


Abstract
Compared to the available protein sequences of different organisms, the number of revealed protein–protein interactions (PPIs) is still very limited. So many computational methods have been developed to facilitate the identification of novel PPIs. However, the methods only using the information of protein sequences are more universal than those that depend on some additional information or predictions about the proteins. In this article, a sequence-based method is proposed by combining a new feature representation using auto covariance (AC) and support vector machine (SVM). AC accounts for the interactions between residues a certain distance apart in the sequence, so this method adequately takes the neighbouring effect into account. When performed on the PPI data of yeast Saccharomyces cerevisiae, the method achieved a very promising prediction result. An independent data set of 11 474 yeast PPIs was used to evaluate this prediction model and the prediction accuracy is 88.09%. The performance of this method is superior to those of the existing sequence-based methods, so it can be a useful supplementary tool for future proteomics studies. The prediction software and all data sets used in this article are freely available at http://www.scucic.cn/Predict_PPI/index.htm.
INTRODUCTION
Identification of protein–protein interactions (PPIs) is crucial for elucidating protein functions and further understanding various biological processes in a cell. It has been the focus of the post-proteomic researches. In recent years, various experimental techniques have been developed for the large-scale PPI analysis, including yeast two-hybrid systems (1,2), mass spectrometry (3,4), protein chip (5) and so on. Because experimental methods are time-consuming and expensive, current PPI pairs obtained from experiments only cover a small fraction of the complete PPI networks (6). Hence, it is of great practical significance to develop the reliable computational methods to facilitate the identification of PPIs.
So far, a number of computational methods have been proposed for the prediction of PPIs. Some methods are based on the genomic information, such as phylogenetic profiles (7), gene neighbourhood (8) and gene fusion events (9,10). Methods using the structural information of proteins (11–13) and the sequence conservation between interacting proteins (14,15) have been reported. Previously predicted (known protein) domains that are responsible for the interactions between proteins have also been considered too (16–20). However, all these methods cannot be implemented if such pre-knowledge about the proteins is not available. Several sequence-based methods (21–25) have shown that the information of amino acid sequences alone may be sufficient to identify novel PPIs, but the highest accuracy of these methods is only ∼80%, such as the methods by Martin et al. (22), and Chou and Cai (25). So Shen et al. (26) have developed an alternative method that yields a high prediction accuracy of 83.9%, when applied to predicting human PPIs. This method considers the local environments of residues through a conjoint triad method, but it only accounts for the properties of one amino acid and its proximate two amino acids. However, the interactions usually occur in the discontinuous amino acids segments in the sequence, and the information of these interactions may be able to further improve the prediction ability of the existing sequence-based methods.
In this article, a new method based on support vector machine (SVM) and auto covariance (AC) was proposed. AC accounts for the interactions between amino acids within a certain number of amino acids apart in the sequence, so this method takes neighbouring effect into account and makes it possible to discover patterns that run through entire sequences. The amino acid residues were translated into numerical values representing physicochemical properties, and then these numerical sequences were analysed by AC based on the calculation of covariance. Finally, the SVM model was constructed using the vectors of AC variables as input. The optimization experiment demonstrated that the interactions of one amino acid and its 30 vicinal amino acids would contribute to characterizing the PPI information. The method was tested by the PPI data of yeast Saccharomyces cerevisiae and yielded a prediction accuracy of 87.36%. At last, this model was further evaluated by an independent data set of other yeast PPIs with the prediction accuracy of 88.09%.
MATERIALS AND METHODS
Data collection and data set construction
The PPI data was collected from Saccharomyces cerevisiae core subset of database of interacting proteins (DIP) (27), version DIP_20070219. The reliability of this core subset has been tested by two methods, expression profile reliability (EPR) and paralogous verification method (PVM) (28). At the time of doing the experiments, the core subset contained 5966 interaction pairs. The protein pairs that contained a protein with <50 amino acids were removed and the remaining 5943 protein pairs comprised the final positive data set. All proteins in the data set were aligned using the multiple sequence alignment tool, cd-hit program (29). The aligned result shows that among the 5943 protein pairs, the overwhelming majority of them (5594 PPIs) have <40% pairwise sequence identity to one another. Although there are only 349 pairs with ≥40% identity in the training data set, the classifier will possibly be biased to these homologous sequence pairs.
Since the non-interacting pairs were not readily available, three strategies for constructing negative data set were used in order to compare the effects of different training data sets on the performance of the method. The first strategy has been described by Shen and colleagues (26) in detail. The non-interacting pairs were generated by randomly pairing proteins that appeared in the positive data set. Here the negative data set based on this method is called Prcp. The second is based on such an assumption that proteins occupying different subcellular localizations do not interact. The subcellular localization information of the proteins in the positive data set was extracted from Swiss-Prot (http://www.expasy.org/sprot/). The proteins without subcellular localization information and those denoted as ‘putative’, ‘hypothetical’ were excluded. The remaining proteins were grouped into eight subsets based on the eight main types of localization—cytoplasm, nucleus, mitochondrion, endoplasmic reticulum, golgi apparatus, peroxisome, vacuole and cytoplasm&nucleus. Each subset contained 10 proteins at least. The non-interacting pairs were generated by pairing proteins from one subset with proteins from the other subset. It must be pointed out that proteins from cytoplasm subset and nucleus subset cannot be paired with those from cytoplasm&nucleus subset. Here the negative data set based on subcellular localization information is called Psub. The two strategies must meet three requirements: (i) the non-interacting pairs cannot appear in the whole DIP yeast interacting pairs, (ii) the number of negative pairs is equal to that of positive pairs and (iii) the contribution of proteins in negative set should be as harmonious as possible (24,26).
As a comparison, the third strategy was used for creating non-interacting pairs composed of artificial protein sequences. It has been demonstrated that if a sequence of one interacting pair is shuffled, then the two proteins can be deemed not to interact with each other (30). Thus, the negative data set was prepared by shuffling the sequences of right-side interacting pairs with k-let (k = 1,2,3) counts using the Shufflet program (31).
Feature extraction and AC
Protein–protein interaction can be defined as four interaction modes: electrostatic interaction, hydrophobic interaction, steric interaction and hydrogen bond. Here seven physicochemical properties of amino acids were selected to reflect these interaction modes whenever possible and they are hydrophobicity (32), hydrophicility (33), volumes of side chains of amino acids (34), polarity (35), polarizability (36), solvent-accessible surface area (SASA) (37) and net charge index (NCI) of side chains of amino acids (38), respectively. The original values of the seven physicochemical properties for each amino acid are listed in Supplementary Table S1. They were first normalized to zero mean and unit standard deviation (SD) according to Equation (1):
| 1 |
where Pi,j is the j-th descriptor value for i-th amino acid, Pj the mean of j-th descriptor over the 20 amino acids and Sj the corresponding SD. Then each protein sequence was translated into seven vectors with each amino acid represented by the normalized values of seven descriptors.
Artificial intelligence-based techniques such as SVM and the neural network require a fixed number of inputs for training. However, there are often unequal-length vectors because of protein sequences with different lengths. So auto cross covariance (ACC) was used to transform these numerical vectors into uniform matrices. As a statistical tool for analyzing sequences of vectors developed by Wold et al. (39), ACC has been adopted by more and more leading investigators for protein classification (40–42). ACC results in two kinds of variables, AC between the same descriptor, and cross covariance (CC) between two different descriptors. In this study, only AC variables were used in order to avoid generating too large number of variants, compared to the limited number of PPI pairs. Given a protein sequence, AC variables describe the average interactions between residues, a certain lag apart throughout the whole sequence. Here, lag is the distance between one residue and its neighbour, a certain number of residues away. The AC variables are calculated according to Equation (2), where j represents one descriptor, i the position in the sequence X, n the length of the sequence X and lag the value of the lag.
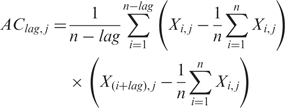 |
2 |
In this way, the number of AC variables, D can be calculated as D = lg × P, where P is the number of descriptors and lg is the maximum lag (lag = 1, 2, … , lg). After each protein sequence was represented as a vector of AC variables, a protein pair was characterized by concatenating the vectors of two proteins in this protein pair.
Model construction
The classification model for predicting PPIs was based on SVM. Vapnik (43) has given a full description about how to use SVM to do classification. The software libsvm 2.84 (http://www.csie.ntu.edu.tw/~cjlin/libsvm/) was employed in this work. A radial basis function (RBF) was chosen as the kernel function. Two parameters, the regularization parameter C and the kernel width parameter γ were optimized using a grid search approach. In statistical prediction, sub-sampling test and jackknife test are often used as two cross-validation methods (44). Jackknife test is deemed more objective and has been widely adopted by many investigators (41,45–56) to test the power of various predictors, but it will take much long time to perform the jackknife test. Although it has been demonstrated that the sub-sampling test cannot avoid arbitrariness according to a recent comprehensive review (45) and a penetrating analysis in (57), it is still a good validation method for the large data set. Considering the numerous samples used in this work, 5-fold cross-validation was used to investigate the training set.
The final data set consisted of 11 886 protein pairs, half from the positive data set and half from the negative data set. Here three-fifths of the protein pairs respectively from the positive and negative data set were randomly chosen as the training set (7130 protein pairs) and the remaining two-fifths (4576 protein pairs) were used as the test set. An SVM model was built using the training set and 5-fold cross-validation, and the performance of this model was evaluated by the test set. In order to test the robustness of the method, this process of random selection of training set and test set, model-building and model-evaluating was repeated five times. Thus, five training sets and five test sets were prepared, so five models were generated. Three parameters, sensitivity, precision and accuracy were used to measure the performance of this method. They are defined as follows:
| 3 |
| 4 |
| 5 |
where TP, TN, FP and FN represent true positive, true negative, false positive and false negative, respectively.
RESULTS AND DISCUSSION
Comparing the prediction performances of different negative data sets
SVM models were constructed using the five negative data sets derived from the three strategies. In this step, lg was initialized to be 25 amino acids. Table 1 gives the average prediction results of SVM models using different negative data sets. Using 1-let, 2-let and 3-let shuffled protein sequences as negative data set, the average prediction accuracy is 79.25, 77.30 and 70.25%, respectively. The trend that the prediction accuracy decreases with the increase of k is resulted from the fact that the shuffling procedure provides more native-like artificial proteins by conserving higher-order biases. The model based on the negative data set Prcp yields very low prediction accuracy of 58.42%, while the model built with the same strategy by Shen et al. (26) achieves a good performance with an accuracy of 83.90%. It is probably due to the different feature representation methods and data sources. Compared to other four models, the model based on the negative data set Psub gives the best performance. The average prediction accuracy, sensitivity and precision are 86.23, 85.22 and 87.83%, respectively, which indicate that this method is successful in predicting PPIs using the non-interacting pairs of non co-localized proteins as the negative data set. However, it is necessary to point out that selecting non-interacting pairs of non co-localized protein will lead to over-optimistic estimates of classifier accuracy, as denoted by Ben-hur and Noble (58).
Table 1.
The comparative results of the prediction performance of the method based on different negative data sets, respectively, using AC with lg of 25 amino acids
| Negative data set | Psub | Prcp | 1-let | 2-let | 3-let |
|---|---|---|---|---|---|
| Sensitivity (%) | 85.22 | 41.76 | 79.29 | 69.81 | 60.74 |
| Precision (%) | 87.83 | 62.64 | 82.67 | 85.14 | 80.15 |
| Accuracy (%) | 86.23 ± 1.95 | 58.42 ± 1.68 | 79.25 ± 7.80 | 77.30 ± 12.38 | 70.25 ± 10.40 |
Psub is the negative data set of non-interacting pairs of non-co-localized proteins; Prcp is the negative data set derived from the method by Shen et al. (26). The three negative data sets, 1-let, 2-let and 3-let are obtained by shuffling the protein sequences with k-let counts, k = 1, 2, 3.
Selecting optimal lg
The use of AC with large lags will result in more variables that account for interactions of amino acids with large distances apart in the sequence. The maximal possible lg is the length of the shortest sequence (50 amino acids) in the data set. In this study, several lgs were optimized in order to achieve the best characterization of the protein sequences. Using Psub as the negative data set, nine models were constructed with nine different lgs, respectively (lg = 5, 10, 15, 20, 25, 30, 35, 40, 45). The prediction results for the nine models are shown in Figure 1. As seen from the curve, the prediction accuracy increases when lg increases from 5 to 30, but it slightly fluctuates when lg increases from 30 up to 45. There is a peak point with an average accuracy of 87.36% and the lg of 30 amino acids. It is concluded that AC with lg less than 30 amino acids would lose some useful features of the protein sequences and larger lgs could introduce noise instead of improving the prediction power of the model. So the optimal lg is 30 amino acids.
Figure 1.

The average prediction accuracy of the method with AC of different lgs respectively.
Comparing the performance of AC with that of ACC
After represented by the seven descriptors, a protein pair was converted into a 420-dimensional (2 × 30 × 7) vector by AC with lg of 30 amino acids. However, when ACC is used, a protein sequence will be a vector of 2940 dimension (2 × 30 × 7 × 7). To reduce the calculating time, only AC variables were used as the input of SVM. Here, we also used ACC to transform the protein sequences and compared the performance of the model based on ACC with that of the model based on AC. From Table 2, we can see that the model based on ACC transform gives good results with the average sensitivity, precision and accuracy of 89.93, 88.87 and 89.33 ± 2.67%, respectively. However, when the dimension of vector space is dramatically reduced from 2940 to 420 using AC transform, the performance of the model based on AC is very close to that of the model based on ACC. It proves that CC variables only have a little contribution to the performance of the model and AC variables are the principal components of ACC variables.
Table 2.
The prediction results of the test sets based on the negative data set Psub and lg of 30 amino acids
| Test set | TP | FN | TN | FP | Sensitivity (%) | Precision (%) | Accuracy (%) | |
|---|---|---|---|---|---|---|---|---|
| ACC | 1 | 2096 | 282 | 2226 | 152 | 88.14 | 93.24 | 90.87 |
| 2 | 2282 | 96 | 1741 | 637 | 95.96 | 78.18 | 84.59 | |
| 3 | 2023 | 355 | 2291 | 87 | 85.07 | 95.88 | 90.71 | |
| 4 | 2181 | 197 | 2099 | 279 | 91.72 | 88.66 | 89.99 | |
| 5 | 2052 | 267 | 2194 | 184 | 88.77 | 91.98 | 90.52 | |
| Average | 2138 | 240 | 2110 | 268 | 89.93 | 88.87 | 89.33 ± 2.67 | |
| AC | 1 | 2161 | 217 | 1944 | 434 | 90.87 | 83.28 | 86.31 |
| 2 | 2215 | 163 | 1890 | 488 | 93.15 | 81.95 | 86.31 | |
| 3 | 2062 | 316 | 2153 | 225 | 86.71 | 90.16 | 88.63 | |
| 4 | 1890 | 488 | 2221 | 157 | 79.48 | 92.33 | 86.44 | |
| 5 | 2052 | 326 | 2185 | 193 | 86.29 | 91.40 | 89.10 | |
| Average | 2076 | 312 | 2079 | 299 | 87.30 | 87.82 | 87.36 ± 1.38 |
TP, true positive; FP, false positive; TN, true negative; FN, false negative; Psub is the negative data set of non-interacting pairs of non-co-localized proteins.
So in this work, the optimal model was based on the negative data set Psub and AC transform with lg of 30 amino acids. The prediction results for five test sets are listed in Table 2. For all five models, the prediction accuracies are all >86% with a relatively low SD of 1.38%. On average, the sensitivity, precision and prediction accuracy of this model are 87.30, 87.82 and 87.36%, respectively. These results are obtained based on the original data set that contains homologous protein pairs. However, for the statistical predictions, it is absolutely necessary to avoid redundancy and homology bias in the training data set (57). In order to determine the homology effects, the non-redundant data set was constructed by removing the protein pairs with ≥40% pairwise sequence identity from the whole original data set. The performance of the five models based on this non-redundant data set is shown in Supplementary Table S2. The average prediction accuracy of the non-redundant data set is 86.55%.
Two SVM parameters, C and γ were optimized as 32 and 0.03125. So using the whole data set, the final prediction model was built with the optimal parameters.
Performance on the independent data set
In order to evaluate the practical prediction ability of the final prediction model, a large independent data set was constructed. In DIP, the yeast data set contained 17 491 interaction pairs, out of which that which contained a protein with <50 amino acids and those appearing in the training data set were all excluded. Among the remaining 11 474 protein pairs, 10 108 PPIs are correctly predicted by the prediction model and the success rate is 88.09%. In this article, the negative training set was generated by selecting non-interacting pairs of non-co-localized proteins. However, Ben-Hur and Noble (58) have denoted that restricting negative examples to non-co-localized protein pairs leads to a biased estimate of the accuracy of a PPI predictor. So it is necessary to generate a test data set of the non-interacting pairs with the same localization to test the effects of this bias. The yeast proteins used in the positive training set were assigned with the seven main types of localization. The non-interacting protein pairs with the same localization were generated and none of them has occurred in the whole DIP yeast interacting pairs. The performance of this method in predicting such negative samples is summarized in Supplementary Table S3. For cytoplasm and nucleus subsets, only 8000 non-interactions were randomly selected from the large-scale data set, respectively. The result shows that the prediction model is able to correctly predict the non-interacting pairs of all subsets with >80% accuracy, except the cytoplasm subset with 77% accuracy and endoplasmic reticulum subset with 69% accuracy. For all 27 204 non-interactions, the total prediction accuracy is 81.46%. In addition, using the model based on the non-redundant data set, the prediction accuracy for 11 474 yeast PPIs is 93.25% and the result of the non-interacting pairs is shown in Supplementary Table S4. All these results demonstrate that this method is also able to predict non-interacting pairs with the same localization.
CONCLUSION
In this article, we developed a new method for predicting PPIs only using the primary sequences of proteins. The prediction model was constructed based on SVM and AC. Shen et al. (26) have denoted that usually the methods with no local environments of amino acids are not reliable and robust, so they proposed a conjoint triad method to consider the properties of each amino acid and its two proximate amino acids. However, in most cases, the long-range interactions are also important for representing the PPI information. In this article, AC was used to involve the information of interactions between amino acids a longer distance apart in the sequence. A protein sequence was characterized by a series of ACs that covered the information of interactions between one amino acid and its 30 vicinal amino acids in the sequence. So this method adequately takes the neighbouring effect into account. As expected, this method improved the prediction accuracy compared with the current methods. Moreover, three different negative data sets were compared and the model trained using non-interacting pairs of non co-localized proteins yielded the best performance with a high accuracy of 87.36%, when applied to predicting the PPIs of S. cerevisiae. Meanwhile, the final prediction model was tested using the independent data set of the yeast PPIs with a good performance. Overall, such a robust method will be a useful tool to elucidate the biological function of newly discovered proteins and to expedite the study of protein networks.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
The authors gratefully thank Eivind Coward for sharing the Shufflet sequence-randomizing code. The work was funded by the National Natural Science Foundation of China (No. 20775052). Funding to pay the Open Access publication charges for this article was provided by the National Natural Science Foundation of China (No. 20775052).
Conflict of interest statement. None declared.
REFERENCES
- 1.Fields S, Song O. A novel genetic system to detect protein–protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 2.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl Acad. Sci. USA. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gavin AC, Boche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick J, Michon A, Cruciat C. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 4.Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams S, Millar A, Taylor P, Bennett K, Boutilier K, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 5.Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, Lan N, Jansen R, Bidlingmaier S, Houfek T, et al. Global analysis of protein activities using proteome chips. Science. 2001;193:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- 6.Han JD, Dupuy D, Bertin N, Cusick ME, Vidal M. Effect of sampling on topology predictions of protein–protein interaction networks. Nat. Biotechnol. 2005;23:839–844. doi: 10.1038/nbt1116. [DOI] [PubMed] [Google Scholar]
- 7.Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc. Natl Acad. Sci. USA. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Overbeek R, Fonstein M, D'Souza M, Pusch GD, Maltsev N. Use of contiguity on the chromosome to predict functional coupling. In Silico Biol. 1999;1:93–108. [PubMed] [Google Scholar]
- 9.Marcotte EM. Detecting protein function and protein–protein interactions from genome sequences. Science. 1999;285:751–753. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- 10.Enright AJ, Iliopoulos I, Kyrpides NC, Ouzounis CA. Protein interaction maps for complete genomes based on gene fusion events. Nature. 1999;402:86–90. doi: 10.1038/47056. [DOI] [PubMed] [Google Scholar]
- 11.Aloy P, Russell RB. Interrogating protein interaction networks through structural biology. Proc. Natl Acad. Sci. USA. 2002;99:5896–5901. doi: 10.1073/pnas.092147999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aloy P, Russell RB. InterPreTS: protein interaction prediction through tertiary structure. Bioinformatics. 2003;19:161–162. doi: 10.1093/bioinformatics/19.1.161. [DOI] [PubMed] [Google Scholar]
- 13.Ogmen U, Keskin O, Aytuna AS, Nussinov R, Gursoy A. PRISM: protein interactions by structural matching. Nucleic Acids Res. 2005;33:W331–W336. doi: 10.1093/nar/gki585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang TW, Tien AC, Huang WS, Lee YC, Peng CL, Tseng HH, Kao CY, Huang CY. POINT: a database for the prediction of protein–protein interactions based on the orthologous interactome. Bioinformatics. 2004;20:3273–3276. doi: 10.1093/bioinformatics/bth366. [DOI] [PubMed] [Google Scholar]
- 15.Espadaler J, Romero-Isart O, Jackson RM, Oliva B. Prediction of protein–protein interactions using distant conservation of sequence patterns and structure relationships. Bioinformatics. 2005;21:3360–3368. doi: 10.1093/bioinformatics/bti522. [DOI] [PubMed] [Google Scholar]
- 16.Sprinzak E, Margalit H. Correlated sequence-signatures as markers of protein–protein interaction. J. Mol. Biol. 2001;311:681–692. doi: 10.1006/jmbi.2001.4920. [DOI] [PubMed] [Google Scholar]
- 17.Kim WK, Park J, Suh JK. Large scale statistical prediction of protein–protein interaction by potentially interacting domain (PID) pair. Genome Inform. 2002;13:42–50. [PubMed] [Google Scholar]
- 18.Han DS, Kim HS, Jang WH, Lee SD, Suh JK. PreSPI: a domain combination based prediction system for protein–protein interaction. Nucleic Acids Res. 2004;32:6312–6320. doi: 10.1093/nar/gkh972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morrison JL, Breitling R, Higham DJ, Gilbert DR. A lock-and-key model for protein–protein interaction. Bioinformatics. 2006;22:2212–2019. doi: 10.1093/bioinformatics/btl338. [DOI] [PubMed] [Google Scholar]
- 20.Singhal M, Resat H. A domain-based approach to predict protein–protein interactions. BMC Bioinformatics. 2007;8:199. doi: 10.1186/1471-2105-8-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bock JR, Gough DA. Predicting protein–protein interactions from primary structure. Bioinformatics. 2001;17:455–460. doi: 10.1093/bioinformatics/17.5.455. [DOI] [PubMed] [Google Scholar]
- 22.Martin S, Roe D, Faulon JL. Predicting protein–protein interactions using signature products. Bioinformatics. 2005;21:218–226. doi: 10.1093/bioinformatics/bth483. [DOI] [PubMed] [Google Scholar]
- 23.Lo SL, Cai CZ, Chen YZ, Chung MCM. Effect of training datasets on support vector machine prediction of protein–protein interactions. Proteomics. 2005;5:876–884. doi: 10.1002/pmic.200401118. [DOI] [PubMed] [Google Scholar]
- 24.Pitre S, Dehne F, Chan A, Cheetham J, Duong A, Emili A, Gebbia M, Greenblatt J, Jessulat M, Krogan N, et al. PIPE: a protein–protein interaction prediction engine based on the re-occurring short polypeptide sequences between known interacting protein pairs. BMC Bioinformatics. 2006;7:365. doi: 10.1186/1471-2105-7-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chou KC, Cai YD. Predicting protein–protein interactions from sequences in a hybridization space. J. Proteome Res. 2006;5:316–322. doi: 10.1021/pr050331g. [DOI] [PubMed] [Google Scholar]
- 26.Shen JW, Zhang J, Luo XM, Zhu WL, Yu KQ, Chen KX, Li YX, Jiang HL. Predicting protein–protein interactions based only on sequences information. Proc. Natl Acad. Sci. USA. 2007;104:4337–4341. doi: 10.1073/pnas.0607879104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xenarios I, Salwinski L, Duan XJ, Higney P, Kim SM, Eisenberg D. DIP: the database of interacting proteins. A research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002;30:303–305. doi: 10.1093/nar/30.1.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deane CM, Salwinski L, Xenarios I, Eisenberg D. Protein interactions: two methods for assessment of the reliability of high throughput observations. Mol. Cell. Proteomics. 2002;1:349–356. doi: 10.1074/mcp.m100037-mcp200. [DOI] [PubMed] [Google Scholar]
- 29.Li WZ, Jaroszewski L, Godzik A. Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics. 2001;17:282–283. doi: 10.1093/bioinformatics/17.3.282. [DOI] [PubMed] [Google Scholar]
- 30.Kandel Y, Matias R, Unger R, Winkler PM. Shuffling biological sequences. Discrete Appl. Math. 1996;71:171–185. [Google Scholar]
- 31.Coward E. Shufflet: shuffling sequences while conserving the k-let counts. Bioinformatics. 1999;15:1058–1059. doi: 10.1093/bioinformatics/15.12.1058. [DOI] [PubMed] [Google Scholar]
- 32.Tanford C. Contribution of hydrophobic interactions to the stability of the globular conformation of proteins. J. Am. Chem. Soc. 1962;84:4240–4274. [Google Scholar]
- 33.Hopp TP, Woods KR. Prediction of protein antigenic determinants from amino acid sequences. Proc. Natl Acad. Sci. USA. 1981;78:3824–3828. doi: 10.1073/pnas.78.6.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krigbaum WR, Komoriya A. Local interactions as a structure determinant for protein molecules: II. Biochim. Biophys. Acta. 1979;576:204–228. doi: 10.1016/0005-2795(79)90498-7. [DOI] [PubMed] [Google Scholar]
- 35.Grantham R. Amino acid difference formular to help explain protein evolution. Science. 1974;185:862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 36.Charton M, Charton BI. The structure dependence of amino acid hydrophobicity parameters. J. Theor. Biol. 1982;99:629–644. doi: 10.1016/0022-5193(82)90191-6. [DOI] [PubMed] [Google Scholar]
- 37.Rose GD, Geselowitz AR, Lesser GJ, Lee RH, Zehfus MH. Hydrophobicity of amino acid residues in globular proteins. Science. 1985;229:834–838. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- 38.Zhou P, Tian FF, Li B, Wu SR, Li ZL. Genetic algorithm-base virtual screening of combinative mode for peptide/protein. Acta Chim. Sinica. 2006;64:691–697. [Google Scholar]
- 39.Wold S, Jonsson J, Sjöström M, Sandberg M, Rännar S. DNA and peptide sequences and chemical processes mutlivariately modelled by principal component analysis and partial least-squares projections to latent structures. Anal. Chim. Acta. 1993;277:239–253. [Google Scholar]
- 40.Guo YZ, Li ML, Lu MC, Wen ZN, Huang ZT. Predicting G-protein coupled receptors-G-protein coupling specificity based on autocross-covariance transform. Proteins. 2006;65:55–60. doi: 10.1002/prot.21097. [DOI] [PubMed] [Google Scholar]
- 41.Wen ZN, Li ML, Li YZ, Guo YZ, Wang KL. Delaunay triangulation with partial least squares projection to latent structures: a model for G-protein coupled receptors classification and fast structure recognition. Amino Acids. 2007;32:277–283. doi: 10.1007/s00726-006-0341-y. [DOI] [PubMed] [Google Scholar]
- 42.Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics. 2007;8:4. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vapnik V. Statistical learning theory. New York: Wiley; 1998. [Google Scholar]
- 44.Chou KC, Zhang CT. Review: prediction of protein structural classes. Crit. Rev. Biochem. Mol. Biol. 1995;30:275–349. doi: 10.3109/10409239509083488. [DOI] [PubMed] [Google Scholar]
- 45.Chou KC, Shen HB. Review: recent progresses in protein subcellular location prediction. Anal. Biochem. 2007;370:1–16. doi: 10.1016/j.ab.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 46.Zhou GP, Doctor K. Subcellular location prediction of apoptosis proteins. Proteins. 2003;50:44–48. doi: 10.1002/prot.10251. [DOI] [PubMed] [Google Scholar]
- 47.Huang Y, Li Y. Prediction of protein subcellular locations using fuzzy k-NN method. Bioinformatics. 2004;20:21–28. doi: 10.1093/bioinformatics/btg366. [DOI] [PubMed] [Google Scholar]
- 48.Du PF, Li YD. Prediction of protein submitochondria locations by hybridizing pseudo-amino acid composition with various physicochemical features of segmented sequence. BMC Bioinformatics. 2006;7:518. doi: 10.1186/1471-2105-7-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mondal S, Bhavna R, Mohan-Babu R, Ramakumar S. Pseudo amino acid composition and multi-class support vector machines approach for conotoxin superfamily classification. J. Theor. Biol. 2006;243:252–260. doi: 10.1016/j.jtbi.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 50.Guo J, Lin YL, Liu XJ. GNBSL: a new integrative system to predict the subcellular location for Gram-negative bacteria proteins. Proteomics. 2006;6:5099–5105. doi: 10.1002/pmic.200600064. [DOI] [PubMed] [Google Scholar]
- 51.Kedarisetti KD, Kurgan L, Dick S. Classifier ensembles for protein structural class prediction with varying homology. Biochem. Biophys. Res. Commun. 2006;348:981–988. doi: 10.1016/j.bbrc.2006.07.141. [DOI] [PubMed] [Google Scholar]
- 52.Guo YZ, Li ML, Lu MC, Wen ZN, Wang KL, Li GB, Wu J. Classifying G protein-coupled receptors and nuclear receptors based on protein power spectrum from fast Fourier transform. Amino Acids. 2006;30:397–402. doi: 10.1007/s00726-006-0332-z. [DOI] [PubMed] [Google Scholar]
- 53.Zhang TL, Ding YS. Using pseudo amino acid composition and binary-tree support vector machines to predict protein structural classes. Amino Acids. 2007;33:623–629. doi: 10.1007/s00726-007-0496-1. [DOI] [PubMed] [Google Scholar]
- 54.Pugalenthi G, Tang K, Suganthan PN, Archunan G, Sowdhamini R. A machine learning approach for the identification of odorant binding proteins from sequence-derived properties. BMC Bioinformatics. 2007;8:351. doi: 10.1186/1471-2105-8-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan FY, Feng XY, Fang Z, Li ML, Guo YZ, Jiang L. Prediction of mitochondrial proteins based on genetic algorithm: partial least squares and support vector machine. Amino Acids. 2007;33:669–675. doi: 10.1007/s00726-006-0465-0. [DOI] [PubMed] [Google Scholar]
- 56.Diao YB, Ma DC, Wen ZN, Yin JJ, Xiang J, Li ML. Using pseudo amino acid composition to predict transmembrane regions in protein: cellular automata and Lempel-Ziv complexity. Amino Acids. 2008;34:111–117. doi: 10.1007/s00726-007-0550-z. [DOI] [PubMed] [Google Scholar]
- 57.Chou KC, Shen HB. Cell-PLoc: a package of web-servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc. 2008;3:153–162. doi: 10.1038/nprot.2007.494. [DOI] [PubMed] [Google Scholar]
- 58.Ben-Hur A, Noble WS. Choosing negative examples for the prediction of protein–protein interactions. BMC Bioinformatics. 2006;7:S2. doi: 10.1186/1471-2105-7-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.