

SUMMARY
Glycoprotein gH, together with its chaperone gL and a third glycoprotein gB, is essential to cell cell fusion and virus cell fusion mediated by herpesviruses. Epstein-Barr virus (EBV), the prototype human lymphocryptovirus, requires a fourth glycoprotein gp42 to support fusion with B cells in addition to epithelial cells. Two other lymphocryptoviruses, the rhesus lymphocryptovirus (Rh-LCV) and the common marmoset lymphocryptovirus (CalHV3), have been sequenced in their entirety and each has a gp42 homolog. Combinations of proteins from EBV, Rh-LCV and CalHV3 were able to mediate fusion of epithelial cells, but, even when complexed with EBV gp42, only RhLCV and not CalHV3 proteins were able to mediate fusion with human B cells. CalHV3 gL was also unable to function effectively as a chaperone for EBV or Rh-LCV gH. The Rh-LCV gH homolog supported more fusion than EBV gH with an epithelial cell and supported the highest levels of fusion with a B cell. Chimeric constructs made from Rh-LCV gH and EBV gH which have 85.4% sequence identity should prove useful for mapping the regions of gH that are of importance to fusion as a whole and to B cell fusion in particular.
INTRODUCTION
Epstein-Barr virus (EBV) is the most extensively studied of the herpesviruses classified in the gamma-1 or lymphocryptovirus genus. It is orally transmitted and persists in the majority of adults worldwide (reviewed in (Rickinson & Kieff, 2001)). Primary infections acquired in childhood are usually asymptomatic, but with increasing age they are more likely to result in a self-limiting infectious mononucleosis. Of greatest concern, however, is that long term carriage can be associated with development of malignancies. These are predominantly of lymphoid and epithelial origin reflecting the fact that although the primary reservoir of virus in the infected host is in the B lymphocyte population, infection of epithelial cells also appears to continue throughout life and trafficking of virus between B cells and epithelial cells may be a common feature of persistence (Jiang et al., 2006, Pegtel et al., 2004, Sitki-Green et al., 2003).
Initiation of infection of B cells and epithelial cells, perhaps not surprisingly, involves different viral and cellular proteins. Fusion of virus with both cell types, as for all herpesviruses (Spear & Longnecker, 2003), requires the activity of the conserved glycoproteins gB and gHgL. These proteins are not only necessary but sufficient for EBV fusion with an epithelial cell (McShane & Longnecker, 2004). Fusion with a B cell, however, additionally requires glycoprotein gp42, a protein that is restricted to the lymphocryptoviruses. Epithelial cell fusion is triggered by a direct interaction between gHgL and an as yet unidentified coreceptor, gHgLR (Borza et al., 2004, Wang et al., 1998). B cell fusion is triggered by an interaction between gp42 and HLA class II which functions as a B cell coreceptor (Li et al., 1997b). The trigger is presumably transmitted to gHgL with which gp42 forms a complex. Activation of fusion by the two different coreceptors is mutually exclusive, requiring carriage of two different complexes in the virion. Only gHgL complexes that include gp42 can trigger B cell fusion (Wang & Hutt-Fletcher, 1998) and only complexes that lack gp42 can trigger epithelial cell fusion because the presence of gp42 blocks the interaction of gHgL with gHgLR (Wang et al., 1998). In an HLA class II-positive B cell, but not in an HLA class II-negative epithelial cell, some three part complexes are lost as they interact with and accompany HLA class II to the protease-rich peptide-loading compartment. As a result epithelial cell virus is more B-cell tropic and B cell virus is more tropic for epithelial cells, perhaps contributing to the movement of virus between the two cell types (Borza & Hutt-Fletcher, 2002).
The differences in the use of the gHgL complex for cell entry can be seen at the level of the gH sequence itself. Residues at both the amino terminus and the carboxyl terminus of the protein are important to fusion of B cells and epithelial cells, but those that are involved in B cell and epithelial cell fusion differ (Omerovic et al., 2005, Wu et al., 2005, Wu & Hutt-Fletcher, 2007). The limited mutational analysis of EBV gH that has been done to date has focused on regions of the protein that have chosen either because of computer driven predictions (Omerovic et al., 2005) or because of mapping of sequences that are recognized by a monoclonal antibody (Mab) to gH that blocks epithelial cell but not B cell fusion (Wu et al., 2005). Several non-human primate lymphocryptoviruses have been identified and the gH homologs of these viruses may offer additional insight into regions that are important to function. The rhesus lymphocryptovirus (Rh-LCV) isolated from the rhesus macaque, (Cho et al., 2001) and the Callitrichine herpesvirus 3 (CalHV-3) isolated from the common marmoset (Rivailler et al., 2002a) have been sequenced in their entirety (Rivailler et al., 2002a, Rivailler et al., 2002b). Rh-LCV is most similar to EBV. Each predicted open reading frame has a counterpart in EBV and the average homology between the two viruses is 76.6%; the homology of the lytic cycle genes is even higher. EBV gH and Rh-LCV gH have 85.4% identity and 92.6% similarity. The average homology of EBV and CalHV-3 genes is less at only 47.3% and EBV gH and CalHV3 gH have 46.2% identity and 64.6% similarity. The sequence homology of Rh-LCV and CalHV3 proteins is also not high. The Rh-LCV gH has 45.8% identity and 64.6% similarity to CalHV3 gH. The gL homologs parallel this trend. EBV gL and Rh-LCV gL have 81.8% identity and 89.8% similarity, EBV gL and CalHV3 gL have 51.4% identity and 63.2 % similarity and Rh-LCV gL and CalHV3 gL have 51.0% identity and 62.2% similarity. We explore here whether conserved sequences of the gH and gL homologs of EBV, Rh-LCV and CalHV3 are sufficient to enable them to substitute functionally for each other.
METHODS
Cells
CV-1 monkey kidney cells were grown in Dulbecco's modified Eagle's medium. AGS, a human gastric carcinoma cell line and Chinese hamster ovary (CHO-K1) cells were grown in Ham's F12 medium. Daudi 29 cells (a gift of Richard Longnecker, Northwestern University), B cells that stably express T7 RNA polymerase from the pOS2 vector (Whetter et al., 1994), were grown in RPMI medium. All culture media were obtained from Gibco-BRL Life Technologies (Grand Island NY) and supplemented with 10% heat inactivated fetal bovine serum.
Antibodies
Antibodies used were monoclonal antibodies (Mabs) E1D1 to gHgL, CL59 and CL40 to gH (Molesworth et al., 2000), CL55 to gB and F-2-1 to gp42 (Li et al., 1995) and an anti-peptide antibody to EBV gL (Yaswen et al., 1993). All antibodies were affinity purified on protein A-agarose.
Plasmids
Plasmids pCAGGS-EBV gH, pCAGGS-EBV gL, pCAGGS-EBV gB and pCAGGS-EBV g42 (Wu et al., 2005), were made by cloning PCR amplified EBV sequences into the pCAGGS/MCS vector (a gift of Martin Muggeridge, LSUHSC) for expression under control of the β-actin promoter in cooperation with the HCMV-IE enhancer (Niwa et al., 1991). Plasmid pCAGGS-RhLCV gH was made by cloning sequences amplified by PCR from the cosmid DK12 (Rivailler et al., 2002b). Plasmid pCAGGS-RhLCV gL was made by cloning sequences amplified by PCR from the cosmid LV28. Plasmids pCAGGS-CalHV3 gH and pCAGGS-CalHV3 gB were made by cloning sequenced amplified from cosmid A10 (Rivailler et al., 2002a), pCAGGS-CalHV3 gL was made by cloning sequences amplified from cosmid D6 and plasmid pCAGGS-CalHV3 gp42 was made by cloning sequences amplified from cosmid D4. All cosmids were a gift of Fred Wang (Harvard University). All plasmids were confirmed by sequencing. Cosmid A10 was discovered to include a single base pair change in the gH open reading frame at base pair 34451of the genome that converted a tyrosine residue to premature stop codon. This was repaired by cloning an Xho1-SacI fragment into the very small pSP72 vector (Promega, Madison, WI) for repair of the mutation using the Quickchange II protocol (Strategene, La Jolla, CA). The repair was confirmed by sequencing and the repaired sequences were recloned into pCAGGS. All Plasmids pTM1-EBV gH, pTM1-EBV gL and pTM1-EBV gp42 were made as previously described (Li et al., 1995) by cloning PCR amplified sequences into the pTM1 vector (Moss et al., 1990) which contains a T7 promoter, the encephalomyocarditis virus cap-independent translation signal, a multiple cloning site and a T7 transcriptional terminator. Plasmid pTM1-CalHV3 gH was made by cloning PCR amplified sequences into the same vector. The premature stop codon was not repaired in this construct.
Immunofluorescence
For internal staining, slides bearing air-dried acetone-fixed cells were incubated at 370C for 30 min in a humidified atmosphere with Mabs, washed three times with phosphate-buffered saline for 5 min each, reincubated for 30 min with the appropriate dilution of fluorescein isothiocyanate (FITC)-conjugated sheep anti-mouse immunoglobulin (ICN/Cappel, Irvine, CA), washed three times and mounted in Mounting Medium (Kirkegaard Perry Laboratories, Gaithersburg, MD).
Levels of cell surface expression
The levels of expression at the cell surface of EBV gH and Rh-LCV gH and CalHV3 gH were measured by modification of previously described methods (Wu et al., 2005). Briefly, CHO-K1 cells were transfected with the appropriate pCAGGS-based plasmid together with pCAGGS-EBV gB and either pCAGGS-EBV gL or pCAGGS- Rh-LCV gL. 3.2 μg DNA was mixed with 4 μl Target transfectin F2 and 12 μl Target peptide enhancer (Targeting Systems, Santee, CA) in high glucose DMEM media. Thirty six hours later, transfected cells were removed from the plastic by scraping and recovered for 1 hour at 370 C. The cells were washed with ice-cold phosphate-buffered saline containing 2% fetal bovine serum and serially reacted with Mab E1D1 or Mab CL40 and phycoerythrin conjugated anti-mouse immunoglobulin (Jackson ImmunoResearch Laboratories, West Grove, PA) with washing between each step and analyzed by flow cytometry.
AGS cell-cell fusion assay
Fusion of AGS was measured as previously described (Wu et al., 2005). Cells were seeded in two-well chamber slides and were transfected at 70-80% confluency for 4 hours with of 0.25 μg pCAGGS-based plasmids expressing EBV gH, Rh-LCV gH or CalHV3 gH, 0.25 μg plasmids expressing EBV gL, Rh-LCV gL or Cal HV3 gL and 0.6 μg of a plasmid expressing EBV gB or CalHV3 gB. Plasmids were mixed with 1 μl Target transfectin F2 and 2 μl Target peptide enhancer in high glucose DMEM media. Twenty four hours post transfection cells were fixed with ice-cold acetone. Cells transfected with EBV gB were stained with Mab CL55. Cells expressing CalHV3 gB, CalHV3 gH and CalHV3 gL were visualized as a result of expression of green fluorescent protein from plasmid pmax-GFP (Amaxa). Fluorescent cells containing four or more nuclei were considered to have undergone fusion and were recorded as one fusion event. The extent of fusion was calculated as the number of fusion events as a percentage of the total number of fluorescent cells. The combination of EBV gH, gL and gB which supported fusion of 49+/−12 % of the total number of fluorescent transfected cells was set at 100% and fusion mediated by other combinations was expressed relative to this.
Epithelial cell-B cell fusion assay
B cell fusion was measured as previously described (Wu et al., 2005). CHO-K1 cells were seeded in 6-well plates and were transfected at 70-80% confluency for 4 hours with 0.8 μg pCAGGS-based plasmids expressing gH together with 0.8 μg pCAGGS-based plasmids expressing gL, 1.4 ug plasmids expressing gB, and 1.2 μg plasmids expressing gp42 and 0.8 μg pST7-Luc containing the T7 promoter upstream of the luciferase gene (Ferrer et al., 1999) (a gift of Martin Muggeridge). Each well of transfected cells was then overlayed with two million Daudi 29 cells that expressed T7 RNA polymerase. 24 hours later, cells were washed twice with phosphate buffered saline and lysed with 500 μl Passive Lysis Buffer (Promega). Luciferase substrate (100 μl) was added to 20 μl supernatant of lysate. Luminosity readings were obtained by using a TD-20/20 luminometer (Promega).
Transfection infection protocol
Expression from the pTM1-derived vectors was done as described previously (Li et al., 1995). Briefly, CV1 cells were grown to 90% confluency in 100-mm-diameter Petri dishes and infected with vvT7 at a multiplicity of infection of 5. Thirty minutes later the inoculum was removed, and the cells were washed twice in medium without serum and transfected with pTM1 plasmids. 10 μg of DNA was mixed with 30 μl of Lipofectamine (Gibco/BRL) made to a total volume of 2.5 ml with serum-free medium.
Radiolabeling and immunoprecipitation
CV-1 cells that had been infected with vvT7 and transfected with pTM1 plasmids were labeled biosynthetically with 100 μCi of Pro-Mix (70% [35S]methionine, 30% [35S]cysteine) and 300 μCi of [35S]cysteine (>1,000 Ci/mmol; Amersham Biosciences, Arlington IL) per dish (approximately 107 cells) as previously described (Li et al., 1995). Labeled cells were solubilized in radioimmunoprecipitation buffer (50 mM Tris-HCl, pH 7.2, 0.15 M NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 0.1 mM phenylmethyl-sulfonylfluoride, and 100 U of Aprotinin per ml) and immunoprecipitated with antibody and protein A-Sepharose CL4B (Sigma). Immunoprecipitated proteins were washed, dissociated by boiling for 2 min in sample buffer with 2-mercaptoethanol, and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) in 10% polyacrylamide cross-linked with 0.28% N,N'-diallyltartardiamide followed by fluorography.
RESULTS
Monoclonal antibodies to EBV gHgL cross react with Rh-LCV and CalHV3 proteins
We have three conformationally specific Mabs to EBV gH or gHgL. Two of these Mabs, CL59 and CL40, recognize gH in the absence of gL and one, E1D1, recognizes gH only in its presence (Molesworth et al., 2000); it does not recognize gL alone (Li et al., 1995). We have previously reported that Rh-LCV gH cannot be recognized by CL59, but can be seen by CL40 and, in the presence of EBV gL, by E1D1 (Wu et al., 2005). We further explored the ability of the Mabs to recognize different combinations of EBV, Rh-LCV and CalHV3 proteins by indirect immunofluorescence of permeablized cells. EBV gH retained the ability to be recognized by E1D1, CL40 and CL59 if expressed with Rh-LCV gL (Table 1). It retained the ability to be recognized by CL40 and CL59 if expressed with CalHV-3 gL, but lost the ability to be seen by E1D1. Rh-LCV gH retained the ability to be recognized by E1D1 and CL40 if expressed with Rh-LCV gL. It retained the ability to be recognized by CL40, but lost the ability to be seen by E1D1 if expressed with CalHV3-gL. CalHV-gH was seen under no circumstances by either CL40 or CL59. It was recognized by E1D1 if expressed with either EBV gL or Rh-LCV gL but not if expressed with CalHV-3 gL. Thus, expression of the E1D1 epitope was conditional on expression of either EBV or Rh-LCV gL.
Table 1.
Indirect immunofluorescence assay of the reactivity of monoclonal antibodies with acetone-permeabilized cells transfected with different combinations of EBV, Rh-LCV and CalHV3 gH and gL homologs.
| gHgL complex | Reactivity of Mab: |
||
|---|---|---|---|
| E1D1 | CL40 | CL59 | |
| EBV gH + EBV gL | ++ | ++ | ++ |
| EBV gH + Rh-LCV gL | ++ | ++ | ++ |
| EBV gH + CalHV3 gL | − | ++ | ++ |
| Rh-LCV gH + EBV gL | ++ | ++ | − |
| Rh-LCV gH + Rh-LCV gL | ++ | ++ | − |
| Rh-LCV gH + CalHV3 gL | − | ++ | − |
| CalHV3 gH + EBV gL | ++ | − | − |
| CalHV3 gH + Rh-LCV gL | ++ | − | − |
| CalHV3 gH + CalHV3 gL | − | − | − |
Surface expression of combinations of EBV and Rh-LCV or CalHV3 proteins
The interaction of gL and gH is essential for efficient transport of gH to the cell surface. We have previously shown that the gL homologs of EBV and the distantly related varicella zoster virus can serve as chaperones for the gH homologs of either virus (Li et al., 1997a). To determine the effects of different combinations of gH and gL homologs on trafficking of gH to the cell surface, the appropriate vectors, together with vectors expressing EBV gB, were cotransfected into CHO-K1 cells and surface expression of gH was measured by flow cytometry of nonpermeabilized cells that had been stained either with Mab E1D1, which could detect any combination that included EBV gL, or Mab CL40 which could detect EBV gH and Rh-LCV gH in the absence of gL or in the presence of CalHV3-gL. Rh-LCV gL was not as efficient as EBV gL at facilitating transport of either EBV gH or its own gH to the cell surface, although in each case expression levels were at least 60% of that of an EBV gHgL complex (Fig. 1). In contrast, CalHV3 gL was unable to increase levels of either EBV gH or Rh-LCV gH at the cell surface over the low levels seen when the gL expression plasmid was replaced by empty vector. We were unable to determine the expression of CalHV3-gH in the presence of CalHV3-gL, since none of our antibodies recognized this combination, but, surprisingly, EBV gL resulted in surface expression of CalHV-3 gH at levels very similar to those of EBV gH.
Figure 1.

Surface expression of virus proteins measured by flow cytometry of CHO-K1 cells transfected with plasmids expressing the indicated combinations of EBV gH (EgH), EBV gL (EgL), Rh-LCV gH (RgH) and Rh-LCV gL (RgL), CalHV3 gH (CgH), CalHV3 gL (CgL) and empty vector (vec) together with EBV gB. Combinations were stained with Mab E1D1 (combinations that included EBV gL or RH-LCV gL ) or CL40 (combinations without EBV or Rh-LCV gL) and fluorescein-conjugated sheep anti-mouse antibody. Values are the mean level of fluorescence. Values for EBV gH and EBV gL, stained with Mab E1D1 or CL40 as appropriate, were set at 100 and values for each other combination are expressed as a percent of this. Vertical lines indicate the standard deviation of three experiments for each antibody.
Epithelial cell fusion mediated by combinations of EBV and Rh-LCV or CalHV3
The abilities of EBV gH and Rh-LCV gH to mediate fusion of AGS epithelial cells in conjunction with EBV gB were first compared. Transfected cells were stained with a monoclonal antibody to EBV gB to determine the number of cells that had successfully taken up plasmids and the percentage of this population that contained four or more nuclei was counted. Rh-LCV gH together with either Rh-LCV gL or EBV gL mediated robust levels of fusion equal to or greater than EBV gH and gL (Fig. 2). EBV gH and gL together with EBV gB induce fusion in approximately 50% of transfected cells (Wu et al., 2005). A combination of EBV gH and Rh-LCV gL, which was expressed at close to 90% of the levels of EBV gH and gL, also mediated approximately 90% of the levels of fusion. However, CalHV-3 gL, which had facilitated expression of Rh-LCV gH at levels no higher than those of Rh-LCV gH in the absence of gL, supported less than 20% of the levels of fusion.
Figure 2.
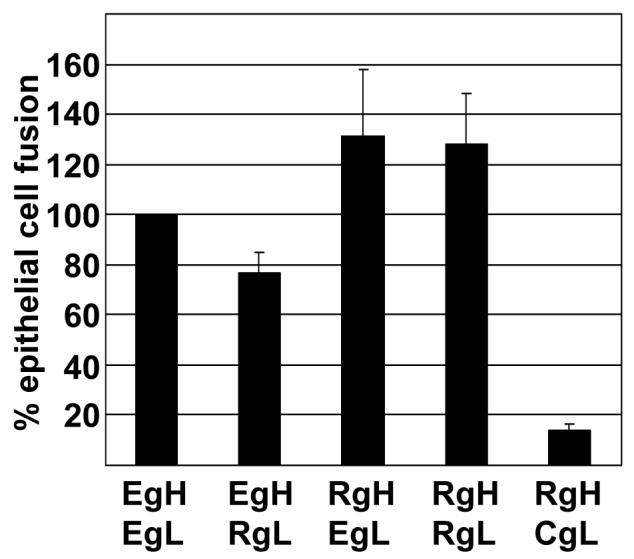
Comparison of fusion of AGS cells supported by EBV and Rh-LCV gH. Cells were transfected with the indicated combinations of plasmids expressing EBV gH (EgH), EBV gL (EgL), Rh-LCV gH (RgH), Rh-LCV gL (RgL) and CalHV3 gL (CgL) together with EBV gB. Cells were stained with Mab CL55 to gB and fusion events were counted by fluorescence microscopy. Numbers of cells containing four or more nuclei were considered as having undergone fusion and the percent of cells that had undergone fusion with each combination of proteins is expressed as a percentage of those that had undergone fusion with EBV gH and EBV gL. Vertical lines indicate the standard deviation of five experiments.
A comparison was next made of EBV gH and CalHV3 gH. A combination of EBV gH and CalHV3 gL, in keeping with the very low level of EBV gH at the cell surface under this circumstance, mediated no fusion (Fig. 3). CalHV3 gH in combination with EBV gL, which was expressed at the cell surface at levels close to those of EBV gHgL mediated close to 50% of that supported by EBV gHgL. However, substitution of CalHV3 gL for EBV gL reduced this level of fusion further. To determine if this might reflect an inability of the CalHV3 gH and gL to function fully with EBV gB, we repeated the experiments with CalHV3 gH, gL and gB, including a vector that expressed green fluorescent protein to mark those cells that had been successfully transfected. Although we were unable to determine the expression levels of the CalHV3 proteins, this combination increased fusion levels to at least 60% of those supported by the combination of all EBV proteins.
Figure 3.
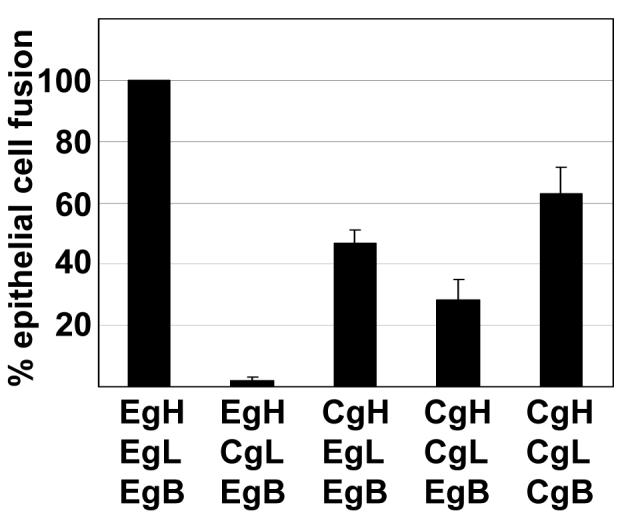
Comparison of fusion of AGS cells supported by EBV and CalHV3 gH. Cells were transfected with the indicated combinations of plasmids expressing EBV gH (EgH), EBV gL (EgL), CalHV3 gH (CgH), CalHV3 gL (CgL) together with EBV gB (EgB) or CalHV3 gB (CgB) and a vector expressing green fluorescent protein. Cells that were transfected with EBV gB were stained with Mab CL55, cells that were transfected with CalHV3-gB were identified by expression of green fluorescent protein and fusion events were counted by fluorescence microscopy. Numbers of cells containing four or more nuclei were considered as having undergone fusion and the percent of cells that had undergone fusion with each combination of proteins is expressed as a percentage of those that had undergone fusion with EBV gH and EBV gL. Vertical lines indicate the standard deviation of five experiments.
B cell fusion mediated by combinations of EBV and Rh-LCV or CalHV3 proteins
A similar analysis was then done of the ability of combinations of EBV gH, and Rh-LCV gH to mediate B cell fusion in the presence of gL homologs, EBV gB and EBV gp42. As, previously reported (Wu et al., 2005), plasmids expressing virus proteins together with a plasmid expressing the luciferase gene under control of a T7 promoter were transfected into CHO-K1 cells, which, unlike AGS cells, do not fuse with each other (McShane & Longnecker, 2004). The transfected cells were overlaid with Daudi cells expressing the T7 polymerase and fusion was measured in terms of an increase in luciferase activity. EBV gH together with Rh-LCV gL mediated between 60 and 70% of fusion mediated by EBV gH and EBV gL (Fig. 4). Rh-LCV gH together with CalHV3 gL also supported an average of 50% of the fusion supported by EBV gH and EBV gL, despite the fact that the Rh-LCV and CalHV3 combination had mediated only very low levels of epithelial cell fusion. Most surprising, however, was the observation that Rh-LCV gH and Rh-LCV gL mediated only 20% of fusion mediated by EBV gH and gL. This could not simply represent a failure to interact with gp42, which binds directly to gH (Wu & Hutt-Fletcher, 2007) because the combination of Rh-LCV gH, EBV gL and gp42, mediated as much as four times the level of fusion as did the combination of EBV gH and EBV gL and EBV gp42. These high levels of B cell fusion were reminiscent of those mediated by an EBV gH construct in which an alanine substitution was made for a glutamic acid residue at position 594 in the full length protein (Wu & Hutt-Fletcher, 2007). This construct could even mediate modest, but significant levels of B cell, though not epithelial cell fusion in the absence of gL. To determine if this were also true for Rh-LCV gH, B cell fusion was measured in the absence of Rh-LCV gL. The combination of Rh-LCV gH, EBV gp42 and EBV gB mediated a variable but significant level of B cell fusion (Fig. 5), although as the EBV gH mutant, Rh-LCV gH mediated no epithelial cell fusion in the absence of gL (not shown).
Figure 4.
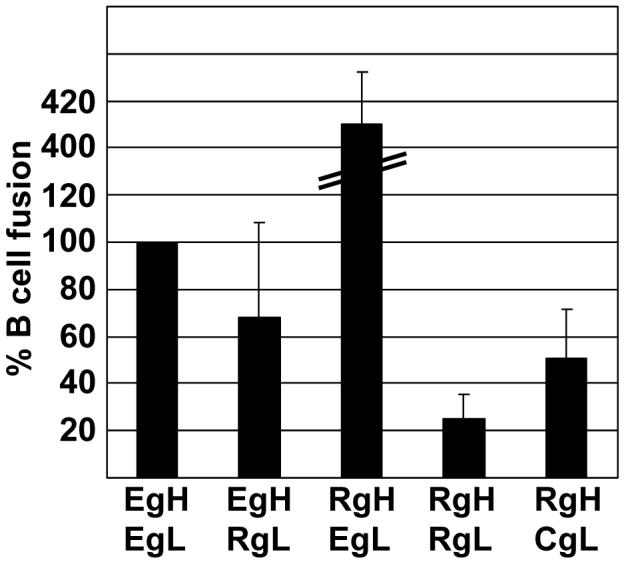
Comparison of fusion of Daudi B cells supported by EBV gH and Rh-LCV gH and measured as relative luciferase activity. CHOK-1 cells were transfected with plasmids expressing the indicated combinations of EBV gH (EgH), EBV gL (EgL), Rh-LCV gH (RgH), Rh-LCV gL (RgL) and CalHV3 gL (CgL) together with EBV gB and EBV gp42 and a plasmid expressing luciferase under control of the T7 promoter. Transfected cells were overlaid with Daudi 29 cells expressing T7 RNA polymerase. Luciferase activity in cells in which empty vector replaced gH was subtracted from activity in the presence of each combination. The remaining value for EBV gH and EBV gL was set at 100 and luciferase activity in the presence of each of the other combinations was expressed as a percentage of this value. Vertical lines indicate the standard deviation of eight experiments.
Figure 5.
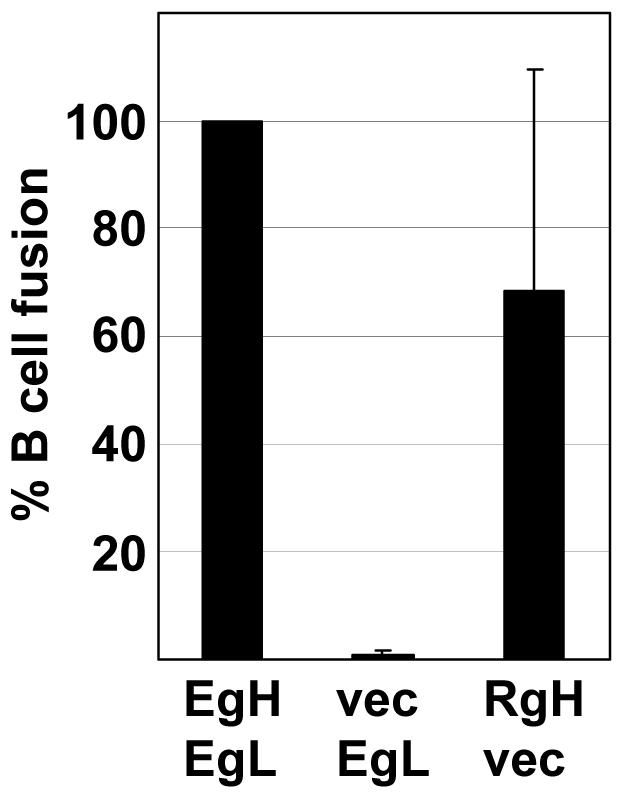
Fusion mediated by Rh-LCV gH in the absence of gL. CHOK-1 cells were transfected with plasmids expressing the indicated combinations of EBV gH (EgH), EBV gL (EgL), Rh-LCV gH (RgH) or empty vector (vec), together with EBV gB and EBV gp42 and a plasmid expressing luciferase under control of the T7 promoter. The value for EBV gH and EBV gL was set at 100 and each of the other combinations was expressed as a percentage of this value. Vertical lines indicate the standard deviation of six experiments.
In contrast, most combinations of EBV gH and CalHV3 proteins mediated little or no fusion at all with B cells (Fig. 6). The only combination that mediated low levels of fusion, close to 20% of EBV gH, gL, gB and gp42, was a combination of CalHV3 gH, CalHV3 gL, EBV gp42 and CalHV3 gB. To confirm that EBV gp42 could interact with CalHV3 gH combinations of CalHV3 gH and EBV gH were expressed for radiolabeling in the pTM1 vector together with either EBV gL or EBV gp42. The CalHV3 gH in this vector still retained the premature stop codon and thus was of a similar size to EBV gH. Both EBV and CalHV3 gH, the only proteins among gH, gL and gp42 that contain methionine residues for labeling beside the start residue, could be immunoprecipitated either with antibody to EBV gL or EBV gp42 (Fig. 7).
Figure 6.
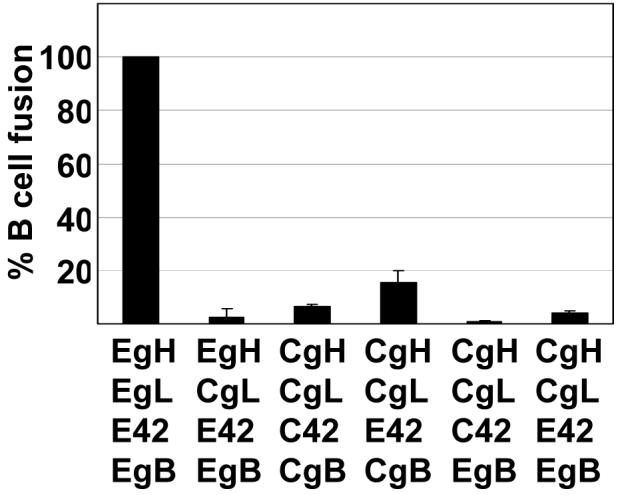
Comparison of fusion of Daudi B cells supported by EBV gH and CalHV3 gH and measured as relative luciferase activity. CHOK-1 cells were transfected with plasmids expressing the indicated combinations of EBV gH (EgH), EBV gL (EgL), CalHV3 gH (CgH), CalHV3 gL (CgL), EBV gB (EgB), CalHV3 gB (CgB), EBV gp42 (E42) and CalHV3 gp42 (C42) together with a plasmid expressing luciferase under control of the T7 promoter. Transfected cells were overlaid with Daudi 29 cells expressing T7 RNA polymerase. Luciferase activity in cells in which empty vector replaced gH was subtracted from activity in the presence of each combination. The remaining value for EBV gH and EBV gL was set at 100 and luciferase activity in the presence of each of the other combinations was expressed as a percentage of this value. Vertical lines indicate the standard deviation of eight experiments.
Figure 7.
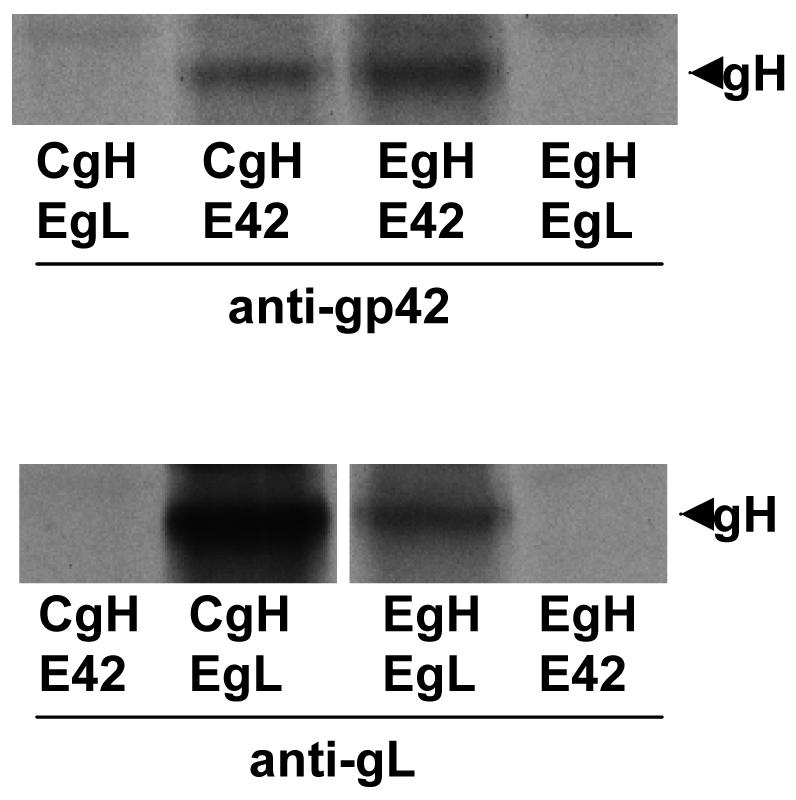
Interaction of EBV gp42 and EBV gL with CalHV3 gH. SDS-PAGE analysis of proteins immunoprecipitated by antibody to gp42 or antibody to gL from CV1 cells labeled with [35S] methionine and transfected as indicated with pTM1-EBV gH (EgH), pTM1-EBV gL (EgL), pTM1-EBV gp42 (E42), and pTM1-CalHV3 gH (CgH). The band indicated with an arrow is gH.
Discussion
Every herpesvirus expresses homologs of gH and gL. The proteins are functionally highly conserved in that they are all required for virus cell fusion, but they differ significantly in sequence, even within subfamilies. Within the lymphocryptovirus genus two viruses, in addition to EBV, have been sequenced and each carries a homolog of the triggering protein, EBV gp42. This provided an opportunity to explore the functional compatibility of the homologs of these more closely related primate viruses.
None of the proteins used was epitope tagged and no antibodies to Rh-LCV or CalHV3 homologs were available. However, all combinations that included EBV gL could be recognized by Mab E1D1, an antibody that fails to react with any gH homolog expressed alone. Although it is still formally possible that the E1D1 epitope is formed by a particular conformation of EBV gH, Rh-LCV gH or CalHV3 gH that can only be attained in the presence of EBV gL, the observation that E1D1 could recognize Rh-LCV gL in concert with EBV gH or CalHv3 gH, but not CalHV3 gL in combination with any of the three gH homologs does also raise the possibility that the epitope recognized by this antibody includes sequences contributed by both gH and gL. This is of interest since E1D1 can partially block binding of EBV virions and soluble forms of gHgL to the epithelial cell receptor/coreceptor gHgLR (Borza et al., 2004, Molesworth et al., 2000). Maruo and colleagues (Maruo et al., 2001) have reported that EBV gL alone can bind, although less robustly, to epithelial cells and this would be consistent with the binding site of gHgLR recognizing a combination of the two proteins.
EBV gH and Rh-LCV gH could both be expressed at the cell surface with their opposite gL homologs at levels close to those of the arbitrary standard of EBV gH in the presence of EBV gL. CalHV3 gL was, however, a poor chaperone for either EBV gH or Rh-LCV gH. This was something of a surprise given the reciprocal ability of EBV gL to mediate high levels of expression of CalHV3 gH and given the ability of EBV gL and varicella zoster virus gL, much more distantly related viruses, to substitute for each other, at least in terms of intracellular transport (Li et al., 1997a).
Of the three gH homologs Rh-LCV gH was the most fusogenic for epithelial cells and EBV and Rh-LCV proteins in any combination were as, or more fusogenic for epithelial cells than CalHV3 gH. Only with its own gL and gB could CalHV3 gH mediate fusion at levels close to the Rh-LCV and EBV combinations. Beyond this, no CalHV3 gH combination could support more than 20% fusion with a B cell. It is possible that CalHV3 gp42 fails to interact with HLA class II, or at least the alleles that are expressed on Daudi B cells, but even the presence of EBV gp42, which could clearly interact with CalHV3 gH, could not rescue B cell fusion to levels equivalent to those seen with epithelial cells. We recognize that the assays used for B cell and epithelial cell fusion may not be measuring identical events. It is formally possible that the luciferase assay is in some cases measuring pore formation in the absence of the pore expansion that would be necessary for production of multinucleate cells that were scored as epithelial fusion (Wu et al., 2005). However, if anything this might overestimate levels of B cell fusion relative to that of epithelial cells. Thus, either EBV gp42 is unable to transmit a triggering signal effectively to CalHV3 gH, gL and gB, the three proteins that have been referred to as the “core fusion machinery” of herpeviruses (Spear & Longnecker, 2003), or there are differences in the way in which the core machinery interacts with B cells and epithelial cells that extend beyond differences in the way it is activated. The failure of combinations of CalHV3 proteins to mediate B cell fusion is, however, consistent with reports that the virus is unable to transform human B cells (Jenson et al., 2002). There is no information with respect to the ability of the virus to infect a human epithelial cell.
Most surprising, perhaps, were the very high levels of B fusion supported by Rh-LCV gH expressed with EBV gL and the low levels that were supported by Rh-LCV gH and its own gL. This again would point to a difficulty in transmission of a signal from EBV gp42 to the Rh-LCV complex since the robust epithelial cell fusion mediated by Rh-LCV gH and Rh-LCV gL confirms its basic integrity. In the absence of Rh-LCV gL a direct interaction of Rh-LCV gH could mediate variable, but significant levels of fusion with a B cell even though its levels of expression at the cell surface were less than half those in the presence of RhLCV gL. This phenotype of Rh-LCV gH is similar, but not identical to that of a point mutant of EBV gH in which a glutamic acid residue at position 595 is replaced with an alanine (Wu & Hutt-Fletcher, 2007). Both proteins mediated higher levels of B cell fusion than wild type EBV gH and both mediated some B cell fusion in the absence of gL. However, Rh-LCV gH also supported significantly higher levels of epithelial cell fusion than EBV gH, whereas the point mutant of EBV gH was significantly inhibited for epithelial cell fusion. The sequences of Rh-LCV gH and EBV gH are identical from residues 592 to 612 so this region is not likely to account for the enhanced activity of Rh-LCV gH.
In view of the limited sequence identity between EBV and Rh-LCV gH homologs and CalHV3 gH it is perhaps not surprising that they do not complement each other well. However, Rh-LCV and EBV proteins do work well together and we have previously shown that chimeras of Rh-LCV gH and EBV gH are viable (Wu et al., 2005). This will provide opportunities to map those regions of the protein that contribute to enhanced B cell fusion and explore the possibility that differences between support of B cell and epithelial cell fusion extend beyond differences in activation of a core fusion machinery.
ACKNOWLEDGMENTS
This work was supported by Public Health Services grants AI20662 from the National Institute of Allergy and Infectious Diseases and DE016669 from the National Institute of Dental and Craniofacial Research. Support was also provided by Cobre grant P20-RR018724 from the National Center for Research Resources of the National Institutes of Health.
We thank Pierre Rivailler and Fred Wang, Harvard University, for rhesus LCV and CalHV3 cosmids, Richard Longnecker, Northwestern University, for Daudi 29 cells and Martin Muggeridge, Louisiana State University Health Sciences Center, for the pCAGGS/MCS and pST7-Luc vectors and for helpful discussion.
REFERENCES
- Borza CM, Hutt-Fletcher LM. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nature Med. 2002;8:594–599. doi: 10.1038/nm0602-594. [DOI] [PubMed] [Google Scholar]
- Borza CM, Morgan AJ, Turk SM, Hutt-Fletcher LM. Use of gHgL for attachment of Epstein-Barr virus to epithelial cells compromises infection. J Virol. 2004;78:5007–5014. doi: 10.1128/JVI.78.10.5007-5014.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y-G, Ramer J, Rivailler P, Quink C, Garber RL, Beier DR, Wang F. An Epstein-Barr virus related herpesvirus from marmoset lymphomas. Proc Natl Acad Sci USA. 2001;98:1224–1229. doi: 10.1073/pnas.98.3.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer M, Kapoor TM, Strassmaier T, Weissenhorn W, Skehel JJ, Oprian D, Schreiber SL, Wiley DC, Harrison SC. Selection of gp41 mediated HIV-1 cell entry inhibitors from biased combinatorial libraries of non-natural binding elements. Nture Struct Biol. 1999;6:953–960. doi: 10.1038/13324. [DOI] [PubMed] [Google Scholar]
- Jenson HB, Ench Y, Zhang Y, Gao SJ, Arrand JR, Mackett M. Characterization of an Epstein-Barr virus related gammaherpesvirus from common marmoset (Callithrix jacchus) J Gen Virol. 2002;83:1621–1633. doi: 10.1099/0022-1317-83-7-1621. [DOI] [PubMed] [Google Scholar]
- Jiang R, Scott RS, Hutt-Fletcher LM. Epstein-Barr virus shed in saliva is high in B cell tropic gp42. J Virol. 2006;80:7281–7283. doi: 10.1128/JVI.00497-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QX, Buranathai C, Grose C, Hutt-Fletcher LM. Chaperone functions common to nonhomologous Epstein-Barr virus gL and Varicella-Zoster virus gL proteins. J Virol. 1997a;71:1667–70. doi: 10.1128/jvi.71.2.1667-1670.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QX, Spriggs MK, Kovats S, Turk SM, Comeau MR, Nepom B, Hutt-Fletcher LM. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J Virol. 1997b;71:4657–4662. doi: 10.1128/jvi.71.6.4657-4662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QX, Turk SM, Hutt-Fletcher LM. The Epstein-Barr virus (EBV) BZLF2 gene product associates with the gH and gL homologs of EBV and carries an epitope critical to infection of B cells but not of epithelial cells. J Virol. 1995;69:3987–3994. doi: 10.1128/jvi.69.7.3987-3994.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruo S, Yang L, Takada K. Roles of Epstein-Barr virus glycoproteins gp350 and gp25 in the infection of human epithelial cells. J Gen Virol. 2001:2373–2383. doi: 10.1099/0022-1317-82-10-2373. [DOI] [PubMed] [Google Scholar]
- McShane MP, Longnecker R. Cell-surface expression of a mutated Epstein-Barr virus glycoprotein B allows fusion independent of other viral proteins. Proc Natl Acad Sci USA. 2004;101:17474–17479. doi: 10.1073/pnas.0404535101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molesworth SJ, Lake CM, Borza CM, Turk SM, Hutt-Fletcher LM. Epstein-Barr virus gH is essential for penetration of B cell but also plays a role in attachment of virus to epithelial cells. J Virol. 2000;74:6324–6332. doi: 10.1128/jvi.74.14.6324-6332.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B, Elroy-Stein O, Mizukami T, Alexander WA, Fuerst TR. New mammalian expression vectors. Nature. 1990;348:91–92. doi: 10.1038/348091a0. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression tansfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Omerovic J, Lev L, Longnecker R. The amino terminus of Epstein-Barr virus glycoprotein gH is important for fusion with B cells and epithelial cells. J Virol. 2005;79:12408–12415. doi: 10.1128/JVI.79.19.12408-12415.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegtel DM, Middeldorp J, Thorley-Lawson DA. Epstein-Barr virus infection in exvivo tonsil epithelial cell cultures of asymptomatic carriers. J Virol. 2004;78:12613–12624. doi: 10.1128/JVI.78.22.12613-12624.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickinson AB, Kieff E. Epstein-Barr Virus. In: Knipe DM, Howley PM, editors. Fields Virology. Lippincott Williams and Wilkins; Philadelphia: 2001. pp. 2575–2627. [Google Scholar]
- Rivailler P, Cho Y-G, Wang F. Complete genomic sequence of an Epstein-Barr virus related herpesvirus naturally infecting a new world primate: a defining poin in the evolution of oncogenic lymphocryptoviruses. J Virol. 2002a;76:12055–12068. doi: 10.1128/JVI.76.23.12055-12068.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivailler P, Jiang H, Cho Y-G, Quink C, Wang F. Complete nucleotide sequence of the rhesus lymphocryptovirus: genetic validation for an Epstein-Barr virus animal model. J Virol. 2002b;76:421–426. doi: 10.1128/JVI.76.1.421-426.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitki-Green D, Covington M, Raab-Traub N. Compartmentalization and transmission of mutliple Epstein-Barr virus strains in asymptomatic carriers. J Virol. 2003;77:1840–1847. doi: 10.1128/JVI.77.3.1840-1847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear PG, Longnecker R. Herpesvirus entry: an update. J Virol. 2003;77:10179–10185. doi: 10.1128/JVI.77.19.10179-10185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hutt-Fletcher LM. Epstein-Barr virus lacking glycoprotein gp42 can bind to B cells but is not able to infect. J Virol. 1998;72:158–163. doi: 10.1128/jvi.72.1.158-163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Kenyon WJ, Li QX, Mullberg J, Hutt-Fletcher LM. Epstein-Barr virus uses different complexes of glycoproteins gH and gL to infect B lymphocytes and epithelial cells. J Virol. 1998;72:5552–5558. doi: 10.1128/jvi.72.7.5552-5558.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whetter LE, Day SP, Brown EA, Elroy-Stein O, Lemon SM. Analysis of hepatitis A virus translation in a T7 polymerase-expressing cell line. Arch Virol. 1994;(Suppl 9):291–298. doi: 10.1007/978-3-7091-9326-6_29. [DOI] [PubMed] [Google Scholar]
- Wu L, Borza CM, Hutt-Fletcher LM. Mutations of Epstein-Barr virus gH that are differentially able to support fusion with B cells or epithelial cells. J Virol. 2005;79:10923–10930. doi: 10.1128/JVI.79.17.10923-10930.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Hutt-Fletcher LM. Point mutations in EBV gH that abrogate or differentially affect B cell and epithelial cell fusion. Virology. 2007 doi: 10.1016/j.virol.2007.01.025. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaswen LR, Stephens EB, Davenport LC, Hutt-Fletcher LM. Epstein-Barr virus glycoprotein gp85 associates with the BKRF2 gene product and is incompletely processed as a recombinant protein. Virology. 1993;195:387–396. doi: 10.1006/viro.1993.1388. [DOI] [PubMed] [Google Scholar]