

Abstract
Genetic depletion of macrophages in Polyoma Middle T oncoprotein (PyMT)‐induced mammary tumors in mice delayed the angiogenic switch and the progression to malignancy. To determine whether vascular endothelial growth factor A (VEGF‐A) produced by tumor‐associated macrophages regulated the onset of the angiogenic switch, a genetic approach was used to restore expression of VEGF‐A into tumors at the benign stages. This stimulated formation of a high‐density vessel network and in macrophage‐depleted mice, was followed by accelerated tumor progression. The expression of VEGF‐A led to a massive infiltration into the tumor of leukocytes that were mostly macrophages. This study suggests that macrophage‐produced VEGF regulates malignant progression through stimulating tumor angiogenesis, leukocytic infiltration and tumor cell invasion.
Keywords: Vascular endothelial growth factor, Macrophages, Mammary, Tumor, Angiogenesis, Malignancy, Mouse, PyMT, Progression, Transgenic
1. Introduction
The development of a vascular network in tumors is a crucial step for the transition of the tumor to malignancy and is thought to be rate‐limiting. However, the mechanism regulating this process, known as the angiogenic switch, is still largely unknown. Several angiogenic factors including acidic fibroblast growth factor (bFGF), vascular endothelial growth factor (VEGF), transforming growth factor‐α (TGF‐α), TGF‐β, hepatocyte growth factor, and tumor necrosis factor‐α (TNF‐α) have all been identified to play an important role in regulating tumor angiogenesis. Among these factors, VEGF‐A is a potent angiogenesis promoter for blood vessel growth in both physiological and pathological conditions (Ferrara, 2004). It regulates the survival and proliferation of endothelial cells as well as being a potent inducer of vascular permeability (Dvorak, 2002; Ferrara et al., 2003).
VEGF‐A is a member of a gene family that includes placenta growth factor (PIGF), VEGF‐B, VEGF‐C and VEGF‐D (Ferrara et al., 2003; Ferrara, 2004). In the mouse, alternative splicing leads the generation of four protein isoforms of VEGF‐A (Shima et al., 1996) and among them, VEGF164 is the predominant molecular species found in variety of normal and transformed cells (Keyt et al., 1996). The activity of VEGF‐A is mediated by VEGF‐R1 or FMS‐like tyrosine kinase 1 (Flt‐1) and VEGF‐R2 (Flk‐1), both of which are expressed by endothelial cells (Ferrara, 2004). VEGF‐A is an endothelial mitogen (Leung et al., 1989). In addition to promoting the development of blood vessels, VEGF‐A has also been reported as a chemoattractant for endothelial cells, osteoclasts and monocytes (Yoshida et al., 1996), the latter of which express VEGF‐R1.
The expression of VEGF‐A has been found in various human cancers and it is associated with disease progression and decreased survival rate, suggesting that it plays an important role in malignant progression (Jain et al., 2006). In addition to tumor cells, VEGF is also expressed in the tumor stroma, raising the question of the relative importance of VEGF produced in each compartment in tumor angiogenesis and progression. Dong et al. (2004) reported that VEGF‐null fibrosarcomas were tumorigenic and angiogenic in vivo in spite of the absence of tumor‐derived VEGF. This group further demonstrated that VEGF‐producing stromal fibroblasts played a crucial role in regulating tumor angiogenesis in the transplanted VEGF‐null fibrosarcomas. Using a mouse model of cervical cancer, Giraudo and coworkers also reported that macrophages in the tumor microenvironment regulating tumor angiogenic switch by producing MMP‐9, a protease implicated in mobilization of VEGF (Giraudo et al., 2004). These studies indicate that in certain tumor microenvironments, stromal cells play a dominant role in promoting the angiogenic switch through producing VEGF or affecting the function of VEGF.
We have previously shown that VEGF‐A is produced by tumor‐associated macrophages in a mouse model of breast cancer, PyMT mice (Lin et al., 2006). Depletion of macrophage growth factor, CSF‐1, which reduced the infiltration of macrophages into the tumor stroma, resulted in a delayed tumor angiogenic switch and malignant transition in this model (Lin et al., 2001, 2006). These observations suggest that in the mammary tumors of PyMT mice, macrophage‐produced VEGF‐A regulates the growth and development of blood vessels during the angiogenic switch. The fact that CSF‐1 is able to stimulate the expression of VEGF‐A in cultured macrophages also supports this notion (Eubank et al., 2003). Based on these studies, we hypothesize that VEGF‐A produced by CSF‐1 dependent‐macrophages is required for the onset of the angiogenic switch in PyMT model of breast cancer. To test this hypothesis, we established a strain of transgenic mouse in which the expression of VEGF‐A was expressed in the mammary epithelium in a temporal specific manner. We demonstrate that restoring VEGF‐A expression in the premalignant mammary lesions of CSF‐1 depleted mice promotes their tumor progression to the level of their wild‐type counterparts. Therefore, the lack of macrophage‐produced VEGF‐A is a key element contributing to the delayed tumor progression to malignancy in CSF‐1 depleted PyMT tumors and can be compensated for by tumor produced VEGF‐A.
2. Results
2.1. Regulated expression of VEGF in the mammary gland
To achieve conditional expression of VEGF‐A specifically in the mammary epithelium, we have established a bi‐transgenic mouse line using the tetracycline‐regulated repressor‐transactivator system. In this system, the expression of the transgene, VEGF‐A‐encoding cDNA, is under the control of a tetracycline‐dependent promoter (Figure 1A). The activity of this promoter is regulated by the reverse tetracycline‐dependent transcriptional activator, rtTA, whose expression is under the control of mouse mammary tumor virus (MMTV) promoter, as reported previously, that shows specific expression in the mammary epithelium and to a lesser degree the salivary gland but not elsewhere (Mok et al., 1992). In the bi‐transgenic mice, transcription from the transgenic VEGF is induced in the presence of doxycycline (Dox). To monitor the expression of the transgene in vivo, a foot‐and mouth disease virus (FMDV) 2A DNA segment (Donnelly et al., 2001) was added in‐frame at the 3′ of the VEGF‐A and 5′ of GFP cDNAs as illustrated in Figure 1A. This construct permits multi‐cistronic expression of VEGF‐A, 2A and GFP mRNA. However, as the 2A sequence directs cleavage of the polypeptide during translation (de Felipe, 2004), VEGF‐A and GFP are synthesized in the same cell but as two separate proteins. This approach marks sites of expression while at the same time preventing the potential aberrant function of a GFP–VEGF‐A fusion protein. Furthermore, to reduce leaky transcription from the transgene, a common problem of the tetracycline regulatory system, a silencer construct was co‐injected with the transgene (data not shown). No leakage was found in untreated transgenic mice (data not shown).
Figure 1.

Transgenic expression of VEGF in the mammary gland. (A) The expression of transgenic VEGF‐A and GFP was regulated by the tetracycline inducible system in which the expression of rtTA was under the control of the mammary‐specific promoter, MMTV‐LTR. Upon exposure to doxycycline (Dox), mRNA consisting of both VEGF‐A and GFP was synthesized in mammary epithelium. Subsequently, upon translation, 2A induced cleavage leads to the formation of the two distinct proteins, VEGF and GFP. (B) Western analysis of the transgenic expression. Western blots are prepared from mammary tissues of mice at 6weeks of age carrying the VEGF transgene, lanes 1 and 2 (lane 1, PyMT−; and lane 2, PyMT+) and control mice 3–5 (lane 3: PyMT+, rtTA−, VEGF+; lane 4: PyMT−, rtTA+, VEGF−; lane 5: PyMT+, rtTA−, VEGF+). Mice were treated with 1mg/ml Dox in the drinking water for 2weeks. The filter was probed with anti‐VEGF, ‐GFP and ‐β‐tubulin antibodies. (C) Tissue specific expression of the VEGF transgene. Western analysis of transgenic VEGF expression in various tissues as indicated. M. gland+, mammary gland from a VEGF transgenic mouse; M. gland−, mammary gland from a non‐transgenic control. β‐Tubulin was used as the loading control.
One founder carrying the Vegfa‐2A‐Gfp transgene (Tg(TetopVegfa‐2A‐Gfp)1Jwp) that had co‐integrated with the Tet‐silencer was identified (referred as VEGF‐A bi‐transgenic) and bred onto mice carrying the MMTV LTR/rtTA transgene (Tg(MMTV‐rtTA)Lach). The expression of VEGF‐A and GFP in the mammary glands of the bi‐transgenic mice was compared with mice carrying one of the transgenes using Western blotting analysis (Figure 1B). Both the transgenic and control mice were treated with 1mg/ml doxycycline (Dox) in drinking water for 2weeks from age 6 to 8weeks. The levels of both VEGF‐A and GFP protein were strongly induced in the mammary glands of the bi‐transgenic mice (Figure 1B, lanes 1 and 2) compared to undetectable signals in mice carrying either one of the transgenes (Figure 1B, lanes 3–5).
To confirm that the expression of VEGF‐A is restricted to the mammary gland, different tissues from bi‐transgenic mice with the same treatment were analyzed. Figure 1C shows that the level of VEGF‐A protein in mammary gland isolated from a bi‐transgenic mouse is also strongly induced compared to the signals detected in other tissues from the same mouse as well as the mammary gland from a non‐transgenic control (Figure 1C, M. gland+ vs. M. gland−).
The original MMTV promoter is expressed in several tissues other than the mammary epithelium including some hematopoietic cells (Ross et al., 1990; Kumaraswamy et al., 2003). However, the promoter used in the MMTV‐rTta construct is the MMTV‐206 promoter that contains sequences from V‐H‐ras that confers more restricted expression to the mammary epithelium. In fact in an extensive analysis Gunther et al. (2002) indicated that it was only expressed in the mammary and salivary epithelium. However, because of its importance for our analysis that it is not expressed in hematopoietic cells, we further characterized the MMTV expression using the VEGF‐A–2A–GFP as a reporter. To ensure that GFP was preserved properly during the tissue preparation and to test expression in macrophages, tissues from a Csf1r‐GFP transgenic mouse (tg(Csf1r‐Gfp)Hume), in which GFP was expressed specifically in macrophages (Sasmono et al., 2003), were processed in the same experiment under the same condition as tissues isolated from the VEGF‐A bi‐transgenic mice (Figure 2). In this experiment, Texas‐Red conjugated dextran was also used to mark phagocytic macrophages, as described in Section 4. In the spleen of Csf1r‐GFP mouse, the dextran and GFP‐positive cells were found in the stroma surrounding the white pulp which consists mainly of lymphocytes (Figure 2a and b, blue arrows). No GFP‐positive cells were found in the white pulp (Figure 2a and b, white arrows). Dextran marked macrophages were found in the stroma surrounding the white pulp in the spleen isolated from the VEGF‐A bi‐transgenic mice; however, no GFP‐positive cells were observed in the tissue section (Figure 2c and d). This result indicates that the VEGF‐A transgene is not expressed in either macrophages or lymphocytes in the VEGF‐A bi‐transgenic mice. To further confirm this finding, thymus was prepared from both transgenic strains. Most of the GFP‐positive cells in the thymus of the Csf1r‐Gfp mouse were co‐localized with dextran‐positive cells (Figure 2e and f), indicating that the GFP expression in macrophages was well preserved in this experiment. In contrast, no GFP‐positive cells were found in either dextran‐positive or negative area in the thymus isolated from the VEGF‐A bi‐transgenic mice (Figure 2g and h), confirming that the transgenic VEGF‐A/GFP was not expressed in either myeloid or lymphoid cells.
Figure 2.

Transgenic expression of VEGF/GFP is mammary epithelium specific. Tissue sections prepared from Csf1r‐GFP and VEGF‐A bi‐transgenic mice. To mark the phagocytic macrophages, both mice were i.v. injected with Texas‐Red conjugated dextran 2h before the tissue isolation as described in Section 4. (a–d) Spleen sections from the Csf1r‐GFP (a and b) and the VEGF‐A bi‐transgenic (c and d) mice. The insets in a and c are shown at high magnification in b and d. Blue arrows point to the area consisting of mainly Texas‐Red dextran marked macrophages. Notice that in the spleen from Csf1r‐Gfp mice, most of the dextran‐labeled cells are also GFP‐positive (b). White arrows point to white pulp in the spleen which are the collections of lymphocytes. Notice that no GFP‐positive cells are in these areas. (e–h) Sections of thymus from the Csf1r‐Gfp (e and f) and the VEGF‐A bi‐transgenic (g and h) mice. The insets in e and g are shown at high magnification in f and h. Arrows point to cells that are dextran‐positive. Note that most of the dextran‐positive cells in Csf1r‐GFP thymus are also GFP‐positive (f). (i, j) Mammary gland sections from the Csf1r‐Gfp (i) and the VEGF‐A bi‐transgenic (j) mice. Arrows in j point to GFP‐positive mammary ducts and arrows in i point to GFP‐positive stromal cells. Scale bars for a, c, e, g, i, j: 100μm; for b, d, f, h: 20μm.
Furthermore, and consistent with the Western blot analysis, the expression of GFP was found exclusively in mammary ducts in the VEGF‐A bi‐transgenic mice (Figure 2j) in contrast to the GFP‐positive cells found in the mammary stroma in the Csf1r‐Gfp mouse (Figure 2i). To further confirm that the expression of the transgenic VEGF‐A/GFP is mammary epithelium specific, various tissues and cells including bone marrow derived macrophages, pancreas, liver and skin from the VEGF‐A bi‐transgenic mice were analyzed and no GFP‐positive cells were found in these tissue examined although macrophages could easily be identified by their phagocytosis of Texas‐Red labeled dextran. Taken together these studies reported in Figures 1 and 2 indicate that the expression of the transgenic VEGF‐A is inducible by Dox treatment and the expression is restricted to the mammary and salivary epithelium. Importantly there was no expression in any hematopoietic cells including any tissue macrophages examined.
2.2. Transgenic expression of VEGF‐A induces the development of a high‐density vessel network in the mammary gland
To examine the effect of VEGF‐A in mammary tumor biology, the bi‐transgenic system was crossed onto a mouse model of breast cancer where tumors are induced by the mammary restricted expression of the Polyoma Middle T oncoprotein (PyMT mice) (Guy et al., 1992). Consistent with the increased expression of VEGF‐A, inguinal mammary glands isolated from mice carrying either two or three (with the MMTV–PyMT transgene) transgenes compared to the single‐transgenic controls (Figure 3A, i) were filled with blood vessels (Figure 3A, ii) following Dox treatment. Using fluorescent microscopy, we observed that in contrast to the non‐transgenic controls (Figure 3B, i), the entire mammary epithelial ductal tree and tumors of the bi‐ or tri‐transgenic mice were brightly marked by the green fluorescent protein (Figure 3B, ii). GFP expression was not observed elsewhere in the mammary gland or in other tissues. This result confirms that the transgenic construct is expressed specifically in the mammary epithelium.
Figure 3.

Transgenic expression of VEGF in the mammary gland induces vessel network formation. (A) Mammary whole mounts showing an increase of vessel density in the mammary gland isolated from a mouse carrying the VEGF transgene (ii) compared to a non‐transgenic control (i). (B) GFP expression in mouse mammary glands. Fluorescent micrograph of mammary whole mounts from a VEGF transgenic PyMT mouse (ii) and a non‐transgenic control (i) shows that both the mammary ducts and tumor lesions were marked by GFP. Bar: 1mm. (C) Transgenic VEGF induces vessel formation around the mammary ducts. The images were prepared from mammary whole mounts from a VEGF transgenic mouse i.v. injected with Texas‐Red dextran to label the blood vessels (ii). The mammary ducts are marked by the transgenic expression of GFP (i). The vessel network is formed surrounding the GFP expressing mammary ducts (iii). Bar: 0.3mm.
To examine the effect of the VEGF‐A transgene expression on vessel development in the mammary gland, Dox treated rtTA/VEGF‐A bi‐transgenic mice were i.v. injected with Texas‐Red conjugated dextran 5min before mammary glands were isolated for whole‐mount analysis, as described previously (Lin et al., 2006). Figure 3C shows representative images from such a whole mount preparation. We observed that a vessel network developed that surrounded the GFP‐positive mammary duct (Figure 3C, i–iii). No such vessel network was found in the mammary glands isolated from mice carrying a single transgene (data not shown). This observation shows that the transgenic expression of VEGF‐A in the mammary epithelium can act as an angiogenic promoter to induce vasculature development in the adjacent stroma.
2.3. Transgenic expression of VEGF‐A promotes tumor progression to malignancy in CSF‐1 depleted PyMT mice
We have previously reported that, compared to the wild‐type PyMT mice, tumor progression in the CSF‐1 depleted PyMT mice, Csf1op/Csf1op, was inhibited following a delayed onset of the angiogenic switch in the tumor (Lin et al., 2001, 2006). We also reported that such delayed tumor progression was associated with reduced infiltration of macrophage in the primary tumors (Lin et al., 2001). Since VEGF‐A expression was mainly found in tumor associated macrophages in PyMT mammary tumors (Lin et al., 2006), we hypothesized that the delayed tumor angiogenic switch in Csf1op/Csf1op tumors was due to a lack of macrophage‐produced VEGF‐A.
An increase of macrophage infiltration in mammary tumors was observed in wild‐type PyMT mice at 7–8weeks of age in mixed genetic background (Lin et al., 2001) and 1–2weeks earlier in FVB background (data not shown). The angiogenic switch and malignant transition occurred 1–2weeks following such an increase in macrophage density in the primary tumors. To determine whether the lack of macrophage‐produced VEGF‐A is the cause of delayed tumor progression in Csf1op/Csf1op tumors in which the increase of macrophage infiltration did not occur, we sought to induce the expression of the transgenic VEGF‐A in the Csf1op/Csf1op mammary tumors close to the time when macrophage infiltration occurs in the wild‐type tumors.
To determine whether Dox treatment was able to induce angiogenesis in mammary lesions, we first tested the effect of the transgenic VEGF‐A on vessel network formation in premalignant tumors in wild‐type PyMT mice (Figure 4A). As reported previously, tumors developed in PyMT mice have defined stages of progression (Lin et al., 2002). Premalignant lesions were classified as hyperplasia that progressed to adenoma/MIN, the stage that increased macrophage infiltration was observed (Lin et al., 2001). As mentioned above, we found that tumor progression in mice with FVB background was approximately 2weeks faster than the out‐bred mice. Since the bi‐transgenic mice used in this study were the first generation of outbred mice from FVB background, the rate of tumor progression and the timing of macrophage infiltration in these mice were found to be close to mice with a FVB background (data not shown). Therefore, we treated the transgenic PyMT mice from 4 to 6weeks of age when the increase of macrophage infiltration was observed in the mammary tumors of FVB PyMT mice. At the end of the treatment, a high density vasculature was developed in tumors as determined by IHC staining with an anti‐vWF antibody that reliably marks vessels (Lin et al., 2006) (Figure 4Ab and d). These newly formed capillaries are located throughout the tumor lesion (Figure 4Ab). Many of them have enlarged lumens filled with RBC (Figure 4Ad *). No such capillaries were developed in the PyMT control mice treated in the same manner (Figure 4Aa and c). In the non‐transgenic mammary glands, the vessel density in the lesions appears similar or even lower than the surrounding fat pad (Figure 4Aa, arrows) and the lumens of the vessels are smaller (Figure 4Ac, arrows) compared to the transgenic counterparts (Figure 4Ad). These results indicate that the transgenic VEGF‐A is able to induce the formation of a high‐density vascular network in premalignant lesions that is never observed in non‐transgenic mice.
Figure 4.

Transgenic expression of VEGF induces angiogenesis in pre‐malignant lesions. (A) Induction of a high density vessel network in pre‐malignant tumors from wild‐type PyMT mice. IHC using anti‐vWF antibodies. Section of mammary lesions from wild‐type PyMT mice at 6weeks of age carrying the VEGF transgene (right panel) or the non‐transgenic control (left panel) counter‐stained with hematoxylin. Mice were treated with Dox for 2weeks. The insets in the top panel are shown in the lower panel. The vessel lumen is labeled with an asterisk. Arrows in a and c point to vessel positively stained with the anti‐vWF antibody in the lesion. Scale bars for a and b: 100μm; for c and d: 30μm. (B) Induction of vessel network formation in adenoma stage tumors from Csf1op/Csf1op PyMT mice. (i) Sections of tumor lesions from Csf1op/Csf1op PyMT mice at 8weeks of age. Mice were treated with Dox for 2weeks. Blood vessels were marked by i.v. injection of Texas‐Red conjugated dextran and visualized by fluorescent microscopy. (ii) A tumor from VEGF transgenic mouse and (i), a non‐transgenic control. Arrows point to autofluorescent RBCs in capillaries. Bar: 30μm.
Next we wanted to determine whether the transgenic VEGF‐A is also able to induce vessel network formation in the premalignant lesions of Csf1op/Csf1op mice in which the increase of macrophage infiltration during tumor progression is inhibited. The majority of Csf1op/Csf1op PyMT mice at 6weeks of age had tumors at adenoma/MIN stage, thus we treated these mice with Dox starting at 6weeks of age for 2weeks. Similar to the observation in the wild‐type PyMT mice, a high density of vessel network was developed in the transgenic Csf1op/Csf1op tumors at the end of the Dox treatment (Figure 4B, ii). The vessels in the network had large lumens which were filled with RBC and wrapped around the GFP‐positive acini (Figure 4B, ii, arrows). In contrast, after the Dox treatment of the non‐transgenic lesions, the vessel density was much lower and the size of vessels was smaller. Moreover, the vessel lumens in the non‐transgenic tumors were much smaller than the counter parts in the transgenic tumors (Figure 4B, i, arrows).
To determine the effect of the transgenic expression of VEGF‐A in tumors, we next examined the tumor stages for these treated mice. All of the wild‐type PyMT mice with or without the VEGF‐A transgene examined at the end of the Dox treatment from 6 to 8weeks of age already had lesions that had progressed to malignant stages (Figure 5Ac and d, B) suggesting that the angiogenic switch had occurred in the wild‐type PyMT mice between 6 to 8weeks of age regardless of the expression of the transgenic VEGF‐A. In contrast and consistent with previous reports, tumors from ∼50% of non‐transgenic Csf1op/Csf1op PyMT mice examined were still at pre‐malignant stages (Figure 5Aa and B, op‐non). However, all of the VEGF‐A transgenic Csf1op/Csf1op PyMT mice developed malignant lesions at the end of the treatment showing a significantly enhanced tumor progression versus the non‐transgenic counterparts (Fisher Exact test: p=0.033). These results indicate that increased expression of VEGF‐A in these macrophage‐depleted Csf1op/Csf1op PyMT mice promotes the angiogenic switch and accelerates tumor progression to malignancy to the level similar in a similar manner to the wild‐type PyMT mice.
Figure 5.

Transgenic expression of VEGF in mammary tumors accelerates tumor progression to malignancy. (A) H&E staining of tumor sections from Csf1op/Csf1op (a and b) and wild‐type (c and d) PyMT mice at 8weeks of age. Mice carrying the VEGF transgene: b and d; and the non‐transgenic controls: a and c. All of the mice were treated with 1μg/ml Dox for 2weeks. Bar: 100μm. (B) Quantitative analysis of tumor progression in VEGF transgenic and control PyMT mice. Genotypes: op‐non, Csf1op/Csf1op non‐transgenic mice; op trans, Csf1op/Csf1op mice carrying the MMTV‐rtTA/VEGF transgene; wt‐non, wild‐type non‐transgenic mice; wt trans, wild‐type mice carrying the MMTV‐rtTA/VEGF transgene. Statistical analysis: Fisher's Exact test.
2.4. Transgenic expression of VEGF‐A induces an increase of macrophage infiltration in mammary gland
The transgenic expression of VEGF‐A in mammary glands not only increased the development of a high density vascular network but also increased the infiltration of leukocytes in the stroma of mammary ducts and tumors. Figure 6A shows representative images of mammary ducts from non‐transgenic (Figure 6Aa and c) and VEGF‐A transgenic (Figure 6Ab and d) mice at 8weeks of age which have been treated with Dox for 2weeks. Multiple layers of stromal cells were found in the stroma adjacent to the ducts of VEGF‐A transgenic mammary gland (Figure 6Ad, arrows). This stroma consists of several cell types including fibroblasts and leukocytes. Such heavy leukocytic infiltration is not seen in normal mammary glands in which very few leukocytes are seen in the adjacent stroma of mature ducts (Figure 6Ac) (Gouon‐Evans et al., 2000). In fact, such massive leukocytic infiltration is often seen around the developing terminal end buds (TEBs) of the mammary duct, which consists of a large number of proliferating epithelial cells surrounded by a dense stroma. Similarly, a high density of leukocytic infiltration was also observed in the primary tumors of VEGF‐A transgenic mice treated with Dox. A majority of these infiltrated leukocytes were macrophages, determined by IHC using antibody recognizing the pan macrophage marker, F4/80 (Figure 6Bb and d). Very few F4/80 positive cells were observed at a similar stage of mammary lesions in non‐transgenic littermates (Figure 6Ba and c).
Figure 6.

Transgenic expression of VEGF in mammary glands induces leukocytic infiltration. (A) a–d, H&E staining of mammary ducts. b and d, mammary gland from a VEGF and MMTV‐rtTA double transgenic mouse; a and c, a non‐transgenic control. The insets in a and d are shown at high magnification in c and d. Notice that multiple layers of stromal cells are surrounding the mammary duct in the VEGF expressing transgenic mammary gland (d, arrows). Size bar for a and b: 100μm; for c and d: 30μm. (B) a–d, IHC of mammary lesions stained by an anti‐F4/80 antibody. b and d, mammary lesions from a VEGF transgenic mouse; a and c, lesions from a non‐MMV‐rtTA transgenic control. The insets in a and b are shown in c and d, respectively. RBC filled vessels are labeled by an asterisk. Scale bars for a and b: 100μm; for c and d: 30μm.
These data suggest that VEGF‐A is acting as a chemoattractant to macrophages that are also known to express VEGFR1 (Ferrara, 2004). Thus to determine the phenotype of the recruited macrophages we analyzed the expression of VEGF R1 in myeloid cells isolated from tumor stroma using FACS. Cell suspensions prepared from primary mammary tumors were labeled with anti‐CD11b and F4/80 to identify myeloid cells. This shows that three populations of myeloid cells were identified in VEGF‐A transgenic PyMT mice (Figure 7A). Population R2 and R3 were identified as macrophages according to their relative high expression of macrophage marker F4/80. On the other hand, population R4 was identified as granulocytes determined by their relative low expression of F4/80 and high expression of the neutrophil marker Gr1 (data not shown).
Figure 7.
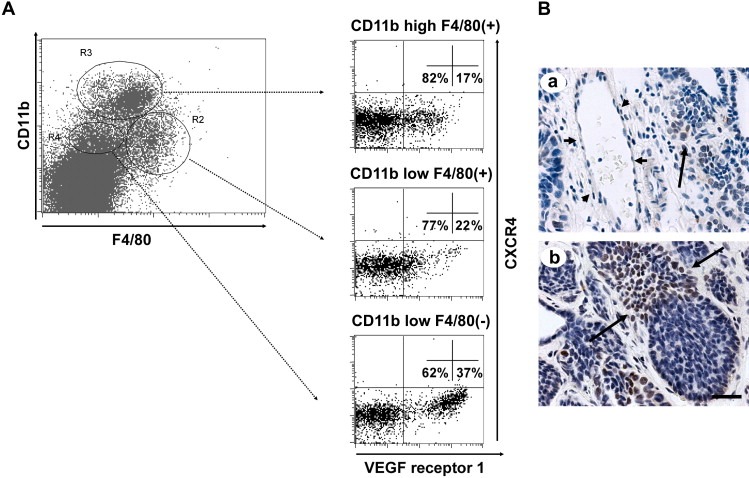
VEGF R1 and SDF‐1/CXCR4 expression in mammary tumors. (A) FACS analysis of VEGF R1 and CXCR4 expressing myeloid cells isolated from VEGF transgenic tumors. Myeloid cells isolated from mammary tumors of MMTV‐rtTA/VEGF transgenic mice were identified by FACS using anti‐CD11b and F4/80 antibodies (A, left). The expression of CXCR4 and VEGF receptor 1 in three different populations of myeloid cells, R2, R3 and R4, were further analyzed using FACS (A, right). (B) IHC staining of VEGF transgenic tumors for SDF1. A blood vessel is pointed out by short arrows (a) and positive stained cells in the tumor are pointed out by long arrows. Bar: 30μm.
Previous studies have suggested that VEGF‐A causes the infiltration of bone marrow‐derived circulating cells (RBCCs) through activating perivascular myofibroblasts to produce the chemokine SDF1 (CCL12) that induces the infiltration of RBCCs (Grunewald et al., 2006). To determine the possible effects of VEGF‐A and SDF1 on the infiltration of myeloid cells in PyMT tumors, we analyzed the expression of VEGF R1 and SDF1 receptor, CXCR4, in the myeloid cells (CD11b+ve) isolated from the tumor stroma. We found that in the VEGF‐A transgenic PyMT tumors, ∼20% of macrophages isolated were VEGF R1 positive (Figure 7A, R3 and R2). Less than 2% of these macrophages expressed SDF1 receptor, CXCR4 (Figure 7A, R2 and R3). On the other hand, a much higher percentage of granulocytes (37%) expressed VEGF receptor 1 (Figure 7A, R4). However, in a similar fashion to macrophages, less than 2% of these cells expressed CXCR4. A similar distribution of VEGF R1 and CXCR4 expressing Cd11b+ve cells were found in non‐transgenic PyMT tumor (data not shown).
These results suggest that SDF1 does not play a major role in regulating the infiltration and/or activation of tumor associated myeloid cells in PyMT tumors. However, VEGF‐A appears to have a direct influence on the myeloid cells in PyMT tumors.
To determine whether SDF1 is expressed in the mammary tumors in PyMT mice, anti‐mouse SDF1 antibody was used to stain the tumor section. As shown in Figure 7B, perivascular myofibroblasts in the tumor appeared to be negative for SDF1 (Figure 7Ba, short arrows) whereas many tumor cells are positive for this factor (Figure 7Ba and b, long arrows). This result suggests that, different from the effect of transgenic VEGF‐A expression in the heart and lung (Grunewald et al., 2006), VEGF‐A expressed in the PyMT tumor did not appear to induce SDF1 expression in perivascular myofibroblasts.
2.5. Transgenic expression of VEGF‐A in mammary tumors promotes tumor invasion
Correlated with the increase of macrophage infiltration in VEGF‐A transgenic mammary tumors, we observed an increase of tumor invasion into the mammary stroma in the CSF‐1 depleted PyMT mice (Figure 8Aa, black arrows). A specific histology referred to as “Medusa” structures was seen in many VEGF‐A transgenic Csf1op/Csf1op tumors (Figure 8A, white arrows). In this structure, tumor cells appeared to grow aggressively and actively invade into the stroma (Figure 8Ab, arrows). Using the expression of Ki67 as a surrogate marker for cells in DNA synthesis, IHC staining with anti‐Ki67 antibody indicated that the cells in these medusa structures are highly proliferative with most of the cell being Ki67 positive (Figure 8Ac) especially at the invasive fronts (Figure 8Ad, arrows). In contrast to this, a large percentage of tumor cells in the tumors of non‐transgenic Csf1op/Csf1op mice are Ki67 negative (Figure 8Ba and b, arrow).
Figure 8.

Transgenic expression of VEGF in Csf1op/Csf1op induces the formation of a “Medusa” structure. (A) Invasive tumor growth. (a, b) H&E staining of mammary tumors from a VEGF transgenic Csf1op/Csf1op PyMT mice at 8weeks of age. Black arrows in image a point to the invasive lesions and white arrows point to the “Medusa” structure. The invasive front of the tumor cells in the “Medusa” structure is shown in b, indicated by the arrows. (c, d) IHC using anti‐Ki67 antibody showing that a large percentage of tumor cells, especially these cells at the invasive front (arrows in d), in the “Medusa” structure is Ki67 positive. The insets in a and c are shown in b and d. (B) Ki67 IHC of mammary lesion from a non‐transgenic Csf1op/Csf1op mouse at 8weeks of age. The inset in a is shown in b. Notice that ∼50% of cells in the lesion are Ki67 negative. Scale bar for the left panel: 100μm; for the right panel: 30μm.
3. Discussion
Establishment of a vasculature is essential for the development of organs during embryogenesis. In this respect, tumors are no exception since they also need an adequate vascular supply to grow effectively (Folkman, 2002). Indeed, there is substantial evidence that as tumors progress to the malignant stage there is a sharp increase in vascular density in a process known as the angiogenic switch (Hanahan and Folkman, 1996; Naumov et al., 2006). When this angiogenic switch is impeded either through the use of angiogenesis inhibitors or by genetic deprivation of essential factors, tumor progression to malignancy is severely impaired (Lin et al., 2006; Naumov et al., 2006).
Many factors are involved in the complex process of angiogenesis that results in the formation of a coherent network of vessels. Prime amongst these factors is VEGF‐A, a growth factor that binds two trans‐membrane receptor tyrosine kinases, VEGFR1 and R2. VEGFR2 is restricted to endothelial cells while VEGFR1 is also expressed upon myeloid cells (Ferrara, 2004). Numerous studies have indicated the importance of VEGF‐A in tumor development and it, together with its receptor system, are the targets for therapy and several anti‐VEGF therapeutics are now in clinical practice (Naumov et al., 2006). Detailed analysis of human breast cancer samples show that VEGF‐A can be produced by both tumor cells and infiltrating macrophages in some cases in mutually exclusive patterns (Leek et al., 2000). These data suggest that tumors absolutely require VEGF‐A but they can obtain it from a variety of sources including infiltrating host macrophages.
To explore the role of macrophages in breast cancer we have used genetic depletion of these cells in a mouse model where the tumors are caused by the transgenic expression of the Polyoma Middle T oncoprotein in the mammary epithelium. This macrophage depletion had multiple consequences including a reduction in the progression of tumors to malignancy and a lowering of metastatic capacity (Lin et al., 2001). Detailed analysis of the transition period when the benign lesions become malignant indicated a classical angiogenic switch in wild‐type mice but a delayed or absent transition in the macrophage depleted mice (Lin et al., 2006). In fact, even in those tumors from macrophage depleted mice that had progressed to late malignancy, the vascular density was reduced by ∼40% compared to wild‐type mice (Lin et al., 2006). In contrast to these data, early recruitment of macrophages to benign lesions by the transgenic expression of CSF‐1 in the mammary epithelium in wild‐type, macrophage replete mice caused premature angiogenesis even in hyperplastic lesions and their acceleration to malignancy (Lin et al., 2006). These data strongly suggest that macrophages press the angiogenic switch at the malignant transition (Lin et al., 2006; Lin and Pollard, 2007).
Immunohistochemistry using anti‐VEGF‐A antibody indicated that as in human breast cancer, in these mouse mammary tumors macrophages express VEGF‐A (Lin et al., 2006). These expression data suggest that the loss of macrophage VEGF‐A is causal in the delay of the angiogenic switch and reduced vascularization observed in the macrophage‐depleted mice. To test the hypothesis that VEGF‐A plays a critical role in this macrophage‐mediated angiogenic switch we developed a tetracycline regulated VEGF‐A expression system where this secreted molecule is expressed in the mammary epithelial cells at times when the macrophage‐depleted tumors are at the hyperplasia to adenoma/MIN stages and thus approaching the malignant transition.
In order to perform these experiments we used a novel method to unequivocally mark VEGF‐A‐expressing cells. This method used the FMDV virus 2A sequence ligated in‐frame between VEGF‐A and GFP cDNAs in a doxycycline‐inducible transgenic construct. This method works to allow concurrent expression of GFP and VEGF‐A from the same mRNA because the 2A sequence directs a proteolytic cleavage during translation between the VEGF‐A and GFP polypeptides such that they are expressed as discreet proteins (de Felipe, 2004). In this manner only cells that express VEGF‐A can be GFP‐positive. This polycistronic transgenic construct was co‐integrated with a tetracycline silencer element, a strategy that resulted in a non‐leaky expression in the absence of doxycycline. Using this method in combination with Western blotting we showed that VEGF‐A was expressed throughout the mammary epithelium and not elsewhere except the salivary epithelium and that it was not expressed unless the mice were treated with doxycycline. Importantly no expression could be detected in any hematopoietic cells. Thus this transgenic strain was suitable to test our hypothesis.
We treated transgenic PyMT mice and their controls for 2weeks with doxycycline at the time when the malignant transition occurs as well as control non‐PyMT mice. This inducible expression of VEGF‐A in the mammary epithelium resulted in a dramatic increase in vascular density in the adjacent stroma in both the normal mammary gland during development and in the mammary tumors. This increase in density was observed in both macrophage replete and depleted mice. Analysis of tumor progression in the primary tumors of the inguinal mammary gland indicated that this was unaffected in wild‐type mice with all tumors showing malignant characteristics. This was probably because in this strain background all the mice even in the absence of transgenically expressed VEGF‐A had already progressed to malignancy. This is most likely to be the explanation since constitutive expression of VEGF‐A in this PyMT model on an FVB background from the MMTV promoter that is expressed in the mammary epithelium from early in life decreased tumor latency, increased tumor burden and accelerated malignancy (Schoeffner et al., 2005). However, in macrophage depleted Csf1op/Csf1op.PyMT mice where tumor progression is significantly delayed compared to wild‐type mice, the over‐expression of VEGF‐A in pre‐malignant lesions accelerated tumor progression to malignancy such that they resembled wild‐type PyMT mice. In addition, it induced a more malignant phenotype characterized by medusa‐like structures with a very high proliferative index and with invasive characteristics.
These data suggest that one of the major functions of macrophages is to bring VEGF‐A into the tumor microenvironment to promote angiogenesis as benign tumors progress towards malignancy. This is consistent with the dramatic recruitment of macrophages to the pre‐malignant lesions through an as yet unknown mechanism (Lin et al., 2006). It is interesting to note that transgenic over‐expression of CSF‐1 in the PyMT model is also sufficient to recruit macrophages to mammary tumors and this in itself is sufficient to promote angiogenesis and the malignant transition (Lin et al., 2001). These data suggest a causal association between the observed over‐expression of CSF‐1 in human breast cancers and poor prognosis (Scholl et al., 1994). This CSF‐1 expression is associated with leukocytic infiltrates containing a large numbers of macrophages (Scholl et al., 1994) and the density of these cells in turn is correlated with increased micro‐vascular density and poor prognosis (Leek et al., 1996). Interestingly, VEGF‐A is a CSF‐1 regulated molecule in cultured macrophages (Eubank et al., 2003) and in macrophages associated with CSF‐1 expressing melanoma cells (Varney et al., 2005), suggesting that this growth factor might not only recruit macrophages but also regulate their expression of VEGF‐A. Indeed the similarity in phenotypes between VEGF‐A and CSF‐1 over‐expression in this and our previous study (Lin et al., 2001) suggests that one of the major functions for macrophages in tumors is CSF‐1 regulated VEGF‐A expression.
It was also striking that VEGF‐A‐expression recruited many macrophages into the mammary tumor microenvironment even in Csf1op/ Csf1op mice. In this latter case we have shown that either local infusion or transgenic expression of CSF‐1 is sufficient to recruit tissue macrophages suggesting that the pool of circulating monocytes is abundant enough to allow recruitment and differentiation of macrophages given the correct local environment. Macrophage recruitment was also observed by Grunewald et al. using a similar tetracycline‐regulated transgenic approach to express VEGF‐A in the heart and liver (Grunewald et al., 2006). These recruited bone marrow‐derived circulating cells (RBCC) had the characteristic of myeloid cells including the expression of Cd11b (Grunewald et al., 2006). The cells recruited by VEGF‐A to tumors in this study had similar characteristics but upon further characterization they were as classified as both macrophages (F4/80hi Cd11b+ve) and neutrophils (F4/80−ve, Gr1+ve). Grunewald et al. (2006) also showed that the RBCCs were VEGFR1+ve and CXCR4+ve and presented evidence that VEGF‐A induced expression of the CXCR4 ligand (CXCL12 or SDF‐1) in perivascular cells was required for their recruitment. In our study however, we showed the recruited myeloid cells were CXCR4−ve and furthermore we could not detect peri‐vascular expression of CXCL12, indicating that this was not the mechanism for their VEGF‐A recruitment into the mammary gland. However, 20% of the recruited myeloid cells were VEGFR1 positive, suggesting that this might be a mechanism for recruitment. VEGF‐A is chemotactic for monocytes (Barleon et al., 1996) data that suggest that the VEGFR1 population might be first recruited, and that these cells then produce other myeloid growth factors that recruit or stimulate the proliferation of the remaining 80% of myeloid cells that are VEGFR1 negative.
The RBCC population found after VEGF‐A expression in liver and heart were angiogenic in an in vitro aortic ring vessel‐sprouting assay (Grunewald et al., 2006). Indeed there is considerable data that macrophages produce angiogenic factors in a variety of settings (Leek and Harris, 2002; Sunderkotter et al., 1994; Lobov et al., 2005; Lewis and Pollard, 2006; Cho et al., 2007). Despite these data and the association of macrophage recruitment in the present experiments with angiogenesis it is still uncertain whether macrophage‐derived factors that are responsible for the increased angiogenesis or the VEGF‐A itself acting upon endothelial cells or a combination of both. This will not be resolved until macrophages can be depleted in the VEGF‐A‐over‐expressing mice.
In this study we have shown that VEGF‐A is sufficient to recruit macrophages to mammary tumors and it also to promote angiogenesis either alone or in combination with macrophage derived factors. VEGF‐A can correct for the deficiency of macrophages in CSF‐1 nullizygous mice in the promotion of PyMT tumor progression to malignancy and increasing metastasis. Since immunohistochemical data suggest that the majority of VEGF‐A in this model is provided by macrophages, this suggests that a major function of macrophages is to bring VEGF‐A into the tumor microenvironment at a time when tumors need angiogenesis for their transition to malignancy. Identification of the recruitment factors for macrophages and/or the signal transduction pathways that regulate the production of VEGF‐A might therefore constitute novel and attractive therapeutic targets.
4. Experimental procedures
4.1. Mice
All procedures involving mice were conducted in accordance with National Institutes of Health (NIH) regulations concerning the use and care of experimental animals. The study of mice was approved by the Albert Einstein College of Medicine animal use committee. The PyMT transgenic mice were kindly provided by Dr. W.J. Muller (McGill University, Canada). The origin, care and identification of CSF‐1 null mutant (Csf1op/Csf1op) mice have been described previously (Lin et al., 2001). Since +/Csf1op mice have normal serum concentration of CSF‐1, normal tissue population of macrophages and, are in all aspects tested equivalent to wild‐type (+/+) mice (Pollard and Stanley, 1996), these +/Csf‐1op are used as controls. The transgenic mice carrying the MMTVLTR‐rtTA transgene (Tg(MMTV‐rtTA)Lach) were kindly provided by Dr. Lewis Chodosh (University of Pennsylvania). The mice used were the first generation of FVB back‐crossed to C3H/B6/FVB. Transgenic mice were generated using standard techniques. The silencer plasmid, pTet‐tTS (BD Biosciences/Clontech) was co‐injected with the VEGF‐A transgenic construct during the preparation transgenic mice. VEGF‐A cDNA (Mus musculus vascular endothelial growth factor A, clone #9895968) was obtained from American Type Culture Collection (ATCC, Manassas, VA). Plasmid carrying FMDV 2A sequence (pSTA/31+39AA 1D) was kindly provided by Dr. Martin D. Ryan (University of St Andrews, UK). The insert carrying the 2A sequence was obtained by restriction digestion using XbaI and ApaI and then ligated in‐frame at the 3′ end of VEGF‐A cDNA and 5′ end of GFP. The transgenic mice were generated using standard method in the Transgenic Facility at Albert Einstein College of Medicine. One founder with co‐integrated transgenes was identified (Tg(Vegfa‐2A‐Gfp)1Jwp) and the expression of the transgene was characterized using standard methods. A PCR based method for genotyping the mice carrying the transgene was established with standard PCR procedure and the following primers: F1, AAGTTCATCTGCACCACCG; F2, TGCTCAGGTAGTGGTTGTCG.
4.2. Induction of transgene expression
Doxycycline (Dox; Clontech) was added to drinking water at 1mg/ml. The Dox solution was replaced every three days.
4.3. Histological analysis
The in vivo vessel labeling method with Texas‐Red conjugated dextran has been described previously (Lin et al., 2006). Briefly, Texas‐Red conjugated dextran (Molecular Probes, Eugene, OR) was i.v. injected into a mouse 5min before tumors were dissected. The tumor tissue was fixed with 10% Formalin (in PBS) at 4°C overnight and then paraffin‐embedded and sectioned. To visualize the location of the staining, tissue sections were counter‐stained with DAPI. The sections were photographed to TIFF images using an Olympus IX70 microscope and a Sesicam QEcooled CCD camera. IHC of macrophage with rat anti‐mouse F4/80 antibody was described previously (Lin et al., 2001). A standard procedure was used for IHC of anti‐vWF antibody (DAKO, Denmark), anti‐Ki67 antibody (LAB VISION Corp., CA) and anti‐DSF antibody (eBioscience, CA). The procedure used to preserved GFP in tissue was described previously (Lin et al., 2006).
To label the phagocytic macrophages with Lysine fixable Texas‐Red conjugated dextran, the reagent was i.v. injected into a mouse 2h before various tissues were dissected. To preserve GFP, tissues were fixed with 5% Formalin in 20% sucrose/PBS solution at 4°C overnight and then embedded for frozen section. To visualize the location of the staining, tissue sections were counter‐stained with DAPI. The sections were photographed to TIFF images using an Olympus IX70 microscope and a Sesicam QEcooled CCD camera.
4.4. Western blotting analysis
Western blotting analysis was performed following a standard procedure. For VEGF‐A protein, rabbit anti‐mouse VEGF‐A, 0.4μg/ml (Serotec Cat. #AAM51, Oxford, UK) and for GFP, anti‐GFP, 0.4μg/ml (Roche Cat. #11 814 460 001), were used. For internal control of protein loading, anti‐Tubulin antibody (Santa Cruz, CA) was used.
4.5. FACS analysis
Mammary tumors were harvested from VEGF‐A transgenic and control mice at 6–14weeks of age. Tissues were minced in 1ml of MEM medium before adding Liberase at 0.028Wunsch units/ml (Sigma) and DNAse I at 20μg/ml (Sigma). The mixture was incubated 0.5h at 37°C under gentle agitation. Cells were separated on a discontinuous Percoll gradient (70/50/40/30%) and the leukocytes were harvested at the 70/50% and 50/40% Percoll interfaces. Cells were stained with antibodies for FACS following standard procedures. Antibodies used for FACS are as follows: Alexa 488 conjugated rat anti‐mouse F4/80 (CALTAG Lab, CA), PE‐Cy7 conjugated rat anti‐mouse CD11b (Becton Dickinson, CA), PE conjugated rat monoclonal anti‐mouse CXCR4 (R&D Systems Inc., MN) and APC conjugated rat‐anti‐mouse VEGF R1 (R&D Systems).
Acknowledgments
We would like to thank Dr. Chris Contag, Stanford University, for his advice on the use of the virus 2A sequence in transgenic mice; Dr M.D. Ryan, University of Glasgow, for the plasmid containing the 2A sequence; Dr. Lewis Chodosh, University of Pennsylvania, for generously supplying the MMTV‐rtTA mice; and Dr. Ken Chen and Binzhi Qian for the useful discussion. We also thank Jim Lee and Mark Thompson for excellent technical support and the technical staff of the Transgenic and Gene Targeting Facility, Analytical Imaging Facility and Histotechnology and Comparative Pathology Facility at Albert Einstein College of Medicine for advice. This work was supported by NCI grants CA RO1 94173, CA PO1 100324 and the Albert Einstein Cancer Center Core grant P30 CA 13330. J.W.P. is the Betty and Sheldon Feinberg senior faculty scholar in cancer research. E.Y.L. is a Miriam Mandel Scholar for 2006–2007.
Lin Elaine Y., Li Jiu-feng, Bricard Gabriel, Wang Weigang, Deng Yan, Sellers Rani, Porcelli Steven A., Pollard Jeffrey W., (2007), Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages, Molecular Oncology, 1, doi: 10.1016/j.molonc.2007.10.003.
References
- Barleon, B. , Sozzani, S. , Zhou, D. , Weich, H.A. , Mantovani, A. , Marme, D. , 1996. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 87, 3336–3343. [PubMed] [Google Scholar]
- Cho, C.H. , Koh, Y.J. , Han, J. , Sung, H.K. , Jong Lee, H. , Morisada, T. , Schwendener, R.A. , Brekken, R.A. , Kang, G. , Oike, Y. , Choi, T.S. , Suda, T. , Yoo, O.J. , Koh, G.Y. , 2007. Angiogenic role of LYVE-1-positive macrophages in adipose tissue. Circ. Res.. 100, e47–e57. [DOI] [PubMed] [Google Scholar]
- Dong, J. , Grunstein, J. , Tejada, M. , Peale, F. , Frantz, G. , Liang, W.C. , Bai, W. , Yu, L. , Kowalski, J. , Liang, X. , Fuh, G. , Gerber, H.P. , Ferrara, N. , 2004. VEGF-null cells require PDGFR alpha signaling-mediated stromal fibroblast recruitment for tumorigenesis. EMBO J. 23, 2800–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly, M.L. , Hughes, L.E. , Luke, G. , Mendoza, H. , ten Dam, E. , Gani, D. , Ryan, M.D. , 2001. The ‘cleavage’ activities of foot-and-mouth disease virus 2A site-directed mutants and naturally occurring ‘2A-like’ sequences. J. Gen. Virol.. 82, 1027–1041. [DOI] [PubMed] [Google Scholar]
- Dvorak, H.F. , 2002. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J. Clin. Oncol.. 20, 4368–4380. [DOI] [PubMed] [Google Scholar]
- Eubank, T.D. , Galloway, M. , Montague, C.M. , Waldman, W.J. , Marsh, C.B. , 2003. M-CSF induces vascular endothelial growth factor production and angiogenic activity from human monocytes. J. Immunol.. 171, 2637–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Felipe, P. , 2004. Skipping the co-expression problem: the new 2A “CHYSEL” technology. Genet. Vaccines Ther.. 2, 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara, N. , 2004. Vascular endothelial growth factor: basic science and clinical progress. Endocr. Rev.. 25, 581–611. [DOI] [PubMed] [Google Scholar]
- Ferrara, N. , Gerber, H.P. , LeCouter, J. , 2003. The biology of VEGF and its receptors. Nat. Med.. 9, 669–676. [DOI] [PubMed] [Google Scholar]
- Folkman, J. , 2002. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol.. 29, 15–18. [DOI] [PubMed] [Google Scholar]
- Giraudo, E. , Inoue, M. , Hanahan, D. , 2004. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Invest.. 114, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouon-Evans, V. , Rothenberg, M.E. , Pollard, J.W. , 2000. Postnatal mammary gland development requires macrophages and eosinophils. Development. 127, 2269–2282. [DOI] [PubMed] [Google Scholar]
- Grunewald, M. , Avraham, I. , Dor, Y. , Bachar-Lustig, E. , Itin, A. , Jung, S. , Chimenti, S. , Landsman, L. , Abramovitch, R. , Keshet, E. , 2006. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell.. 124, 175–189. [DOI] [PubMed] [Google Scholar]
- Gunther, E.J. , Belka, G.K. , Wertheim, G.B.W. , Wang, J. , Hartman, J.L. , Boxer, R.B. , Chodosh, L.A. , 2002. A novel doxycycline-inducible system for the transgenic analysis of mammary gland biology. FASEB J.. 16, 283–292. [DOI] [PubMed] [Google Scholar]
- Guy, C.T. , Cardiff, R.D. , Muller, W.J. , 1992. Induction of mammary tumors by expression of polyomovirus middle T oncogenes: a transgenic mouse mode of a metastatic disease. Mol. Cell. Biol.. 12, 954–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan, D. , Folkman, J. , 1996. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 86, 353–364. [DOI] [PubMed] [Google Scholar]
- Jain, R.K. , Duda, D.G. , Clark, J.W. , Loeffler, J.S. , 2006. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat. Clin. Pract. Oncol.. 3, 24–40. [DOI] [PubMed] [Google Scholar]
- Keyt, B.A. , Berleau, L.T. , Nguyen, H.V. , Chen, H. , Heinsohn, H. , Vandlen, R. , Ferrara, N. , 1996. The carboxyl-terminal domain (111–165) of vascular endothelial growth factor is critical for its mitogenic potency. J. Biol. Chem.. 271, 7788–7795. [DOI] [PubMed] [Google Scholar]
- Kumaraswamy, E. , Carlson, B.A. , Morgan, F. , Miyoshi, K. , Robinson, G.W. , Su, D. , Wang, S. , Southon, E. , Tessarollo, L. , Lee, B.J. , Gladyshev, V.N. , Hennighausen, L. , Hatfield, D.L. , 2003. Selective removal of the selenocysteine tRNA[Ser}Sec gene (Trsp) in mouse mammary epithelium. Mol. Cell. Biol.. 23, 1477–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek, R.D. , Harris, A.L. , 2002. Tumor-associated macrophages in breast cancer. J. Mamm. Gland Biol. Neoplasia. 7, 177–189. [DOI] [PubMed] [Google Scholar]
- Leek, R.D. , Lewis, C.E. , Whitehouse, R. , Greenall, M. , Clarke, J. , Harris, A.L. , 1996. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res.. 56, 4625–4629. [PubMed] [Google Scholar]
- Leek, R.D. , Hunt, N.C. , Landers, R.J. , Lewis, C.E. , Royds, J.A. , Harris, A.L. , 2000. Macrophage infiltration is associated with VEGF and EGFR expression in breast cancer. J. Pathol.. 190, 430–436. [DOI] [PubMed] [Google Scholar]
- Leung, D.W. , Cachianes, G. , Kuang, W.J. , Goeddel, D.V. , Ferrara, N. , 1989. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 246, 1306–1309. [DOI] [PubMed] [Google Scholar]
- Lewis, C.E. , Pollard, J.W. , 2006. Distinct role of macrophages in different tumor microenvironments. Cancer Res.. 66, 605–612. [DOI] [PubMed] [Google Scholar]
- Lin, E.Y. , Pollard, J.W. , 2007. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res.. 67, 5064–5066. [DOI] [PubMed] [Google Scholar]
- Lin, E.Y. , Nguyen, A.V. , Russell, R.G. , Pollard, J.W. , 2001. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med.. 193, 727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, E.Y. , Gouon-Evans, V. , Nguyen, A.V. , Pollard, J.W. , 2002. The macrophage growth factor, CSF-1, in mammary gland development and tumor progression. J. Mamm. Gland Biol. Neoplasia. 7, 147–162. [DOI] [PubMed] [Google Scholar]
- Lin, E.Y. , Li, J.F. , Gnatovskiy, L. , Deng, Y. , Zhu, L. , Grzesik, D.A. , Qian, H. , Xue, X.N. , Pollard, J.W. , 2006. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res.. 66, 11238–11246. [DOI] [PubMed] [Google Scholar]
- Lobov, I.B. , Rao, S. , Carroll, T.J. , Vallance, J.E. , Ito, M. , Ondr, J.K. , Kurup, S. , Glass, D.A. , Patel, M.S. , Shu, W. , Morrisey, E.E. , McMahon, A.P. , Karsenty, G. , Lang, R.A. , 2005. WNT7b mediates macrophage-induced programmed cell death in patterning of the vasculature. Nature. 437, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok, E. , Golovkina, T.V. , Ross, S.R. , 1992. A mouse mammary tumor virus mammary gland enhancer confers tissue-specific but not lactation-dependent expression in transgenic mice. J. Virol.. 66, 7529–7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumov, G.N. , Akslen, L.A. , Folkman, J. , 2006. Role of angiogenesis in human tumor dormancy: animal models of the angiogenic switch. Cell Cycle. 5, 1779–1787. [DOI] [PubMed] [Google Scholar]
- Pollard, J.W. , Stanley, E.R. , 1996. Pleiotropic roles for CSF-1 in development defined by the mouse mutation osteopetrotic (op). Adv. Dev. Biochem.. 4, 153–193. [Google Scholar]
- Ross, S.R. , Hsu, C.-L.L. , Choi, Y. , Mok, E. , Dudley, J.P. , 1990. Negative regulation in correct tissue-specific expression of mouse mammary tumor virus in transgenic mice. Mol. Cell. Biol.. 10, 5822–5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasmono, R.T. , Oceandy, D. , Pollard, J.W. , Tong, W. , Pavli, P. , Wainwright, B.J. , Ostrowski, M.C. , Himes, S.R. , Hume, D.A. , 2003. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood. 101, 1155–1163. [DOI] [PubMed] [Google Scholar]
- Schoeffner, D.J. , Matheny, S.L. , Akahane, T. , Factor, V. , Berry, A. , Merlino, G. , Thorgeirsson, U.P. , 2005. VEGF contributes to mammary tumor growth in transgenic mice through paracrine and autocrine mechanisms. Lab. Invest.. 85, 608–623. [DOI] [PubMed] [Google Scholar]
- Scholl, S.M. , Pallud, C. , Beuvon, F. , Hacene, K. , Stanley, E.R. , Rohrschneider, L.R. , Tang, R. , Pouillart, P. , Lidereau, R. , 1994. Anti-colony-stimulating factor-1 antibody staining in primary breast adenocarcinomas correlates with marked inflammatory cell infiltrates and prognosis. J. Natl. Cancer Inst.. 86, 120–126. [DOI] [PubMed] [Google Scholar]
- Shima, D.T. , Kuroki, M. , Deutsch, U. , Ng, Y.S. , Adamis, A.P. , D'Amore, P.A. , 1996. The mouse gene for vascular endothelial growth factor. Genomic structure, definition of the transcriptional unit, and characterization of transcriptional and post-transcriptional regulatory sequences. J. Biol. Chem.. 271, 3877–3883. [DOI] [PubMed] [Google Scholar]
- Sunderkotter, C. , Steinbrink, K. , GoebelerBhardwaj, R. , Sorg, C. , 1994. Macrophages and angiogenesis. J. Leukocyte Biol.. 55, 410–422. [DOI] [PubMed] [Google Scholar]
- Varney, M.L. , Olsen, K.J. , Mosley, R.L. , Singh, R.K. , 2005. Paracrine regulation of vascular endothelial growth factor–a expression during macrophage-melanoma cell interaction: role of monocyte chemotactic protein-1 and macrophage colony-stimulating factor. J. Interferon Cytokine Res.. 25, 674–683. [DOI] [PubMed] [Google Scholar]
- Yoshida, A. , Anand-Apte, B. , Zetter, B.R. , 1996. Differential endothelial migration and proliferation to basic fibroblast growth factor and vascular endothelial growth factor. Growth Factors. 13, 57–64. [DOI] [PubMed] [Google Scholar]