.C. 4.99.1.1, the human enzyme is hereafter referred to as ferrochelatase). The mechanisms of biological iron chelation have proved to be of great interest with a range of studies examining reaction kinetics (1-3), spectroscopy of bound intermediates (4-6), sensitivity of the reaction to structural variation (1, 3), and behavior of the enzyme analogues, including antibodies (4, 7-9) and both DNA and RNA (10-12) that also catalyze metal ion insertion into porphyrins. Additionally crystal structures of free enzyme (13, 14), as well as with bound metal substrate , bound porphyrin substrate (15) and a tight-binding competitive inhibitor (16) should support confident interpretation of the link between structure and function in this system. In fact, controversy remains over the role of individual residues in the reaction mechanism; compare for example the suggested metal ion binding site of Sellers et al. (1) with that proposed by Gora et al. (17); a recent review provides a fair summary (18). In part, this difficulty arises when the functional significance of individual residues is assessed on the basis of steady-state kinetic parameters which generally reflect multiple reaction steps as well as assignment of putative catalytically significant residues based upon an incorrect assumption that the N-alkyl porphyrin inhibitor binds in the same site and orientation as the native protoporphyrin substrate. Our development of a transient kinetic approach to monitor individual reaction steps should help resolve the mechanistic details of this system.
The in vivo delivery of substrates to ferrochelatase is an area of much current activity with recent proposals suggesting a role for chaperones and for substrate channeling. The porphyrin substrate, protoporphyrin IX, is synthesised from protoporphyrinogen IX in a 6-electron oxidation. The enzyme, protoporphyrinogen oxidase (PPO1, E.C. 1.3.3.4), is associated with the periplasmic side of the inner mitochondrial membrane; i.e. the opposite side of the membrane from ferrochelatase (19). A recent report of the crystal structure of a plant PPO reported that the membrane associated face of this enzyme is complementary to that of ferrochelatase (20). This was in accord with previous studies that suggested the two enzymes form a complex and that, in eukaryotic systems at least, protoporphyrin IX is channeled to ferrochelatase across the inner mitochondrial membrane (20, 21). It has also been proposed that iron is transported directly to ferrochelatase from the inner mitochondrial membrane (22, 23) although an alternative theory suggests that ferrous iron is transferred to ferrochelatase from frataxin, a putative mitochondrial iron chaperone (24).
Chelatases catalyze porphyrin metalation by stabilizi`ng a deformed porphyrin intermediate; this conclusion arises from a series of experimental data including resonance Raman spectroscopy of bound porphyrins (4, 5), co-crystallization of the non-planar N-methyl mesoporphyrin with the ferrochelatase from B. subtilis (16), and the use of non-planar porphyrins in generating model catalysts including both antibodies (4, 7-9) and nucleic acids (10-12). Recent theoretical calculations also support this conclusion (25, 26) as does the crystal structure of human ferrochelatase with bound protoporphyrin substrate (18). On the basis of a comparison of the observed rates of metal-ion insertion into a non-planar substrate analogue with steady-state kinetic parameters, porphyrin deformation has recently been proposed to be the key event in determining the metal-ion specificity of a particular metal-ion chelatase (18).
A considerable number of papers describe the steady-state behavior of ferrochelatase (e.g. 1, 2, 3), but there are very few reports of transient kinetics (3, 27). Most functional analysis and assessment of the significance of mutated residues has relied on steady-state information. As ferrochelatase catalyses the biologically critical conversion of a highly absorbing and fluorescent porphyrin into a non-fluorescent metaloporphyrin with a distinct absorbance spectrum, this enzyme is amenable to transient kinetic analyses (1). We have exploited the large optical signal associated with metal chelation in order to directly measure catalytic events in the active site of ferrochelatase.
Materials and Methods
Materials
Unless otherwise stated, chemicals were obtained from Sigma Chemical Co. (Poole, Dorset).
Enzyme purification
Recombinant human ferrochelatase (R115L) was purified essentially as described previously (28). Briefly, E. coli JM109 cells containing recombinant ferrochelatase were suspended in solubilization buffer (50 mM TRIS-MOPS, 0.1M KCl, 1% (w/v) sodium cholate, pH 8.0) containing 1mM 4-(2-Aminoethyl)-benzenesulfonyl fluoride, sonicated on ice for 3×30 s, and centrifuged at 50 000×g for 30 min at 4 °C. The supernatant was loaded onto a 2 ml Talon column (Clontech, Palo Alto, CA), previously equilibrated with solubilization buffer. The column was washed with 20 ml of solublization buffer, and 10 ml solublization buffer containing in addition 1 M KCl. Ferrochelatase was eluted with solublization buffer containing 300 mM imidazole. The column wash containing 1M KCl was used to remove any endogenous porphyrin. Imidazole was removed by applying purified protein to a 40 ml column of P-6DG (Biorad, Hemel Hempstead, UK) equilibrated in solublization buffer. Pure protein was stored under liquid N2 in 50 mM TRIS-MOPS, 0.1M KCl, 1% (w/v) sodium cholate, pH 8.0. Protein concentration was determined spectrophotometrically using the calculated extinction coefficient ε278 46900 M−1cm−1 (28).
Enzyme assays
Steady-state rates of metal ion insertion were determined using ferrous iron and deuteroporphyrin IX (Porphyrin products, Logan, UT) as substrates in a spectrophotometric assay (1, 29) with 0.17 μM active R115L ferrochelatase (determined by active site titration, 0.19 μM total enzyme) or with 0.16 μM active R115L, E343D ferrochelatase (determined by active site titration, 0.19 μM total enzyme). Reactions were carried out in 100 mM Tris-HCl, 0.5% Tween 20, 1 mM β-mercaptoethanol, pH 8.1 and at 30°C. Steady-state kinetic parameters were evaluated by fitting equation 1 to the initial rates using non-linear regression analysis implemented in Igor Pro. (Wavemetrics Inc., Lake Oswego, OR). Values of kcat and kcat/Km were estimated using the concentration of active enzyme derived from the active site titration described below.
| eqn. 1 |
Transient kinetic studies were performed on an Applied Photophysics Pi-star spectrophotometer with a 2 mm light path operating in fluorescence mode or a 10 mm light path operating in absorbance mode. In order to detect substrate fluorescence, excitation light from a Xenon source passed through a monochrometer set at 496 nm and emitted light was detected at 90° through a 515 nm cut-off filter (OG515, Schott). Absorbance traces were recorded at either 496 nm, measuring substrate depletion, or at 548 nm measuring product accumulation. Traces were collected under the conditions described above for steady state kinetics. Averaged traces (n ≥ 6) were evaluated by fitting the time course to equation 2, which describes a pre-steady state burst phase, with amplitude, A, and an observed rate constant, kobs, followed by a linear steady-state conversion of substrate into product characterized by a steady-state rate, vi, using the data analysis software (ProData 1.43; Applied Photophysics, Leatherhead, Surrey) supplied with the instrument. The burst amplitude is proportional to the concentration of intact active sites provided that the rate constant for the burst is sufficiently greater than the rate constant for the subsequent step (30). This relationship was used to estimate the concentration of an enzyme preparation in terms of active sites.
| eqn. 2 |
Results
Human ferrochelatase can be readily over-expressed and purified using established methods. This ferrochelatase (R115L) shows steady-state kinetic behavior and activity (Figure 1) similar to those seen before (1). Steady-state kinetic parameters were obtained by a global fit to equation 1. The apparent kinetic parameters obtained from individual vi vs. [S] curves show good agreement with the theoretical curves calculated from the global fit (Figure 2). The R115L ferrochelatase mutant increases stability without a significant impact on enzyme activity (28); a crystal structure of this mutant is available (PDB code 1HRK, (13)). The R115L, E343D double mutant has a substantially lower kcat than the R115L mutant but similar Km values (1). In both cases, purification using previously described methods (28) yields protein with a small amount of bound porphyrin, which was removed by incorporating a high salt wash (1M KCl) into the purification procedure. This is an essential step as adventitious porphyrin reduces the concentration of available enzyme active sites and thus the observable amplitude of the pre-steady state transient.
Figure 1.
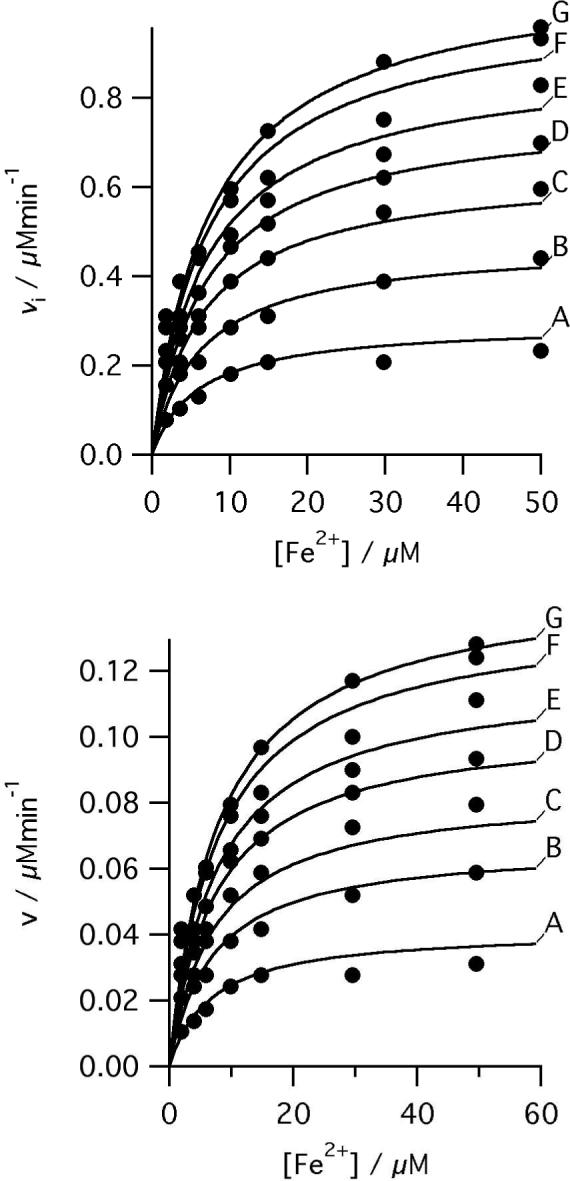
Initial rates of ferrochelatase catalyzed insertion of iron into deuteroporphyrin at pH 8.1, 100mM TRIS-HCl, 250 μM Fe2+, 1mM β mercaptoethanol, 0.5% (v/v) Tween 20, 30°C. Top, 0.17 μM R115L ferrochelatase, deuteroporphyrin concentrations were A 1.8 μM, B 3.6 μM, C 6.3 μM, D 9.9 μM, E 15.3 μM, F 29.7 μM, and G 49.6 μM. The points are experimental and the theoretical lines are described by equation 1 with characterizing parameters KmFe 7.7 ± 1.4 μM, KmDIX 5.5 ± 0.5 μM, KFe.DIX 30.54 ± 8.01 μM2, V 1.21 μMmin−1. Bottom, 0.16 μM R115L, E343D ferrochelatase, deuteroporphyrin concentrations were A 1.9 μM, B 4.0 μM, C 5.9 μM, D 9.9 μM, E 14.9 μM, F 29.7 μM, and G 49.7 μM. The points are experimental and the theoretical lines are described by equation 1 with characterizing parameters KmFe 8.2 ± 1.0 μM, KmDIX 5.8 ± 0.8 μM, KFe.DIX 34.8 ± 8.3 μM2, V 0.16 ± 0.01 μMmin−1.
Figure 2.
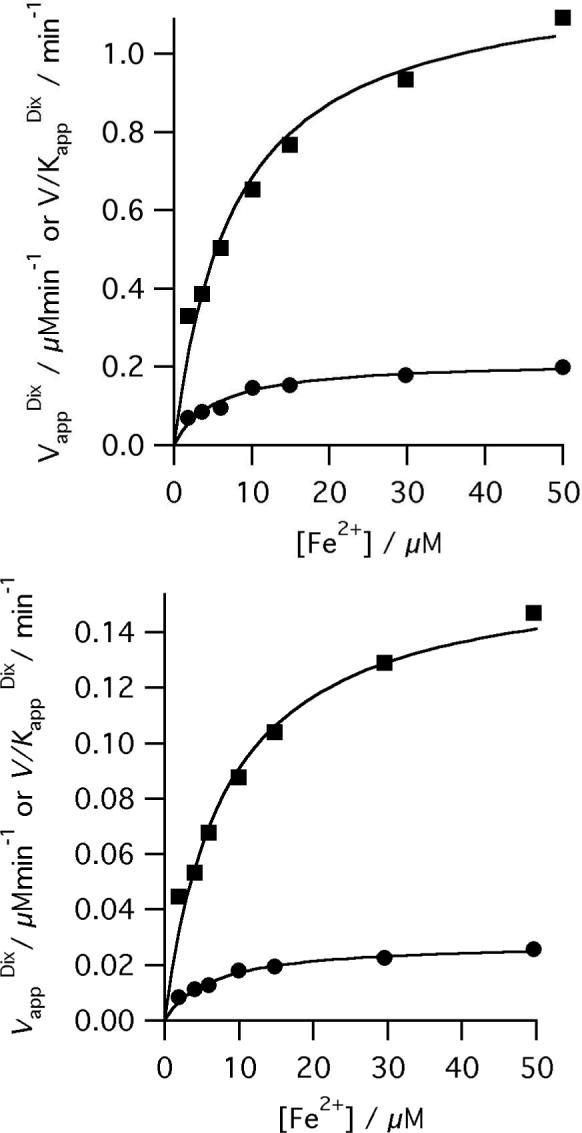
Secondary plots showing the iron dependence of the observed kinetic parameters. The points are the apparent kinetic parameters that arise from fitting the Michaelis-Menten equation to individual vi vs. [S] curves and the lines are described by equation 1 (Vapp filled squares, V/Kapp filled circles) with characterizing parameters, (top) for R115L ferrochelatase KmFe 7.7 ± 1. 4 μM, KmDIX 5.5 ± 0.5 μM, KFe.DIX 30.54 ± 8.01 μM2, V 1.21 μMmin−1 and (bottom) R115L, E343D ferrochelatase KmFe 8.2 ± 1.0 μM, KmDIX 5.8 ± 0.8 μM, KFe.DIX 34.8 ± 8.3 μM2, V 0.16 ± 0.01 μMmin−1.
A pre-steady-state burst phase was measured using either stopped-flow absorbance (data not shown) or fluorescence spectroscopy (Figure 3). The fluorescence measurements monitored substrate consumption. Amplitudes of product formation and substrate consumption were quantified from the absorbance data. Shifts in porphyrin spectra on ferrochelatase binding have been described previously (31) and could potentially interfere with a spectroscopic active site titration. In this case, as the enzyme-porphyrin complex does not accumulate, the burst in substrate consumption arises from loss of free substrate and can be accurately quantified. Additionally, as amplitudes of substrate consumption and product formation agree closely at 548nm there is little difference between the extinction coefficients of bound and free product. The amplitude of the burst in product formation was calculated to correspond to 90% of the total enzyme concentration. This relationship holds as the concentration of ferrochelatase is varied from 0.5 − 5 μM.
Figure 3.
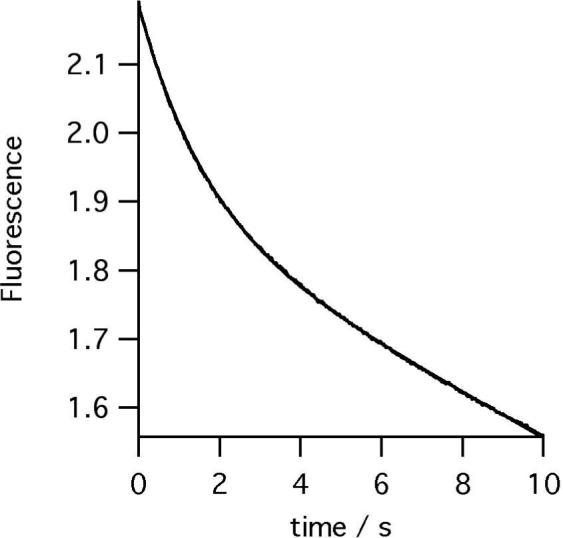
Time-course of DIX (25 μM) consumption by ferrochelatase (0.5 μM) at pH 8.1, 100mM TRIS-HCl, 250 μM Fe2+, 1mM β mercaptoethanol, 0.5% (v/v) Tween 20, 30°C.
The burst in metaloporphyrin formation allows us to directly measure metal ion insertion into the porphyrin ring. Progress curves can be described by a single exponential phase followed by a linear steady-state conversion of substrate into product (equation 2). The observed rate constant (kobs) for metal-ion insertion catalyzed by R115L ferrochelatase shows a hyperbolic dependence on porphyrin concentration with saturating ferrous iron (Figure 4). Similarly, when porphyrin is saturating, the observed rate constant shows a hyperbolic dependence on the concentration of ferrous iron (Figure 5). The R115L, E343D mutant also shows similar hyperbolic concentration dependencies on porphyrin (Figure 6) and ferrous iron concentration (Figure 7). These behaviors are characteristic of a two-step mechanism with rapid substrate binding followed by a slower step, in this case metal ion chelation (Scheme 1). The relationship between the observed rate constant, kobs, and substrate concentration (Equation 3) predicts a positive ordinal intercept if the rate constant for on-enzyme dechelation, is significant. This is not observed, so the on-enzyme metal-ion chelation can be treated as irreversible.
| eqn. 3 |
Figure 4.
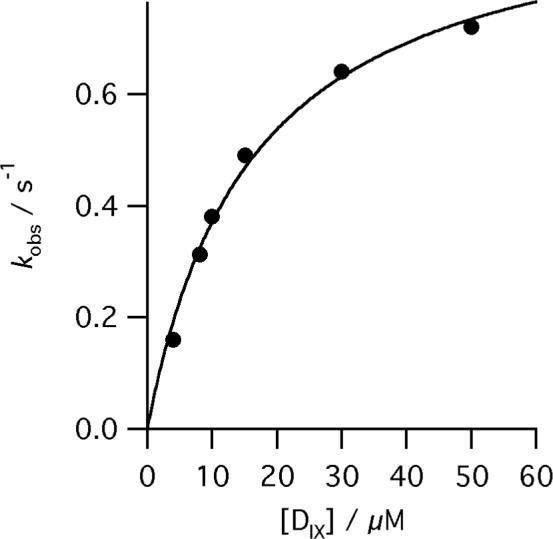
The dependence of kobs for the reaction of R115L human ferrochelatase (0.5 μM) on deuteroporphyrin concentration at pH 8.1, 100mM TRIS-HCl, 250 μM Fe2+, 1mM β mercaptoethanol, 0.5% (v/v) Tween 20, 30°C. The points are experimental and the theoretical line is described by equation 3 and characterizing parameters kchel 0.97 ± 0.05 s−1 and KsDIX 16.3 ± 1.9 μM.
Figure 5.
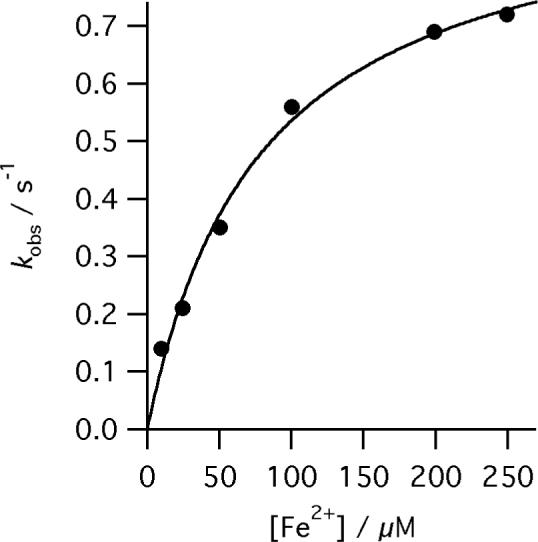
The dependence of kobs for the reaction of R115L human ferrochelatase (0.5 μM) on Fe2+ concentration at pH 8.1, 100mM TRIS-HCl, 50 μM DIX, 1mM β metcaptoethanol, 0.5% (v/v) Tween 20, 30°C. The points are experimental and the theoretical line is described by equation 3 and characterizing parameters kchel 0.96 ± 0.05 s−1 and KsFe 79.1 ± 11.2 μM.
Figure 6.
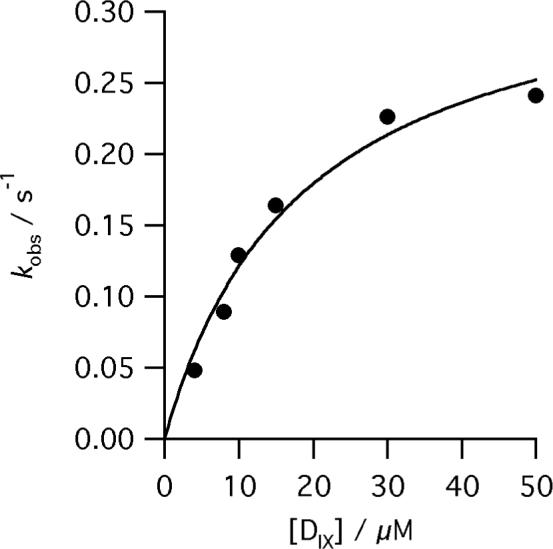
The dependence of kobs for the reaction of E343D, R115L human ferrochelatase (0.5 μM) on deuteroporphyrin concentration at pH 8.1, 100mM TRIS-HCl, 350 μM Fe2+, 1mM β mercaptoethanol, 0.5% (v/v) Tween 20, 30°C. The points are experimental and the theoretical line is described by equation 3 and characterizing parameters kchel 0.35 ± 0.3 s−1 and KsDIX 18.6 ± 4.2 μM.
Figure 7.
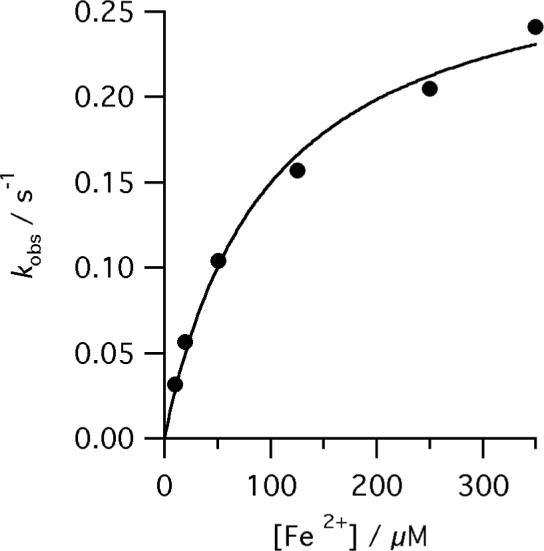
The dependence of kobs for the reaction of E343D, R115L human ferrochelatase (0.5 μM) on Fe2+ concentration at pH 8.1, 100mM TRIS-HCl, 50 μM DIX, 1mM β metcaptoethanol, 0.5% (v/v) Tween 20, 30°C. The points are experimental and the theoretical line is described by equation 3 and characterizing parameters kchel 0.30 ± 0.03 s−1 and KsFe 97.6 ± 15.7 μM.
Scheme 1.
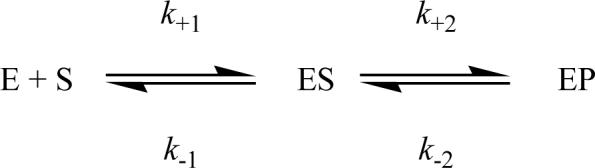
Two step substrate binding to an enzyme, E.
Discussion
Even though extensive steady-state analyses of both wild-type and mutant ferrochelatases are available (1), no systematic transient kinetic analysis of the ferrochelatase catalyzed reaction has been carried out. We therefore investigated the pre-steady state kinetics of the ferrochelatase catalyzed insertion of ferrous iron into a porphyrin macrocycle. A mutant ferrochelatase (R115L) with behavior similar to wild type and a double mutant (E343D, R115L) with a substantially reduced kcat were studied. The observed kinetic behavior of both ferrochelatases allowed us to conclude that chelation is not rate-determining in the steady-state and that an additional slow step is required for product release. The effect of the additional E343D mutation is apparent at multiple stages in the reaction cycle emphasizing the benefits available from a transient kinetic analysis of mutant ferrochelatases.
A pre-steady state burst has recently been observed in the murine ferrochelatase catalyzed insertion of zinc into protoporphyrin IX (27); the transient was used to make a detailed study of binding of the inhibitor N-methyl protoporphyrin. However, rate constants for metal ion insertion in the active site were not determined.
The observed burst in formation of product demonstrates that metaloporphyrin formation is fast compared to the rate-determining processes in the steady-state. The amplitude of the burst phase, ca. 0.9[E], reveals that in the steady-state ferrochelatase exists as an enzyme-product complex. This provides the basis for an active-site titration allowing us to calculate kcat and kcat/Km for ferrochelatase (30, 32). It is possible that some site-directed mutations will have a deleterious effect on enzyme stability, resulting in an underestimation of steady-state kinetic parameters (33).
In the case of the steady-state data presented in this paper, which benefit from knowledge of the active-site concentration, the estimated steady-state parameters do not differ substantially from those described previously. This is most probably a result of both the speed of the purification procedure and the stability of the purified material used for steady-state analysis (1). A recent study revealed that active site mutations can stabilize a series of mutant and wild-type murine ferrochelatases, although this effect may, at least in part, arise from binding to endogenous porphyrin (33).
This paper presents the first determination of the steady state kinetic parameters kcat, kcat/KmDIX and kcat/KmFe for any ferrochelatase based on accurate knowledge of the active enzyme concentration. This allows us to compare rate constants for active site chemistry (kchel) and for the overall reaction (kcat); without this information it could be argued that the fact that kchel is greater than kcat was solely due to an overestimation of active enzyme concentration.
The substrate dependence of kobs allows us to determine k+2 as 0.96 ± 0.05 s−1 in the case of R115L ferrochelatase (Figures 4 and 5) and ca. 0.3 s−1 with the E343D mutation (Figure 6 and 7). This is either the rate constant for metal ion chelation or that of a rapid isomerisation concomitant with the chelation reaction. This rate constant is thus a lower limit for the metal-ion chelation rate constant. As, in both cases, kcat is much less that k+2 another slow step is needed to explain the observed kinetics. We therefore extend our model to include an additional slow step after metal ion insertion (Scheme 2). This step includes product disassociation and may also include a kinetically significant isomerisation of an enzyme-deuteroheme complex. This additional step is rate determining in the steady state and it is the main contributor to kcat (eqn. 4). The rate constant for the on-enzyme dechelation reaction, in Scheme 2, k-chel is negligible in the systems described here. We have included it in our model and our descriptions of steady-state parameters for the assessment of mutant ferrochelatases and alternative substrates, where k–chel ≠ 0 and analysis based on the obvious simplified forms of eqn. 4 and eqn. 5 would be misleading.
| eqn. 4 |
Scheme 2.

A minimal model for the ferrochelatase reaction.
Values of in scheme 1 can be estimated from the relationship between substrate concentration and the observed rate constant for metal ion insertion. These parameters relate to the quasi-equilibrium around the ES complex in scheme 1, and in some cases (i.e. k+2 << k−1) can approximate to Ks, the disassociation constant for the ES complex. It is worth noting that our estimates of Km and are significantly different. This arises from the significant contribution that the EP species makes to Km but not to (32).
This minimal model for the ferrochelatase catalyzed insertion of iron into deuteroporphyrin (Scheme 2) is shown, for convenience, with random binding of the two substrates. The possibility of ordered binding, as seen with the bovine enzyme (2), does not significantly affect our analysis. It has been suggested that in vivo both substrates are channeled to ferrochelatase (20-24). So in vitro studies, carried out without the appropriate partner proteins, may not accurately reflect the sequence of binding events occurring in vivo. In any event, this work is concerned with the behavior of metal ion and porphyrin in the active site of ferrochelatase.
In this model (Scheme 2) we propose that metal ion binding is fast. Previous structural models of metal ion binding have described a multi-stage process where metal binds initially in a remote site and then moves to the active site (34). This model is consistent with the data presented here provided that transport of iron to the active site has a rate constant much greater than 0.96 s−1.
Using our model we can determine the significance of the steady-state parameters kcat and kcat/Km. The turnover number, kcat, contains contributions from both chelation and product release (eqn. 4). The Km values for both substrates contain contributions from chelation and from product disassociation; for example the Km for DIX with random substrate binding and reversible metal ion chelation is . These parameters cannot then be used as indicators of substrate binding. This is particularly important for kinetic analysis of mutant enzymes where Km values have frequently been used for this purpose. The apparent second order rate constant for conversion of free substrate to product, kcat/Km (eqn. 5), is more useful in this regard, as it reflects productive substrate binding (32).
| eqn. 5 |
The kinetic consequences of the additional E343D mutation are a seven-fold reduction in kcat but very little change in Km for either substrate (1). We show that kchel is significantly reduced (ca. three-fold), although not to the same extent as kcat, suggesting that this conservative mutation perturbs the role of this residue in product release as well as metal ion chelation. We observe no changes in suggesting that this conservative mutation does not perturb substrate binding. Previously, this residue has been suggested to have a role in abstraction of protons from the substrate (1) or in metal ion binding (34, 35).
A mutation in the analogous murine residue, E287Q, shows a different form of porphyrin distortion to the wild-type (31). Single turnovers of this mutant and the alanine variant suggest that they bind product more tightly (Kd less than 20 μM) than the wild-type which has a Kd greater than 30 μM (3). The effect of mutations at an analogous position (E264Q, E264V) have been considered in the B. subtilis enzyme (35). Despite an 80% reduction in activity in the E264Q mutant, zinc binding was not perturbed, while the E264V mutant was inactive and zinc binding could not be measured. These results are similar to those described for human ferrochelatase. While the analogous human mutant (E343Q) shows no activity, the conservative mutation E343D has a sharply reduced kcat with no change in substrate Km values (1).
Our results are consistent with these observations. We see no significant change in . This parameter can approximate to the disassociation constant for a substrate from the ternary complex. In contrast, the seven fold drop in kcat can be attributed to events that occur after metal-ion chelation. A role in product release for this residue has been previously been inferred (1, 3). Additionally, the E343D mutation lowers kchel, disrupting the steps that lead to metal ion chelation. Overall these results demonstrate that this residue plays a role in multiple steps in the ferrochelatase mechanism. The dual role of this residue is also apparent from the E343K variant which binds and co-crystallizes with porphyrin (15), but is inactive (1).
In conclusion, we have demonstrated that metal ion chelation is not rate determining in the catalytic cycle of human ferrochelatase. Therefore, steady-state kinetics do not generally provide any information on the significant chemical transformation on the enzyme. In particular Km values do not reflect substrate binding and kcat values are dominated by product release. A simple, transient kinetic approach, however, allows us to measure rate constants for significant steps on the enzyme. This approach allows us to demonstrate that metal-ion chelation, observed with a rate constant of 0.96 s−1, is at least 10 times faster then the rate determining step in the steady-state (kcat 0.1 s−1). Evaluation of structure-function data on site-directed mutants therefore requires both steady state and pre-steady state approaches. As an example ,the E343D mutant, which displays no alterations in , nevertheless shows substantial changes in pre-steady state kinetic behavior at multiple reaction steps with a seven fold drop in kcat and three fold drop in kchel, This conservative mutation primarily affects events occurring after metal ion chelation.
ABBREVIATIONS
- PPO
protoporphyrinogen oxidase
- DIX
deuteroporphyrin IX
Footnotes
Funded by the BBSRC (UK).
Bibliography
- 1.Sellers VM, Wu C-K, Dailey TA, Dailey HA. Human ferrochelatase: characterization of substrate-iron binding and proton-abstracting residues. Biochemistry. 2001;40:9821–9827. doi: 10.1021/bi010012c. [DOI] [PubMed] [Google Scholar]
- 2.Dailey HA, Fleming JE. Bovine ferrochelatase. Kinetic analysis of inhibition by N-methylprotoporphyrin, manganese, and heme. Journal of Biological Chemistry. 1983;258:11453–11459. [PubMed] [Google Scholar]
- 3.Franco R, Pereira AS, Tavares P, Mangravita A, Barber MJ, Moura I, Ferreira GC. Substitution of murine ferrochelatase glutamate-287 with glutamine or alanine leads to porphyrin substrate-bound variants. Biochemical Journal. 2001;356:217–222. doi: 10.1042/0264-6021:3560217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romesberg FE, Santarsiero BD, Spiller B, Yin J, Barnes D, Schultz PG, Stevens RC. Structural and kinetic evidence for strain in biological catalysis. Biochemistry. 1998;37:14404–14409. doi: 10.1021/bi981578c. [DOI] [PubMed] [Google Scholar]
- 5.Blackwood ME, Jr., Rush TS, III, Medlock A, Dailey HA, Spiro TG. Resonance raman spectra of ferrochelatase reveal porphyrin distortion upon metal binding. Journal of the American Chemical Society. 1997;119:12170–12174. [Google Scholar]
- 6.Shi Z, Franco R, Haddad R, Shelnutt JA, Ferreira GC. The Conserved Active-Site Loop Residues of Ferrochelatase Induce Porphyrin Conformational Changes Necessary for Catalysis. Biochemistry. 2006;45:2904–2912. doi: 10.1021/bi051907i. [DOI] [PubMed] [Google Scholar]
- 7.Blackwood ME, Jr., Rush TS, III, Romesberg F, Schultz PG, Spiro TG. Alternative modes of substrate distortion in enzyme and antibody catalyzed ferrochelation reactions. Biochemistry. 1998;37:779–782. doi: 10.1021/bi972616f. [DOI] [PubMed] [Google Scholar]
- 8.Cochran AG, Schultz PG. Antibody-catalyzed porphyrin metallation. Science. 1990;249:781–783. doi: 10.1126/science.2389144. [DOI] [PubMed] [Google Scholar]
- 9.Venkateshrao S, Yin J, Jarzecki AA, Schultz PG, Spiro TG. Porphyrin distortion during affinity maturation of a ferrochelatase antibody, monitored by Resonance Raman spectroscopy. J Am Chem Soc. 2004;126:16361–16367. doi: 10.1021/ja0465395. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Sen D. A catalytic DNA for porphyrin metallation. Nat Struct Biol. 1996;3:743–747. doi: 10.1038/nsb0996-743. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Sen D. Toward an efficient DNAzyme. Biochemistry. 1997;36:5589–5599. doi: 10.1021/bi962694n. [DOI] [PubMed] [Google Scholar]
- 12.Conn MM, Schultz PG. Porphyrin Metalation Catalyzed by a Small RNA Molecule. J Am Chem Soc. 1996;118:7012–7013. [Google Scholar]
- 13.Wu CK, Dailey HA, Rose JP, Burden A, Sellers VM, Wang BC. The 2.0 A structure of human ferrochelatase, the terminal enzyme of heme biosynthesis. Nat Struct Mol Biol. 2001;8:156–160. doi: 10.1038/84152. [DOI] [PubMed] [Google Scholar]
- 14.Al-Karadaghi S, Hansson M, Nikonov S, Jånsson B, Hederstedt L. Crystal structure of ferrochelatase: the terminal enzyme in heme biosynthesis. Structure. 1997;5:1501–1510. doi: 10.1016/s0969-2126(97)00299-2. [DOI] [PubMed] [Google Scholar]
- 15.Medlock A, Swartz L, Dailey TA, Dailey HA, Lanzilotta WN. Substrate interactions with human ferrochelatase. Proc Natl Acad Sci U S A. 2007;104:1789–1793. doi: 10.1073/pnas.0606144104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lecerof D, Fodje M, Hansson A, Hansson M, Al Karadaghi S. Structural and mechanistic basis of porphyrin metallation by ferrochelatase. J.Mol.Biol. 2000;297:221–232. doi: 10.1006/jmbi.2000.3569. [DOI] [PubMed] [Google Scholar]
- 17.Gora M, Grzybowska E, Rytka J, Labbe-Bois R. Probing the active-site residues in Saccharomyces cerevisiae ferrochelatase by directed mutagenesis. In vivo and in vitro analyses. Journal of Biological Chemistry. 1996;271:11810–11816. doi: 10.1074/jbc.271.20.11810. [DOI] [PubMed] [Google Scholar]
- 18.Al-Karadaghi S, Franco R, Hansson M, Shelnutt JA, Isaya G, Ferreira GC. Chelatases: distort to select? Trends Biochem Sci. 2006;31:135–142. doi: 10.1016/j.tibs.2006.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dailey HA, Dailey TA. Ferrochelatase. In: Kadish KM, Smith KM, Guildard R, editors. The Porphyrin Handbook. 2003. pp. 87–114. [Google Scholar]
- 20.Koch M, Breithaupt C, Kiefersauer R, Freigang J, Huber R, Messerschmidt A. Crystal structure of protoporphyrinogen IX oxidase: a key enzyme in haem and chlorophyll biosynthesis. EMBO J. 2004;23:1720–1728. doi: 10.1038/sj.emboj.7600189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Proulx KL, Woodard SI, Dailey HA. In situ conversion of coproporphyrinogen to heme by murine mitochondria: terminal steps of the heme biosynthetic pathway. Protein Sci. 1993;2:1092–1098. doi: 10.1002/pro.5560020703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lange H, Kispal G, Lill R. Mechanism of Iron Transport to the Site of Heme Synthesis inside Yeast Mitochondria. Journal of Biological Chemistry. 1999;274:18989–18996. doi: 10.1074/jbc.274.27.18989. [DOI] [PubMed] [Google Scholar]
- 23.Shaw GC, Cope JJ, Li L, Corson K, Hersey C, Ackermann GE, Gwynn B, Lambert AJ, Wingert RA, Traver D, Trede NS, Barut BA, Zhou Y, Minet E, Donovan A, Brownlie A, Balzan R, Weiss MJ, Peters LL, Kaplan J, Zon LI, Paw BH. Mitoferrin is essential for erythroid iron assimilation. Nature. 2006;440:96–100. doi: 10.1038/nature04512. [DOI] [PubMed] [Google Scholar]
- 24.Yoon T, Cowan JA. Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J Biol Chem. 2004;279:25943–25946. doi: 10.1074/jbc.C400107200. [DOI] [PubMed] [Google Scholar]
- 25.Shen Y, Ryde U. Reaction mechanism of porphyrin metallation studied by theoretical methods. Chemistry. 2005;11:1549–1564. doi: 10.1002/chem.200400298. [DOI] [PubMed] [Google Scholar]
- 26.Sigfridsson E, Ryde U. The importance of porphyrin distortions for the ferrochelatase reaction. Journal of Biological Inorganic Chemistry. 2003;8:273–282. doi: 10.1007/s00775-002-0413-8. [DOI] [PubMed] [Google Scholar]
- 27.Shi Z, Ferreira GC. Modulation of inhibition of ferrochelatase by N-methylprotoporphyrin. Biochem J. 2006;399:21–28. doi: 10.1042/BJ20060753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burden AE, Wu C, Dailey TA, Busch JL, Dhawan IK, Rose JP, Wang B, Dailey HA. Human ferrochelatase: crystallization, characterization of the [2Fe-2S] cluster and determination that the enzyme is a homodimer. Biochim Biophys Acta. 1999;1435:191–197. doi: 10.1016/s0167-4838(99)00196-x. [DOI] [PubMed] [Google Scholar]
- 29.Porra RJ, Vitols KS, Labbe RF, Newton NA. Studies on ferrochelatase. The effects of thiols and other factors on the determination of activity. Biochemical Journal. 1967;104:321–327. doi: 10.1042/bj1040321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bender ML, Begue-Canton ML, Blakeley RL, Brubacher LJ, Feder J, Gunter CR, Kezdy FJ, Killheffer JV, Jr., Marshall TH, Miller CG, Roeske RW, Stoops JK. The determination of the concentration of hydrolytic enzyme solutions: alpha-chymotrypsin, trypsin, papain, elastase, subtilisin, and acetylcholinesterase. J Am Chem Soc. 1966;88:5890–5913. doi: 10.1021/ja00976a034. [DOI] [PubMed] [Google Scholar]
- 31.Franco R, Ma JG, Lu Y, Ferreira GC, Shelnutt JA. Porphyrin interactions with wild-type and mutant mouse ferrochelatase. Biochemistry. 2000;39:2517–2529. doi: 10.1021/bi991346t. [DOI] [PubMed] [Google Scholar]
- 32.Fersht AR. Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. Freeman; New York: 1999. [Google Scholar]
- 33.Franco R, Bai G, Prosinecki V, Abrunhosa F, Ferreira GC, Bastos M. Porphyrin-substrate binding to murine ferrochelatase: effect on the thermal stability of the enzyme. Biochem J. 2005;386:599–605. doi: 10.1042/BJ20040921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karlberg T, Lecerof D, Gora M, Silvegren G, Labbe-Bois R, Hansson M, Al Karadaghi S. Metal Binding to Saccharomyces cerevisiae Ferrochelatase. Biochemistry. 2002;41:13499–13506. doi: 10.1021/bi0260785. [DOI] [PubMed] [Google Scholar]
- 35.Hansson MD, Karlberg T, Rahardja MA, Al-Karadaghi S, Hansson M. Amino Acid Residues His183 and Glu264 in Bacillus subtilis Ferrochelatase Direct and Facilitate the Insertion of Metal Ion into Protoporphyrin IX. Biochemistry. 2007;46:87–94. doi: 10.1021/bi061760a. [DOI] [PubMed] [Google Scholar]