

Abstract
The premature-ageing disease Hutchinson-Gilford Progeria Syndrome (HGPS) is caused by constitutive production of progerin, a mutant form of the nuclear architectural protein lamin A1,2. Progerin is also expressed sporadically in wild-type cells and has been linked to physiological ageing3. Cells from HGPS patients exhibit extensive nuclear defects, including abnormal chromatin structure4,5 and increased DNA damage6. At the organismal level, HGPS affects several tissues, particularly those of mesenchymal origin7. How the cellular defects of HGPS cells lead to the organismal defects has been unclear. Here, we provide evidence that progerin interferes with the function of human mesenchymal stem cells (hMSCs). We find that expression of progerin activates major downstream effectors of the Notch signalling pathway. Induction of progerin in hMSCs changes their molecular identity and differentiation potential. Our results support a model in which accelerated ageing in HGPS patients, and possibly also physiological ageing, is the result of adult stem cell dysfunction and progressive deterioration of tissue functions.
The causal agent in HGPS is the progerin protein, an aberrant form of the nuclear architectural protein lamin A1,2,8. Progerin is generated by a heterozygous C1824T mutation in the LMNA gene, which activates a cryptic splice site and gives rise to an incompletely processed form of lamin A1,2,9. Progerin acts in a dominant gain-of-function fashion by accumulating at the nuclear periphery and altering the nuclear lamina structure4,6,10,11. Low levels of progerin are also produced in healthy individuals by sporadic use of the cryptic splice site, and the presence of progerin in healthy cells has been linked to normal ageing3. To analyse the mechanisms by which progerin exerts its pathological effect, we developed a human telomerase reverse transcriptase (hTERT)-immortalized skin-fibroblast cell line that expressed inducible green fluorescent protein (GFP)–progerin9 (Supplementary Information, Fig. S1). A significant accumulation of GFP–progerin was first observed 5 d after induction and reached a plateau at 10 d (Supplementary Information, Fig. S1a, c). Expression of GFP–progerin at levels similar to those found in cells from HGPS patients produced several of the typical HGPS defects, including aberrant nuclear shape, loss of the lamin-associated polypeptide 2 (LAP2) proteins and increased DNA damage (Supplementary Information, Fig. S1b, c). As a control, expression of GFP-tagged wild-type lamin A (GFP–wt-lamin A) had no effect on these cellular parameters in the majority of the cells, although mild effects on nuclear shape and LAP2 localization were observed in cells that expressed high levels of exogenous protein, suggesting that excessive accumulation of lamin A at the nuclear periphery induces mild HGPS-like defects (Supplementary Information, Fig. S1b, c).
To begin to unravel how progerin causes disease phenotypes, we analysed time-dependent changes in transcriptional profiles in response to progerin expression. Genome-wide microarray analysis (GEO accession number: GSE10123) revealed changes in 194 genes 5 d after induction and 1013 genes after 10 d (Fig. 1a). Misregulation ranged from −14-fold to +17.3-fold (Fig. 1a). About 23% of the genes that responded to progerin were also affected by overexpression of wild-type lamin A, confirming that accumulation of excessive wild-type lamin A partially recapitulates the effects of progerin (Fig. 1b). However, the effect of wild-type lamin A on gene expression was milder than that of progerin. Only 570 genes were affected at 10 d and changes in their expression levels only ranged from −6.5-fold to +10.1-fold (Fig. 1a, b).
Figure 1.

Activation of Notch signalling pathway effectors in response to progerin. (a) Time-dependent changes in gene expression in skin fibroblasts expressing progerin and wild-type lamin A. Normalized intensity of differentially expressed genes (≥ 2-fold) in either cell line (1394 genes) is plotted as a function of time relative to each uninduced state. Values of the two replicates are shown for each sample. The colour of each gene corresponds to its colour in progerin-expressing cells at 10 d. Red–orange are upregulated genes, blue–grey are downregulated genes. (b) Heatmap representing relative expression levels of genes showing a difference of at least 2-fold on induction in either cell line. Colours represent values normalized to the range of intensity for each gene in all conditions. Genes progressively upregulated, progressively downregulated or transiently up- or downregulated in progerin-expressing cells and the corresponding changes in wild-type lamin A-expressing cells are shown. (c–e) Time-dependent changes in expression levels of HES1 (c), HES5 (d) and TLE1 (e) in cells expressing either progerin or wild-type lamin A, as detected by microarrays. Values represent mean ± s. d. from two biological replicates. (f) Time-dependent changes in expression levels of HES1, HES5 and HEY1 during induction in progerin-expressing cells detected by quantitative RT-PCR. Values are normalized to the housekeeping gene cyclophillin A. Asterisks indicate statistically significant differences compared with the uninduced state (P < 0.001). Values represent mean ± s. d. from two biological replicates. (g) Immunofluorescence microscopy of progerin-expressing cells before induction and 4 d after doxycycline removal using an anti-HES1 antibody. The presence of HES1 correlates with expression of GFP–progerin. Scale bar: 20 μm.
Pathway analysis of differentially expressed genes over time revealed that one of the prominent early events after progerin expression is the activation of several components of the Notch cell-signalling pathway, a major regulator of cell fate and stem-cell differentiation12. In particular, the major direct effector of Notch, HES1 (Hairy and enhancer of split 1; ref. 13), was the highest scoring gene among the early responsive ones, showing an increase of approximately 8-fold 5 d after induction of progerin (Fig. 1c). In addition, a second major transcriptional regulator of Notch target genes, HES5 (ref. 13), and TLE1, the transcriptional repressor through which HES proteins exert their repressive action13, were similarly upregulated in the presence of progerin (Fig. 1d, e). Activation of downstream portions of the Notch pathway was further indicated by downregulation of the known HES target genes NEUROG1, ANKRD16, NR2F2 and MYH10 (Supplementary Information, Fig. S2a). Activation of Notch effectors was not mediated by changes in expression levels of upstream components of the pathway, as no change over time was detected in genes encoding Notch receptors, DLL and JAG ligands or components of γ-secretase12 (Supplementary Information, Fig. S2c). Overexpression of wild-type lamin A induced similar changes in HES1 mRNA (Fig. 1c) but not in HES5 (Fig. 1d) and caused a decrease in TLE1 (Fig. 1e), indicating that, despite the similar effect on HES1, pathway activation is a specific response to progerin. Misexpression of these and other Notch pathway components was confirmed by quantitative RT-PCR (Fig. 1f). In addition to HES1 and HES5, the related Notch effector HEY1 (ref. 13) was also found to be upregulated in response to progerin (Fig. 1f). For all affected genes, misregulation was already observed 5 d after induction and reached 10–15-fold after 10 d (Fig. 1f). In addition, upregulation of HES1 protein in response to progerin was confirmed by indirect immunofluorescence microscopy (Fig. 1g). Taken together, these observations suggest that Notch downstream effectors are activated in response to progerin.
To confirm the specific effect of progerin on Notch effectors in a natural system, and to assess whether misregulation of Notch signalling is physiologically relevant for disease, we analysed expression of Notch components in cell lines from HGPS patients. HES1, HES5 and HEY1 were markedly upregulated in four HGPS cell lines, compared with three wild-type cell lines (Fig. 2a–c). Quantitative RT-PCR demonstrated an average upregulation (~ 20-fold) of HES1 in HGPS cells and an average increase (~ 3-fold) of HEY1 mRNA, compared with cells from healthy individuals (Fig. 2a, b). Semi-quantitative RT-PCR analysis similarly indicated significant differences in HES5 mRNA levels, which were undetectable in wild-type cells and markedly upregulated in all HGPS cell lines (Fig. 2c). Upregulation of HES1 in HGPS cells was also confirmed by indirect immunofluorescence microscopy (Fig. 2d): wild-type cells showed only a very weak signal, whereas more than 60% of HGPS cells contained high levels of HES1. As is characteristic of many cellular markers in HGPS cells, the level of HES1 varied among individual cells within the population. However, the strongest upregulation was evident in cells in which progerin-induced defects, such as downregulation of HP1γ4, were prominent (arrowheads in Fig. 2d). In further support, a general Notch pathway reporter14 and a HES1 reporter15 were activated in HGPS cells with progerin-induced nuclear defects (Supplementary Information, Fig. S3). Consistent with the microarray analysis, downregulation of the HEY1 target gene ANKRD16 in several HGPS cells confirmed activation of the pathway (Supplementary Information, Fig. S2b).
Figure 2.
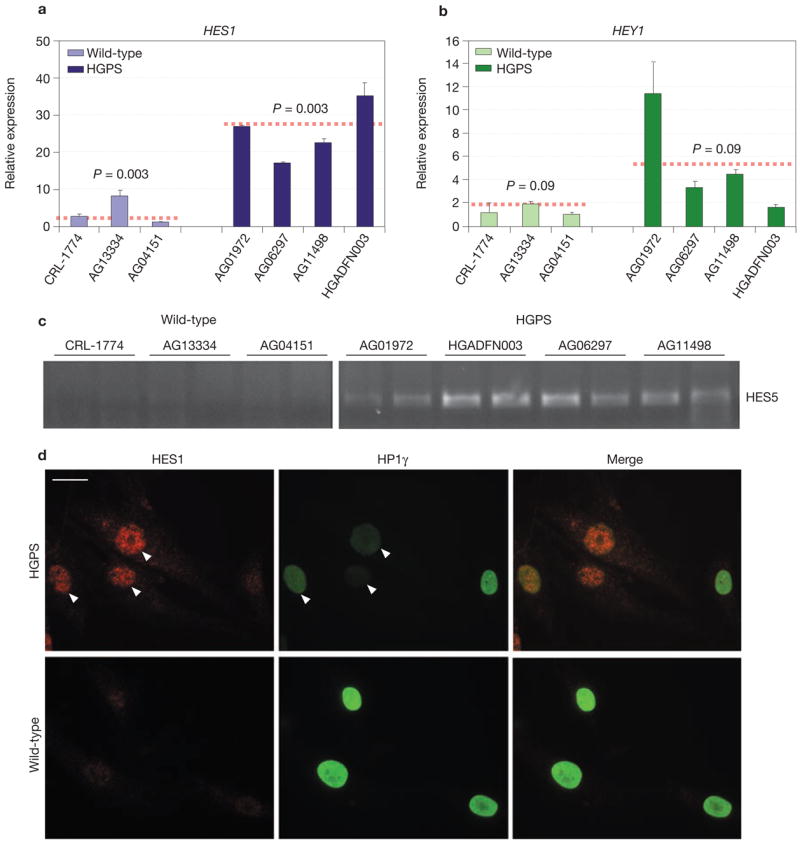
Activation of Notch signalling pathway effectors in cells from HGPS patients. (a, b) Quantitative RT-PCR analysis of HES1 and HEY1 expression levels in four different HGPS cells lines and three wild-type control cell lines. Values are normalized to the housekeeping gene cyclophillin A. Statistical significance of the differences between the two groups of cell lines is shown. Values represent mean ± s. d from at least two experiments. (c) Semi-quantitative RT-PCR analysis of HES5 expression levels in four different HGPS cells lines and three wild-type control cell lines. Each sample was analysed in duplicate and products were detected with Syber Green. (d) Immunofluorescence microscopy of HGPS cells using anti-HES1 and anti-HP1γ antibodies. The presence of HES1 correlates with progerin-induced reduction of HP1γ (arrows). Scale bar: 15 μm.
Despite the marked upregulation of HES1, HES5 and HEY1 in HGPS cells, and in accordance with the microarray analysis, we were unable to detect increased levels of the cleaved NICD (Notch intracellular cleaved domain)12, suggesting that progerin may function by activating Notch effectors downstream of NICD (Supplementary Information, Fig. S2d). To test this possibility, we probed the status of major transcriptional regulators of Notch target genes in HGPS cells. Although protein levels of the DNA-binding, NICD-interacting protein CBF1 (ref. 16) were normal in all HGPS cells, those with progerin-induced nuclear defects showed reduced levels of the transcriptional co-repressor NcoR16 and increased levels of SKIP, a nuclear matrix-associated transcriptional co-activator of Notch target genes16,17 (Supplementary Information, Fig. S4a). Overexpression of SKIP in wild-type cells resulted in activation of a Notch reporter and an increase in the level of endogenous HES1 protein, indicating that enhanced SKIP activity is sufficient to induce expression of Notch effectors (Supplementary Information, Fig. S4b). Interestingly, in situ preparation of nuclear matrix revealed loss of SKIP from the nuclear lamina in HGPS cells (Supplementary Information, Fig. S4c). Taken together, these results suggest that the presence of progerin might affect the normal sequestration of the SKIP co-activator to the nuclear periphery and promote activation of Notch effectors, at least partially, by increasing the pool of SKIP molecules free to bind to their promoters.
One of the major physiological roles of the Notch pathway is regulation of stem-cell differentiation12. As many of the tissues affected in HGPS patient are of mesenchymal origin7, we tested the hypothesis that progerin interferes with mesenchymal stem cell function. To address this issue, we introduced progerin into immortalized hMSCs18 (Supplementary Information, Fig. S5a). As observed in fibroblasts, constitutive expression of progerin induced upregulation of HES1 and HEY1 in hMSCs (Supplementary Information, Fig. S5b). Consistent with alterations in cell-fate regulators, we found that subsets of progerin-positive cells aberrantly expressed general and tissue-specific differentiation markers (Fig. 3a–e, g–j). Approximately 10% of hMSCs expressing progerin were positive for the general differentiation marker SSEA1 (stage-specific embryonic antigen)19, indicating sporadic, spontaneous differentiation in response to progerin expression (Fig. 3a, j). When tested for more tissue-specific markers, similar fractions of the cell population were found to be positive for collagen IV, the major component of epithelium and endothelium basement membrane (Fig. 3b, j), or the neuroprogenitor marker nestin (Fig. 3c, j). Furthermore, the endothelial marker MCAM (Fig. 3d, j) was frequently found in progerin-expressing hMSCs. Quantitative RT-PCR analysis indicated an upregulation (~4-fold) of endothelin (ET-1, an angiogenic factor normally secreted by endothelial cells) and a small but significant upregulation of VEGFR1 (Fig. 3e). Consistent with the presence of endothelial markers and production of a paracrine angiogenic molecule, undifferentiated hMSCs were able to form capillaries when plated on basement-membrane extract, whereas untransduced cells failed to do so (Fig. 3f). In addition to the spurious expression of markers not typically associated with mesenchymal lineages, progerin-expressing, undifferentiated hMSCs also contained high levels of the osteogenic marker osteopontin (Fig. 3g, h). In agreement with the notion that overexpression of wild-type lamin A may cause mild HGPS-like cellular defects, intermediate, but significantly lower (P < 0.05 and P < 0.0005), levels of these markers were detected when wild-type lamin A was overexpressed (Fig. 3j). Expression of GFP had no effect on any of these markers compared with untransduced cells (Fig. 3e, h, j). Consistent with the altered cellular identity of progerin-expressing hMSCs, several differentiation markers were also upregulated in progerin-expressing fibroblasts, as detected by microarray analysis (Supplementary Information, Fig. S6). The unexpected presence of differentiation markers in populations of undifferentiated stem cells indicates that progerin alters the molecular and cellular identity of hMSCs and suggests possible consequences for their differentiation potential and cell fate.
Figure 3.
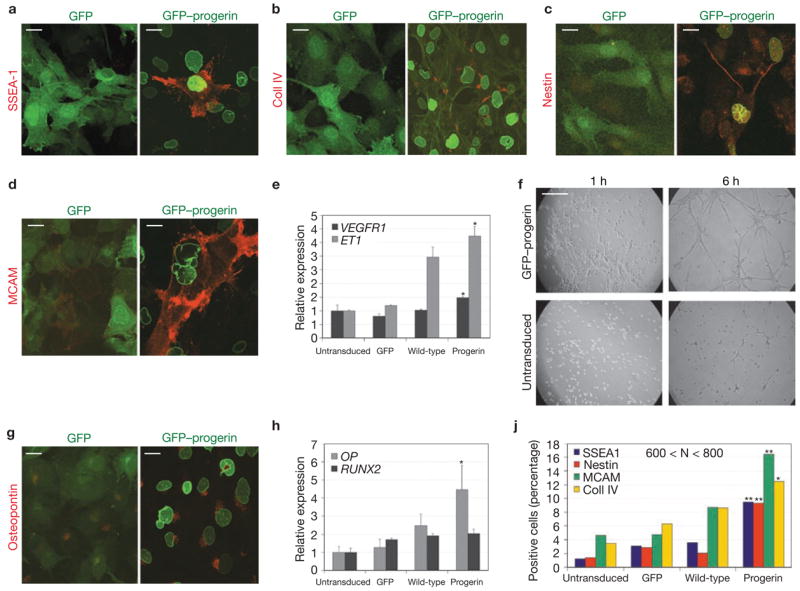
Progerin alters the molecular and cellular identity of hMSCs. (a–d, g) Immunofluorescence microscopy of undifferentiated hMSCs expressing GFP or GFP–progerin using the indicated antibodies. Merge between the GFP signal (green) and the antibody signal (red) is shown. FITC–phalloidin was used together with anti-collagen IV to show the cellular edge. Scale bar: 20 μm. (e, h) Quantitative RT-PCR analysis of expression levels of endothelial (e) and osteogenic (h) markers in untransduced hMSCs and cells expressing GFP, GFP–progerin, or GFP–wt-lamin A. Statistical significance of the differences between progerin-expressing and untransduced cells is indicated by one asterisk (P < 0.05). Values represent mean ± s. d from two experiments. (f) Angiogenic assay. Undifferentiated hMSCs expressing GFP–progerin or untransduced were plated onto basement membrane extract in the presence of 10 ng ml−1 h VEGF. Pictures were taken at the indicated times after plating. Scale bar: 1 mm. (j). Quantification of differentiation markers in undifferentiated hMSCs expressing GFP, GFP–progerin or GFP–wt-lamin A, or untransduced. Statistical significance of the differences between progerin- and wild-type lamin A-expressing cells is indicated by one (P < 0.05) or two (P < 0.0005) asterisks. 600 < N < 800 cells.
Among the major tissues affected in HGPS patients are the mesoderm-derived bone, adipose tissue and vascular epithelium7. Our initial analysis of undifferentiated cells suggested an abnormal tendency of progerin-expressing cells to differentiate spontaneously into endothelial cells. To test directly whether progerin interferes with differentiation of hMSCs along mesenchymal lineages, we assessed differentiation behaviour of hMSCs stably expressing progerin towards fat, bone and cartilage (Fig. 4). We found distinct effects of progerin on the differentiation potential of hMSCs, depending on lineage (Fig. 4). On induction of osteogenesis, the presence of progerin enhanced differentiation, as judged by detection of mineralized matrix (Fig. 4a) and by increased calcium deposition, compared with the controls (Fig. 4b). Expression of wild-type lamin A had an intermediate, but significantly milder, effect (P = 0.0008; Fig. 4b). In addition, upregulation of osteopontin was observed throughout differentiation in progerin-expressing cells, compared with the controls (Fig. 4c). Abnormalities in osteogenesis are consistent with the phenotype of HGPS patients, who have high bone-turnover and severe skeletal dysplasia7.
Figure 4.
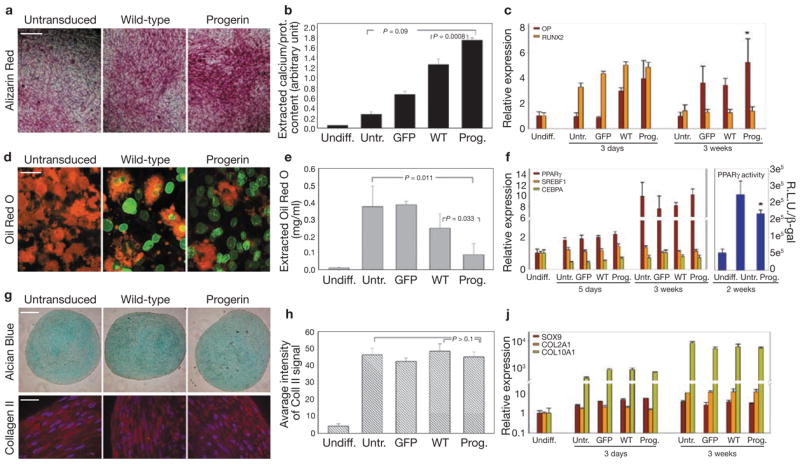
Altered differentiation potential of progerin-expressing hMSCs. (a) Detection of mineralized matrix by Alizarin red staining in hMSCs expressing GFP–progerin or GFP–wt-lamin A or untransduced after 18 d of osteogenic differentiation. Scale bar: 150 μm. (b) Quantitative analysis of calcium deposition by hMSCs expressing GFP, GFP–progerin or GFP–wt-lamin A or untransduced after 18 d of osteogenic differentiation. Values represent mean ± s. d. from three biological replicates. Statistical significance of the differences between progerin-expressing cells and untransduced or wild-type lamin A-expressing cells is indicated. (c, f, j) Quantitative RT-PCR analysis of expression levels of osteogenic (c), adipogenic (f, left panel) and chondrogenic (j) markers in hMSCs expressing GFP, GFP–progerin, or GFP–wt-lamin A or untransduced after 3–5 d and 21 d of differentiation. Values represent mean ± s. d. from three biological replicates. The right panel in f shows luciferase assays measuring PPARγ activity. Values represents mean ± s. d. from three biological replicates. Statistical significance of the differences between progerin-expressing cells and untransduced cells is indicated by the asterisk (P < 0.05). (d) Detection of lipid droplets by Oil red O staining in hMSCs expressing GFP–progerin or GFP–wt-lamin A or untransduced after 19 d of adipogenic differentiation. Merge between the GFP signal (green) and the Oil red O signal (red) is shown. Oil red O was observed using a Texas Red filter. Scale bar: 40 μm. (e) Quantitative analysis of incorporated Oil red O after elution with isopropanol. Values represent mean ± s. d. from three biological replicates. Statistical significance of the differences between progerin-expressing cells and untransduced or wild-type lamin A-expressing cells is indicated. (g) Detection of glycosaminoglycans in cartilage pellets by Alcian Blue staining, and collagen II by indirect immunofluorescence microscopy. Merge between collagen II staining (red) and DAPI staining (blue) is shown. Scale bar: 500 μm. (h) Quantitative analysis of collagen II fluorescent signal in cartilage pellets derived from hMSCs expressing GFP, GFP–progerin or GFP–wt-lamin A or untransduced after 23 d of chondrogenesis. Values represent mean ± s. d. from three different sections. Statistical significance of the differences between progerin- expressing cells and untransduced or wild-type lamin A-expressing cells is indicated. Scale bar: 40 μm.
One of the major hallmarks of HGPS patients is loss of subcutaneous adipose tissue7. In agreement with a direct role of progerin in this process, differentiation of hMSCs along the adipogenic lineage was markedly reduced in hMSCs expressing progerin, as measured by quantitative Oil red O staining (Fig. 4d, e). Analysis of single cells showed that only those expressing very low levels of GFP–progerin accumulated lipid droplets in their cytoplasm. Although no specific change was observed in the expression levels of the three major adipogenic regulators PPARγ, cEBPα and SERBPF, reporter assays showed a significant reduction in PPAR activity in progerin-expressing adipocytes (Fig. 4f). Expression of wild-type lamin A again had an intermediate effect on the cell population (Fig. 4e), but at the single-cell level did not preclude lipid accumulation (Fig. 4d). The effects of progerin on hMSC lineage differentiation was specific and differentiation along the chondrocytic lineage was not affected, as judged by detection of glycosaminoglycans by Alcian Blue, quantitative staining of Collagen II, and quantitative RT-PCR on early (SOX9) and late (COLL2A1 and COLL10A1) cartilage markers (Fig. 4g–j). We conclude that progerin interferes with differentiation of hMSCs in a lineage-specific manner.
To formally demonstrate the involvement of the Notch pathway in the alterations in stem-cell differentiation induced by progerin, we transduced hMSCs with a constitutively active form of Notch (NICD; Supplementary Information, Fig. S5e, f). Similarly to progerin-expressing cells, NICD-expressing hMSCs showed abnormalities in both the undifferentiated and differentiated states (Fig. 5). Undifferentiated NICD-expressing hMSCs cells already expressed high levels of the osteogenic markers osteopontin and alkaline phosphatase, as assessed by fluorescence microscopy and spectrophotometric analysis (Fig. 5a, b). Consistent with a higher osteogenic potential, and mimicking the behaviour of progerin-expressing hMSCs, NICD-expressing hMSCs underwent enhanced osteogenic differentiation and exhibited increased alkaline phosphatase activity and calcium deposition (Fig. 5c, d). In contrast, akin to progerin-expressing hMSCs, expression of NICD led to almost complete inhibition of adipogenesis (Fig. 5e, f). We conclude that activation of the Notch pathway by overexpression of NICD reproduces the lineage-specific effects of progerin on stem-cell differentiation. As further evidence that progerin affects hMSCs lineage-specification by activating the Notch pathway, treatment of progerin-expressing hMSCs with the Notch pathway inhibitor DAPT20 partially blocked the lineage-specific effects of progerin on hMSC differentiation (Supplementary Information, Fig. S5c, d).
Figure 5.
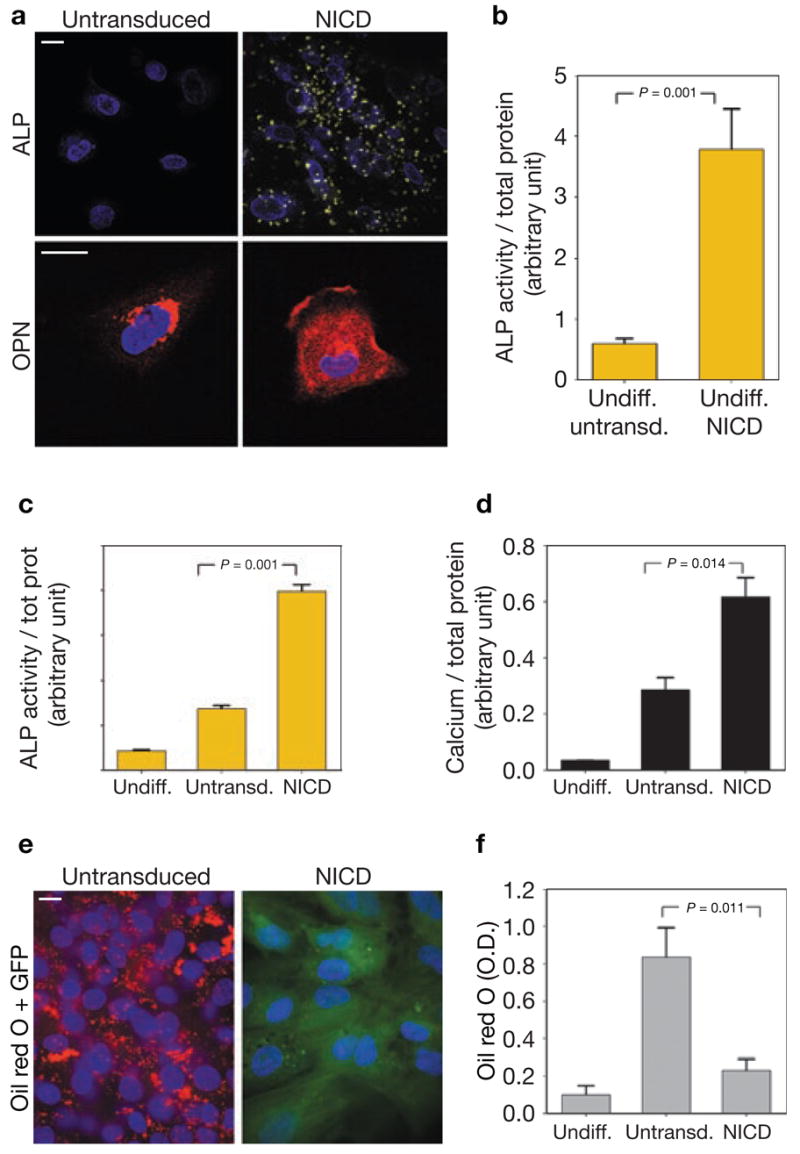
Altered differentiation potential of NICD-expressing hMSCs. (a) Immunofluorescence microscopy of undifferentiated hMSCs untransduced or expressing NICD using a fluorescent substrate of alkaline phosphatase (ALP) and an anti-osteopontin (OPN) antibody. The presence of yellow crystals indicates enzyme activity in NICD-expressing hMSCs. No crystals were detected in untransduced hMSCs. High levels of extracellular osteopontin were detected in undifferentiated NICD-expressing cells but not in untransduced cells. Merge with the DAPI signal (blue) is shown. Scale bar: 10 μm. (b, c) Quantification of alkaline phosphatase activity by spectrophotometric analysis in undifferentiated hMSCs (b) and differentiated osteoblasts (c). Values represent mean ± s. d. from three biological replicates. Statistical significance of the differences is indicated. (d) Quantitative analysis of calcium deposition by hMSCs expressing NICD or untransduced cells after 20 d of osteogenic differentiation. Values represent mean ± s. d. from three biological replicates. The statistical significance of the differences is indicated. (e) Observation of lipid droplets by Oil red O staining in hMSCs expressing NICD or untransduced cells after 21 d of adipogenic differentiation. Merge between the DAPI signal (blue), the GFP signal (green) and the Oil red O signal (red) is shown. Oil red O was shown using a Texas Red filter. Scale bar: 10 μm. (f) Quantitative analysis of incorporated Oil red O after elution with isopropanol. Values represent mean ± s. d. from three biological replicates. Statistical significance of the differences is indicated.
Taken together, these results suggest a possible mechanism by which progerin causes some of the organismal disease symptoms in HGPS patients and they may also have implications for our understanding of physiological ageing3. We found that the presence of progerin caused activation of several downstream effectors in the Notch signalling pathway, a major regulator of hMSCs, which give rise to many of the tissues affected in HGPS21. The observed effects of progerin on hMSC identity, and its interference with differentiation pathways, supports the view that at least some of the organismal defects in HGPS patients are caused by aberrant differentiation of hMSCs and defective tissue homeostasis. In support of this, we have shown that several Notch effectors are misregulated in cell lines from HGPS patients. Furthermore, the observed aberrations of osteogenesis and adipogenesis are consistent with the defects seen in patients7, and endothelial dysfunction caused by Notch pathway misregulation22 may contribute to the development of vascular abnormalities characteristic of the disease.
Our results suggest that progerin functions downstream in the Notch signalling pathway by affecting the expression of the effector proteins without activation of the upstream components of the pathway. However, the presence of undetectable levels of biologically active NICD in HGPS cells cannot be ruled out23. An attractive possibility is that common transcriptional regulators of the downstream effectors are sequestered by the nuclear lamina. This interpretation is consistent with our finding that association of the transcriptional co-activator SKIP with the nuclear lamina is disrupted in the presence of progerin, thus increasing its availability in the nuclear interior. Precedents for transcriptional regulation by lamina-sequestration exist for several major transcriptional regulators, including pRB, c-Fos, SMADs and the sterol regulator SREBPF1 (ref. 24). Alternative, but not mutually exclusive, possibilities are that the Notch effector genes are more directly regulated by their physical association with the lamina or that their misregulation is caused by changes in epigenetic modifications frequently observed in cell lines from HGPS patients and during ageing3,4.
Our observations may also be relevant for normal ageing as progerin is present at low levels in cells from healthy individuals and has been implicated in the normal ageing process3. It can be envisioned that progerin-induced defects in Notch signalling during normal ageing similarly affect hMSCs and their differentiation, possibly contributing to the reduced potential for tissue homeostasis commonly associated with the ageing process.
METHODS
Plasmids and cell lines
hTERT-TetOff-Pro cell lines expressing inducible GFP–wt-lamin A and GFP–progerin were generated by sequentially infecting hTERT-immortalized wild-type skin fibroblasts25 with pRevTetOff and pRevTRE–GFP–lamin A or pRevTRE–GFP–progerin constructs9. Cells were grown in minimum essential medium (MEM) containing15% Tet-free fetal bovine serum (FBS), 2 mM L-glutamine, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 1 μg ml−1 doxycycline at 37 °C in 5% CO2. Tet-off cells were selected with 500 μg ml−1 G418. Cells expressing GFP–fusion proteins were selected with 50 μg ml−1 hygromycin. To induce protein expression, the concentration of doxycycline was reduced to 0.02 ng ml−1. Subpopulations of cells expressing homogenous levels of inducible protein were isolated from two polyclonal cell lines by serial dilution. pBABE-puro–GFP–wt-lamin A and pBABE-puro–GFP–progerin expression plasmids were generated by cloning NheI–BamHI fragments from pEGFP–wt-lamin A and pEGFP–Δ50 lamin A4 into the BamHI site of pBABE-puro vector after filling of the ends. pBABE-puro–GFP was generated by deleting the lamin A coding region from pBABE-puro–wt–GFP–lamin A by digestion with SalI and filling the ends before self-ligation. The retroviral construct pMyc–NICD was kindly provided by T. Takizawa (National Cancer Institute, NIH, Bethesda, MD). The construct encodes a bicistronic RNA expressing Myc-tagged NICD and GFP as transfection markers. pMyc–NICD was generated from pEFBOS–Myc–NICD26
Immortalized hMSCs18 were transduced with pBABE-puro–GFP–wt-lamin A, pBABE-puro–GFP–progerin, pBABE-puro–GFP or pEFBOS–Myc–NICD. For viral infection, approximately 10 million Phoenix ampho cells were transfected with each plasmid by electroporation using 2-mm cuvettes (20 μg of DNA in 200 μl of cell suspension; 200 mV, 1-ms pulse, five pulses, 0.5-s interval between pulses) and then maintained in 7 ml of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS and antibiotics in 10-cm plates. Viral supernatants were collected at 24-h intervals, starting 24 h after electroporation, diluted 1:1 in fresh medium in the presence of polybrene (4 μg ml−1) and added to hMSCs seeded in six-well plates at 50% confluence. To increase infection efficiency, hMSCs were spun at 615 × g for 45 min at room temperature in a Sorvall RT 6000D centrifuge immediately after addition of the viral supernatant. After 24 h at 37 °C, the viral supernatant was replaced with a fresh aliquot. Three sequential rounds of infection were performed for each condition, after which more than 90% of the cells expressed the exogenous proteins. Cells were grown in DMEM supplemented with 10% FBS, 2 mM L-glutamine, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin, at 37 °C in 5% CO2.
Primary dermal-fibroblast cell lines from HGPS patients and wild-type donors were obtained from the National Institute of Ageing collection of the Coriell Cell Repository (Camden), from the American Type Culture Collection (ATCC) and the Progeria Research Foundation (Peabody) Cells and Tissue Bank. HGPS fibroblast cell lines were AG01972, AG06297, AG11498 and HGADFN003. Cell lines from wild-type donors were CRL-1474, AG13334 and AG04151. Cells were grown in MEM supplemented with 15% FBS, 2 mM L-glutamine, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin at 37 °C in 5% CO2.
SKIP cDNA was obtained from the IMAGE. consortium (clone 40006529) and the pFLAG–SKIP expression construct was generated by cloning a PstI–HindIII fragment into the pCMV–Tag2C vector. The PPAR reporter construct Acyl-CoA ×3-tk-Luc, and the Notch reporter constructs pHes1–d2EGFP and p12×CSL–d1EGFP were kindly provided by M. Lazar (University of Pennysylvania, Philadelphia, PA), T. Ohtsuka (Kyoto University, Kyoto, Japan) and U. Lendahl (Karolinska Institute, Stockholm, Sweden) respectively, and have been described previously14,15,27.
Induction and quantitative analysis of hMSCs differentiation
Adipogenesis, osteogenesis and chondrogenesis were induced in transduced and untransduced cells as described previously18, using respectively adipogenic, osteogenic and chondrogenic bullet kits (Lonza). All experiments were performed in triplicate for each cell line. Calcium deposition by osteoblasts was analysed after 18 d of differentiation. Cells were fixed and stained with Alizarin red S (Sigma), as described previously28, or extracted with 0.5 M HCl for 24 h at 4 °C with agitation. Calcium released in the supernatant was quantified using the Liquicolor Kit (StanBio Laboratory) and normalized to the protein content of each culture dish. Alkaline phosphatase activity in undifferentiated hMSCs and osteoblasts was observed by microscopy using the ELF phosphatase detection kit (ATCC) and measured by spectrophotometric analysis using the Sensolyte pNPP Alkaline Phosphatase Assay kit (AnaSpec). Adipocytes were stained with Oil red O (Sigma) after 19 d of differentiation. The incorporated dye was extracted with isopropanol and compared to an Oil red O standard titration curve. Luciferase assays were performed after 2 weeks of adipogenic stimulation using the Promega Luciferase kit. Luciferase activity was normalized to β-galactosidase activity to account for variation in transfection efficiency. Cartilage pellets were fixed in 10% formalin after 23 d of differentiation and embedded in paraffin. Sections were stained with Alcian blue (Sigma) pH 2.5 and with anti-collagen II antibody (Millipore). Quantitative analysis of collagen II staining was performed by measuring the intensity of the fluorescent signal with Metamorph software. The in vitro angiogenenic assay was performed essentially as described previously29, by plating undifferentiated hMSCs onto basement membrane extract (three-dimensional Culture Matrix reduced growth factor, Trevigen) in serum-free medium in the presence of 10 ng ml−1 hVEGF (Fitzgerald). Formation of tubular structures was monitored over 48 h.
To inhibit the Notch signalling pathway in progerin-expressing hMSCs, cells were treated with the γ-secretase inhibitor DAPT (Sigma), as described previously20. Cells were treated with 10 μM DAPT for 10 d and with 15 μM for the following 11 d together with the differentiation cocktails.
Microarrays and quantitative analysis of gene expression
RNA was extracted using the RNAeasy kit (Qiagen). RNA labelling, hybridization on GE Healthcare CodeLink Gene Expression Bioarrays (Human Whole Genome) and data analysis were performed by GenUs Biosystems. RNA samples were collected at 0, 5 and 10 d after doxycycline removal. Two biological replicates for each time point were analysed. Intensity values after hybridization were normalized to the median intensity of the chip and ratios between 5-d and 10-d time points to 0 d were calculated for each gene in either progerin- or wild-type lamin A-expressing cell lines. Genes showing at least two-fold differences in either cell line were selected for further analysis. Relative changes in expression levels for each gene in both conditions were calculated and shown as a heatmap. Validation of microarray data by quantitative RT-PCR was performed as described previously3.
RNA samples to analyse markers of hMSC differentiation were collected either 3–5 d or 3 weeks after induction of differentiation. To extract RNA from cartilage pellets, samples were homogenized in lysis buffer with a TissueRuptor (Qiagen) for 30 s at maximum speed. Primer sequences used to specifically amplify Notch effectors and differentiation markers are available in Supplementary Information, S7. Cyclophillin A was used as reference housekeeping gene. Each sample was analysed in duplicate in at least two different reactions.
Immunofluorescence microscopy and western blotting
Antibody staining for immunofluorescence microscopy was performed as described previously3 using anti-HES1 (Millipore and Santa Cruz), anti-HP1((Millipore), anti-LAP2 (kindly provided by K. Wilson, Johns Hopkins University School of Medicine, Baltimore, MD), anti-γ-H2AX (Millipore), anti-cd146 (MCAM; Santa Cruz, P1H12), anti-SSEA1 (Santa Cruz), anti-collagen IV (DakoCytomation), anti-nestin (kindly provided by R. McKay, NIH), anti-osteopontin (clone MPIIIB10 DSHB), anti-collagen II (Millipore), anti-CBF1 (Abcam), anti-SKIP (Abcam) and anti-NcoR (Abcam) antibodies. Staining of cartilage sections with anti-collagen II antibody was preceded by antigen retrieval (5 min at 100 °C in citric buffer and slow cooling to room temperature). Staining for surface markers was performed on non-permeabilized cells. Quantitative analysis of differentiation markers in undifferentiated hMSCs was performed by scoring positive cells among 600–800 cells. For the surface markers SSEA1 and MCAM, cells were considered positive when showing a fluorescent signal at least 2-fold higher than background staining. For nestin, cells were scored as positive when showing stained cellular processes. For collagen IV, cells were considered positive when showing intense fibrillar structures. Cells were observed on a Nikon E800 microscope. Western blot analysis was performed as described previously4 using anti-GFP (Abcam), anti-lamin A/C (Santa Cruz, N18), anti-β-actin (Sigma) and anti-cleaved Notch (Cell Signaling) antibodies.
In situ nuclear matrix preparation
In situ nuclear matrix preparation was performed as described previously30.
Statistical Analysis
Data are presented as mean ± s. d. For quantitative analysis of hMSC differentiation and quantitative RT-PCR, three biological replicates were analysed and statistical significance of the differences was estimated using the one-tailed t-test. For quantitative analysis of differentiation markers in undifferentiated hMSCs by immunofluorescence microscopy, statistical significance of the differences was estimated by contingency table analysis and χ2 test (600 < N < 800).
Accession number
The microarray data have been deposited in NCBI’s Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE10123) and are accessible through GEO Series accession number GSE10123.
Supplementary Material
Note: Supplementary Information is available on the Nature Cell Biology website.
Acknowledgments
We thank A. Magra for help with adipogenesis, J. Roix for help with microarray analysis, T. Voss for help with microscopy, M. Conboy for technical advice on Notch activation, T. Takizawa, R.G. Faragher, T. Glover, J. Toguchida, M. Olive, M. Lazar, T. Ohtsuka, U. Lendahl and A. Marcello for providing reagents. The MPIIIB10 monoclonal antibody developed by M. Solursh was obtained from the Developmental Studies Hybridoma Bank, NICHD, University of Iowa. Fluorescence imaging was performed at the NCI Fluorescence Imaging Facility. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and by the Progeria Research Foundation.
Footnotes
AUTHOR CONTRIBUTIONS
P.S. and T.M. designed the study and wrote the manuscript. P.S. performed the experiments.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Eriksson M, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Sandre-Giovannoli A, et al. Lamin A truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 3.Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human ageing. Science. 2006;312:1059–1063. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature ageing disease Hutchinson-Gilford progeria syndrome. Nature Med. 2005;11:440–445. doi: 10.1038/nm1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shumaker DK, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature ageing. Proc Natl Acad Sci USA. 2006;103:8703–8708. doi: 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu B, et al. Genomic instability in laminopathy-based premature ageing. Nature Med. 2005;11:780–785. doi: 10.1038/nm1266. [DOI] [PubMed] [Google Scholar]
- 7.Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet. 2006;140:2603–2624. doi: 10.1002/ajmg.a.31346. [DOI] [PubMed] [Google Scholar]
- 8.Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev. 2006;86:967–1008. doi: 10.1152/physrev.00047.2005. [DOI] [PubMed] [Google Scholar]
- 9.Glynn MW, Glover TW. Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum Mol Genet. 2005;14:2959–2969. doi: 10.1093/hmg/ddi326. [DOI] [PubMed] [Google Scholar]
- 10.Dahl KN, et al. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA. 2006;103:10271–10276. doi: 10.1073/pnas.0601058103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang SH, et al. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc Natl Acad Sci USA. 2005;102:10291–10296. doi: 10.1073/pnas.0504641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiba S. Notch signaling in stem cell systems. Stem Cells. 2006;24:2437–2447. doi: 10.1634/stemcells.2005-0661. [DOI] [PubMed] [Google Scholar]
- 13.Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol. 2003;194:237–255. doi: 10.1002/jcp.10208. [DOI] [PubMed] [Google Scholar]
- 14.Hansson EM, et al. Recording Notch signaling in real time. Dev Neurosci. 2006;28:118–127. doi: 10.1159/000090758. [DOI] [PubMed] [Google Scholar]
- 15.Ohtsuka T, et al. Visualization of embryonic neural stem cells using Hes promoters in transgenic mice. Mol Cell Neurosci. 2006;31:109–122. doi: 10.1016/j.mcn.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 16.Bray SJ. Notch signalling: a simple pathway becomes complex. Nature Rev Mol Cell Biol. 2006;7:678–689. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 17.Zhang C, et al. Nuclear coactivator-62 kDa/Ski-interacting protein is a nuclear matrix-associated coactivator that may couple vitamin D receptor-mediated transcription and RNA splicing. J Biol Chem. 2003;278:35325–35336. doi: 10.1074/jbc.M305191200. [DOI] [PubMed] [Google Scholar]
- 18.Okamoto T, et al. Clonal heterogeneity in differentiation potential of immortalized human mesenchymal stem cells. Biochem Biophys Res Commun. 2002;295:354–361. doi: 10.1016/s0006-291x(02)00661-7. [DOI] [PubMed] [Google Scholar]
- 19.Draper JS, Pigott C, Thomson JA, Andrews PW. Surface antigens of human embryonic stem cells: changes upon differentiation in culture. J Anat. 2002;200:249–258. doi: 10.1046/j.1469-7580.2002.00030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vujovic S, Henderson SR, Flanagan AM, Clements MO. Inhibition of -secretases alters both proliferation and differentiation of mesenchymal stem cells. Cell Prolif. 2007;40:185–195. doi: 10.1111/j.1365-2184.2007.00426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Satija NK, et al. Mesenchymal stem cells: molecular targets for tissue engineering. Stem Cells Dev. 2007;16:7–23. doi: 10.1089/scd.2006.9998. [DOI] [PubMed] [Google Scholar]
- 22.Gridley T. Notch signaling in vascular development and physiology. Development. 2007;134:2709–2718. doi: 10.1242/dev.004184. [DOI] [PubMed] [Google Scholar]
- 23.Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393:382–386. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- 24.Heessen S, Fornerod M. The inner nuclear envelope as a transcription factor resting place. EMBO Rep. 2007;8:914–9. doi: 10.1038/sj.embor.7401075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wallis CV, et al. Fibroblast clones from patients with Hutchinson-Gilford progeria can senesce despite the presence of telomerase. Exp Gerontol. 2004;39:461–467. doi: 10.1016/j.exger.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 26.Takizawa T, Ochiai W, Nakashima K, Taga T. Enhanced gene activation by Notch and BMP signaling cross-talk. Nucleic Acids Res. 2003;31:5723–5731. doi: 10.1093/nar/gkg778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. Transcriptional activation by peroxisome proliferator-activated receptor is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J Biol Chem. 1997;272:5128–5132. doi: 10.1074/jbc.272.8.5128. [DOI] [PubMed] [Google Scholar]
- 28.Gregory CA, Gunn WG, Peister A, Prockop DJ. An Alizarin red-based assay of mineralization by adherent cells in culture: comparison with cetylpyridinium chloride extraction. Anal Biochem. 2004;329:77–84. doi: 10.1016/j.ab.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 29.Salani D, et al. Endothelin-1 induces an angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Am J Pathol. 2000;157:1703–1711. doi: 10.1016/S0002-9440(10)64807-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parada LA, Elbi C, Dundr M, Misteli T. In: Essential Cell Biology. Davey J, Lord MJ, editors. Ch 2. Oxford Univ. Press; Oxford: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary Information is available on the Nature Cell Biology website.