

Abstract
The TC-83 vaccine strain of Venezuelan equine encephalitis virus (VEEV) causes encephalitis and death in C3H/HeN mice infected by intranasal instillation. Since TC-83 is exempt as a Select Agent, this mouse model was used in the evaluation of antiviral therapies. Virus titers in the brains of infected mice peaked on 4 dpi and persisted at high levels until death at 9.4 ± 0.5 dpi. Mouse brains appeared histologically normal on 2 dpi, but developed meningoencephalitis, neuropil vacuolation, and gliosis by 8 dpi. Results from a protein cytokine array showed significant elevations over time in IL-1α, IL-1β, IL-6, IL-12, MCP-1, IFNγ, TNFα, MIP-1α, and RANTES in homogenized brain samples of infected mice. Immunohistochemical staining showed a colocalization of viral antigen with neuron markers. Treatment with interferon-α B/D or ampligen significantly improved survival, brain virus titer and cytokine levels, mean day-to-death, and weight change in infected mice. The time-course of infection and disease parameters of mice infected with TC-83 VEEV were similar in many ways to disease parameters in mice infected with other VEEV strains. Thus, infection of C3H/HeN mice with TC-83 VEEV may serve as a suitable model for the evaluation of antiviral compounds for the treatment of this viral disease.
1. Background
Venezuelan equine encephalitis virus (VEEV) is an alphavirus that causes periodic outbreaks in horses and man. Infection in man can cause severe acute disease that is followed by muscle weakness (Weaver et al., 2004). Lethal infection is generally rare and is thought to occur in less than 1% of cases. Histopathology in lethal human cases is observed in the brain, spleen, lymph nodes, gastrointestinal tract, liver, and lungs, which is similar to that seen in rodent models (de la Monte et al., 1985).
Disease in mice infected with wild-type strains of VEEV is biphasic, including an early lymphoid phase that is usually self-limiting followed by a central nervous system (CNS) phase that can result in death (Charles et al., 2001; Davis et al., 1994). The lymphoid phase is accompanied by a prolonged viremia that persists until death, and virus is found in the liver, spleen, pancreas, and various lymphoid tissues including the Peyer’s patches of the small intestine and the thymus (Charles et al., 2001; Jackson et al., 1991). CNS involvement occurs as early as 18 h with detection of virus in olfactory neuroepithelium, which next moves to the olfactory bulb, and then on to other areas of the brain coinciding with clearance of the virus from the periphery (Charles et al., 1995; Davis et al., 1994). Virus is also detected in the grey matter of the spinal cord and may be associated with limb weakness and paralysis (Jackson et al., 1991). Whole brain virus in mice infected with wild-type VEEV strains reaches peak titers at approximately 5 days after peripheral virus challenge (Charles et al., 2001; Schoneboom et al., 2000).
Aside from direct infection of various tissues, immunopathogenesis appears to play a significant role in disease and mortality in VEEV infection. An increase in the induction of many cytokines, including interferon-γ (IFN-γ), interleukin (IL)-6, IL-10, IL-12, and tumor necrosis factor-α (TNF-α), is observed in brain and other tissues after infection with wild-type strains of VEEV (Griffin et al., 1997; Muehlenbein et al., 2006; Schoneboom et al., 2000). Similar cytokine induction, with upregulation of IL-1, IL-4, IL-6, IL-10, leukemia inhibitory factor (LIF), and TGF-β, was observed in the brains mice infected with Sindbis virus, a related alphavirus (Wesselingh et al., 1994). The induction of cytokines and other immunomodulatory genes likely contributes to the pathogenesis of VEEV disease, and understanding of immunopathological mechanisms could lead to strategies for therapeutic intervention (Schoneboom et al., 2000).
TC-83 is an attenuated vaccine strain of VEEV derived from the highly pathogenic Trinidad Donkey (TrD) strain, differing from the parental wild-type strain in 12 nucleotide positions (Johnson et al., 1986; Kinney et al., 1993). Two mutations, one in the 5′-noncoding region and one in the E2 envelope glycoprotein have been shown to be associated with the attenuated phenotype (Kinney et al., 1993). Further, the mutation in the 5′-noncoding area is responsible for greater susceptibility to the effects of endogenous interferon in an infected host (White et al., 2001).
TC-83 and other attenuated strains will replicate to detectable titers in the brain of many mouse strains, but this infection does not result in death of the host (Ludwig et al., 2001; Schoneboom et al., 2000). However, intranasal (i.n.) instillation, aerosol exposure, or direct intracranial (i.c.) injection of the virus in the C3H/HeN mouse strain results in morbidity and mortality (Hart et al., 1997; Ludwig et al., 2001). The i.n. route was selected for these studies because it does not require the special machinery needed for aerosol exposure or the highly invasive techniques of i.c. delivery.
The reason for a difference in morbidity and mortality of C3H/HeN mice after infection with various pathogens as compared with relative resistance in other mouse strains has been addressed, although no comprehensive studies have been conducted to provide any specific mechanisms. A decrease in the rate of clearance of TC-83 from the brains of C3H/HeN mice as compared with BALB/c mice has been observed, which has been suggested to be a result of a reduced cellular immune response (Steele et al., 1998). A decrease in the induction of mucosal IgA response correlated with increased sensitivity to an aerosol challenge with TrD VEEV in TC-83-vaccinated C3H/HeN mice, although there was no difference between IgG (Hart et al., 1997). This lack of mucosal IgA in C3H/HeN mice may at least partially explain the susceptibility of this mouse strain to i.n. challenge with TC-83, despite resistance to peripheral inoculation.
One objective of this study was to further characterize the C3H/HeN mouse model of TC-83 VEEV disease for use in antiviral experiments. TC-83 infection of out bred mouse strains has been used as an indirect model for the evaluation of antiviral compounds (Kuehne et al., 1977). Exogenous interferon has been used in the prevention or treatment of disease in mice infected with wild-type VEEV strains (Lukaszewski and Brooks, 2000). It appears that many VEEV strains are relatively resistant to the effects of interferon in vitro, and the sensitivity or resistance to exogenous interferon-α/β treatment correlated with virulence determinants in different strains of VEEV (Spotts et al., 1998). Ampligen is a single-stranded RNA that induces an antiviral state in the cells of treated animals via interferon production, and prophylactic treatment is protective in many different animal models of virus infection (2004; Julander et al., 2006; Leyssen et al., 2003; Niu et al., 1993; Pinto et al., 1988; Smee et al., 1993). If active, one or both of these agents may serve as positive control compounds in future antiviral experiments with agents of unknown activity. The utility of i.n.-infected C3H/HeN mice with the TC-83 vaccine strain of VEEV for use in antiviral studies will be evaluated to determine possible benefits of a lower Biosafety level model of VEEV disease.
2. Materials and Methods
2.1. Animals
Female C3H/HeN mice were obtained from Charles River Laboratories (Wilmington, MA) between 16 and 18 g. Animals were randomly assigned to cages and individually marked with ear tags. Mice were fed standard mouse chow and tap water ad libitum.
2.2. Facilities
Experiments were conducted in the BSL-3 animal suite at Utah State University Laboratory Animal Research Center (LARC). All personnel continue to receive special training on blood-borne pathogen handling by this university’s Environmental Health and Safety Office. Standard operating procedures for BSL-3 were used.
2.3. Test Articles
Interferon-alpha B/D (IFN--α B/D) was obtained from David Gangemi (Clemson University, Clemson, SC). The compound was diluted in physiological saline and stored at 4°C until use. Ampligen was obtained as a viscous solution from Hemispherx Biopharma (Philadelphia, PA) and was used undiluted at 12 mg/kg/d. Compound was stored at −20°C and was thawed and heated up to 50 C prior to use to ensure a homogenous mixture.
2.4. Venezuelan equine encephalitis virus
The TC-83 vaccine strain of VEE was obtained from USAMRIID (Fort Detrick, MD) and was passaged in Vero cells before use. Virus was prepared in MEM containing no FBS for mouse inoculations. Animals were anesthetized with ketamine + xylazine (100 and 10 mg/kg, respectively) and challenged by intranasal (i.n.) instillation with 0.05 ml of the diluted virus. Virus was previously titrated in mice to determine an optimal infectious dose.
2.5. Infectious cell culture assay
The virus titers in tissues or plasma were assayed using an infectious cell culture assay where a specific volume of either tissue homogenate or plasma was added to the first tube of a series of dilution tubes. Serial dilutions were made and added to Vero 76 cells. Three days later cytopathic effect (CPE) was used to identify the end-point of infection (Reed and Muench, 1938). Four replicates were used to calculate the infectious doses per ml of plasma or gram of tissue.
2.6. Quantitative (Real-time) RT-PCR
Primer-pairs (forward TCTGACAAGACGTTCCCAATCA, reverse GAATAACTTCCCTCCGACCACA) and Taq-man probe (5′ 6-carboxyfluorescein-TGTTGGAAGGGAAGATAAACGGCTACGC-6-carboxy-N,N,N′,N′-tetramethylrhodamine-3′) specific for nucleotides 7931-8005 of the TC-83 VEEV strain (L01443) were used (Qiagen, Valencia, CA). The one-step Brilliant QRT-PCR master mix 1-step kit (Stratagene, La Jolla, CA) was used for RT and amplification of VEEV RNA with primers and probe at 0.2 μM. One μl of total cellular RNA (from a total of 100 μl), extracted from infected or control tissues using Tri Reagent (Sigma Chemical Company, St. Louis, MO), was used. Samples were run on a DNA Engine Opticon 2 (MJ Research Inc, Waltham, MA). Reverse transcription of cellular RNA was performed for 30 min at 50°C followed by PCR, which consisted of 40 cycles of 15 s at 95°C and 60 sec at 61°C. Results were given in terms of relative equivalents (re), reflecting the amount of VEEV present in the sample as extrapolated from a standard curve obtained from amplification of serial dilutions of total RNA obtained 2 dpi from Vero cells infected with VEEV.
2.7. Cytokine and chemokine multiplex profiling
Brain expression of 16 cytokines and chemokines (IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12, MCP-1, IFN-γ, TNF-α, MIP-1α, GM-CSF, and RANTES) was determined using Q-plex mouse cytokine array (Quansys Biosciences, Logan, UT). Brain homogenates from mice infected 3, 4, 5, 6, 7, and 8 days post virus instillation (dpi) were analyzed per manufactures instructions. Cytokine levels obtained from infected mouse brains were compared with a 50% effective dose (ED50) generated in vitro by the supplier (R&D Systems, Inc, Minneapolis, MN) using recombinant mouse cytokines/chemokines, which served as an indication of possible biological significance.
2.8. Plaque assays
A plaque assay was used for comparison of plaque size between stock TC-83 and virus recovered 6 dpi from the brains of C3H/HeN mice infected i.n.. Briefly, stock virus or tissue homogenates taken from infected animals were serially diluted and plated on Vero cells. An agarose overlay was added to each well and plates were incubated for 3 days, after which the cells were stained with neutral red to allow measurement of plaque size. Plaques isolated from mouse brain homogenates were measured and compared with plaques obtained from a serial dilution of stock virus.
2.9. Immunohistochemistry
At the time of necropsy, infected and uninfected animals were perfused directly with PBS and 4% paraformaldehyde during cardiac puncture. Slides made from paraffin embedded samples were subjected to immunohistochemical protocols for staining. Tissue sections were deparafinized and rehydrated with xylene and alcohol series. Sections were immersed in diluted (1:10) DakoCytomation target retrieval solution (DakoCytomation Inc, Carpinteria, CA) in distilled water and boiled in a microwave for 4 cycles of 1 minute or boiled at 125°C for 4 minutes in a decloaking chamber (Bio-care Medical, Walnut Creek CA). Sections were permeabilized with 0.5% Triton X 100 in PBS for 5 minutes, and blocked using 10% normal goat serum in 0.2% Triton X 100 in PBS blocking solution. Slides were simultaneously incubated with mouse anti-VEEV (MAB8767), polyclonal anti-neuron specific enolase (NSE, AB951), and polyclonal anti-glial fibrillary acidic protein (GFAP, G9269) (Chemicon, Temecula, CA) primary antibodies diluted in 5% normal goat serum and 0.2% Triton X 100 in PBS for 2 hr. After washing with PBS, slides were incubated with diluted Alexa-fluor® 568 goat anti-mouse IgG and Alexa-fluor® 488 goat anti-rabbit IgG secondary antibodies (Molecular Probes, Eugene, OR) (1:200). The slides were washed and mounted with VECTASHIELD® mounting medium (Vector Laboratories, Burlingame, CA). Stained slides were visualized using a Nikon Eclipse TE300 microscope (Nikon) attached with Lambda DG4 (Sutter Instrument Company, Novato, CA) and a Bio-Rad MRC 1024 confocal microscope (Bio-Rad, Hercules, CA). Control and experimental images were collected and processed using the same instrument settings. Sections stained with hematoxylin and eosin were observed to determine if morphology of infected animals was altered.
2.10. Experimental design
Initial titration experiments were conducted to determine the LD100 for C3H/HeN mice receiving i.n. instillation of TC-83 VEEV. Five mice per group were lightly anesthetized, and subjected to i.n. instillation of 50 μl of 10-fold dilutions of virus stock (109.1 to 104.1 CCID50/ml). Mortality was monitored daily for 21 days and weight change was recorded on 0, 3, 5, and 7 dpi. A reversion experiment was conducted to determine if a more virulent revertant was generated after a single passage of TC-83 in Vero cells. A group of 5 C57BL/6 or C3H/HeN mice were infected in parallel with 107.1 CCID50 TC-83 and mortality was observed over several weeks.
For model characterization, tissue samples from 10 mice were collected after weight was recorded each day after virus instillation, beginning 2 dpi. Virus titer in brain, liver, spleen, kidney, and serum was determined. Weight was recorded daily from 4 to 8 dpi, and weight change was calculated by comparing daily weights with initial weight on 4 dpi. For pathogenic characterization, two mice were anesthetized and the whole mouse was fixed by transcardial perfusion with freshly prepared 4% paraformaldehyde on 2, 4, 6, and 8 dpi for overall pathology. Brain, intestine, stomach, pancreas, adrenal gland, lung, liver, spleen, kidney, thymus, esophagus, lymph node, heart, uterus, ovary, urinary bladder, skeletal muscle, bone marrow, and bone were observed for any pathologic changes due to viral infection. RNA was obtained from brain and spleen samples from five additional mice for virus titration by QRT-PCR.
Mice were challenged i.n. with an LD90 dose of VEE virus, and groups were treated by intraperitoneal (i.p.) injection of IFN-α B/D at a dose of 107, 106.5, or 106 IU/kg qd for 8 days, beginning 24 h prior to virus challenge or with ampligen at a dose of 12 mg/treatment administered −4 h and 2 dpi. Placebo control mice were treated with saline on the same schedule as interferon treatment. Mortality was checked daily for 21 days. Mice were weighed daily starting on the day of virus challenge through 9 dpi. Toxicity controls (uninfected mice) were treated in parallel and weight changes were noted. Brain samples were taken 6 dpi to determine the effect of IFN and ampligen treatment on virus load and cytokine levels in the brains of treated animals as compared with placebo-treated controls. Surviving mice were re-challenged with TC-83 VEEV by i.n. instillation to determine their immunological status.
2.11. Statistical analysis
Survival data were analyzed using the Wilcoxon log-rank survival analysis (JMP™ software, the Statistical Discovery Software, SAS Institute, Inc). All other statistical analysis was done using one-way Student’s t-test.
3. Results
3.1. Model Characterization
C3H/HeN mice were infected with 10-fold dilutions of stock virus to determine an optimal infectious dose for use in subsequent experiments. Infection of C3H/HeN mice through i.n. instillation with the TC-83 vaccine strain of VEEV resulted in 100% morbidity and mortality at virus concentrations between 109.1 and 107.1 CCID50 by 10 dpi. A dose of 106.1 CCID50 gave a mortality rate between 90 to 100% in two separate titration experiments, but further 10-fold dilutions caused very little mortality (data not shown). A virus concentration of 107.1 CCID50 was chosen for characterization of the model and for use in antiviral experiments. Of mice infected with this dose, the mean day-to-death was 9.4 ± 0.5 days. Infection of C57BL/6 mice with 107.1 CCID50 TC-83 by intranasal instillation did not result in any observed morbidity or mortality (data not shown), while 80% of C3H/HeN mice infected in parallel died.
To characterize the C3H/HeN mouse model, 10 animals were included in each group, and one group was necropsied each day after virus challenge beginning 2 days post-virus instillation (dpi). Virus was detectable by infectious cell culture assay in brain homogenates at the earliest tested time-point of 2 dpi, and virus titer peaked on 4 dpi (Fig. 1A). Brain virus titers were persistently high until death with a slight drop on 8 dpi. Virus time-course in the brain on 2, 4, 6, and 8 dpi was also determined by QRT-PCR, and showed a similar trend as those data generated by infectious cell culture assay (Fig. 1B). Spleens also had elevated titers observed between 2 and 6 dpi (Fig. 1C). Virus titer in liver, kidney, and serum samples was slightly elevated at different times after virus challenge, but generally only half of the samples were virus positive on days where significant elevation was observed (Fig. 1D–F). Weights of infected mice began to decline on 6 dpi, and a significant difference in weight change, as compared with uninfected controls, was observed on 7 dpi (Fig. 2).
Fig. 1.
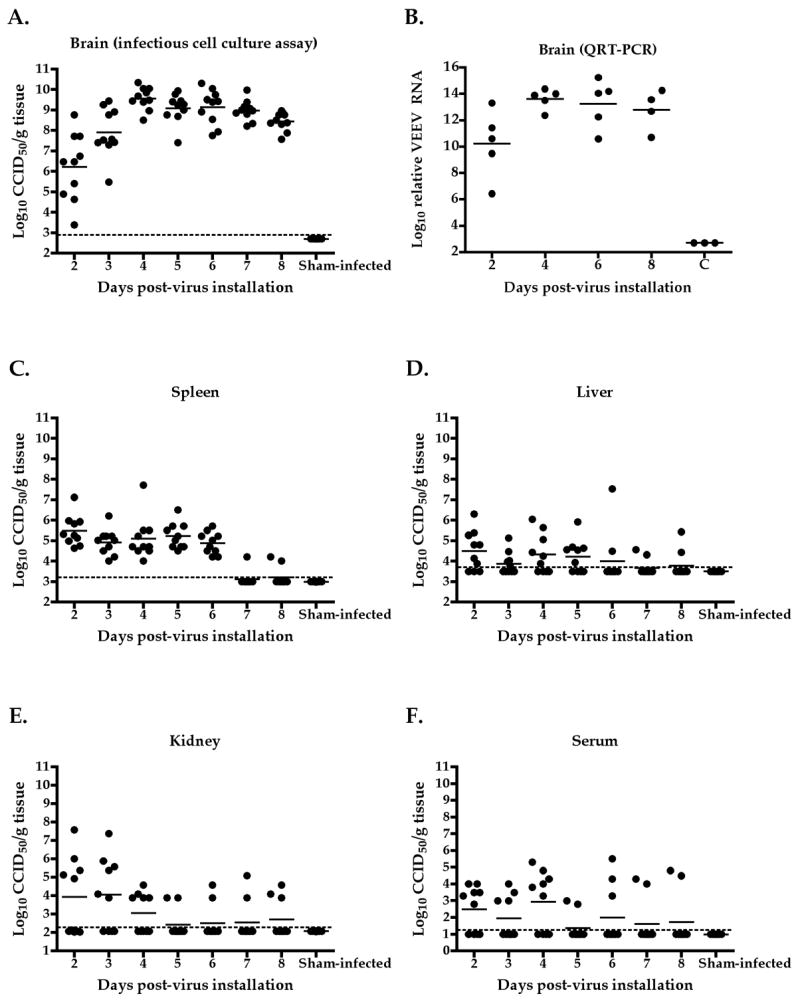
Time-course of TC-83 VEEV titer in brain (A), spleen (C), liver (D), kidney (E), and serum (F) taken from infected mice on various days after i.n. viral challenge and titrated by infectious cell culture assay. Titers were also determined by QRT-PCR in brains (B) of mice infected 2, 4, 6, and 8 dpi.
Fig. 2.
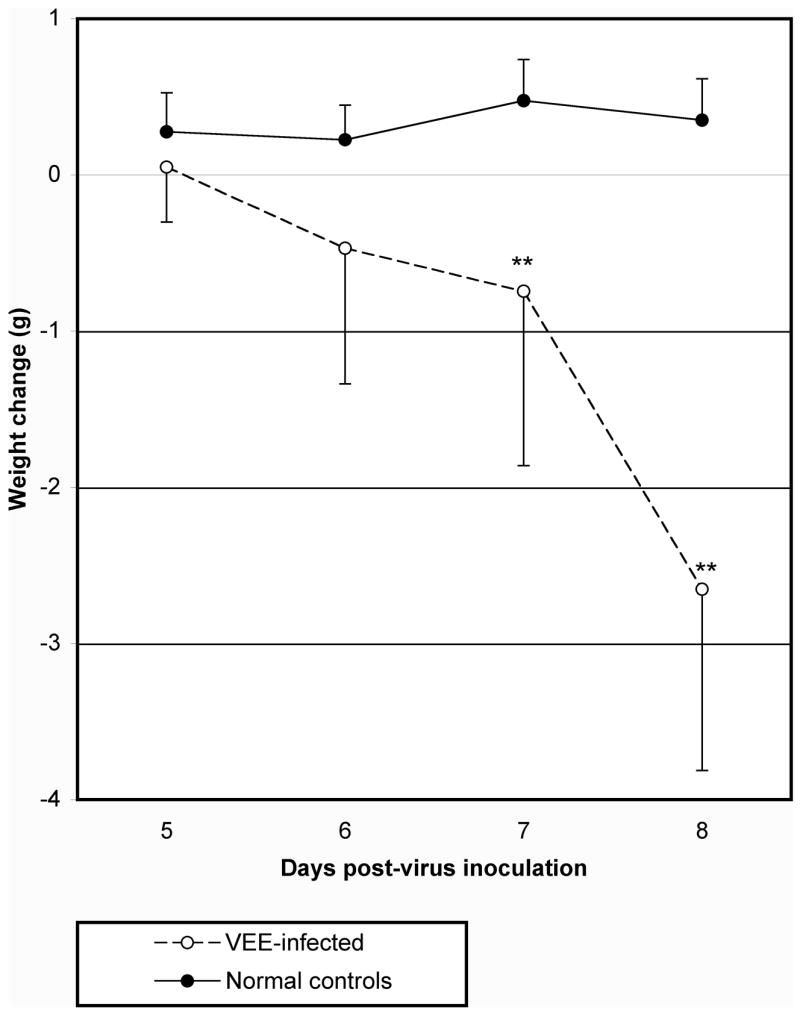
Mean weight change of C3H/HeN mice challenged i.n. with TC-83 VEEV (**P<0.01, as compared with uninfected controls).
A pathological time-course showed a progression of disease in the brains of mice infected with VEEV. A general progression from normal brain on 2 dpi to meningoencephalitis with multifocal neuropil vacuolation and gliosis on 8 dpi was observed in hematoxylin and eosin stained, coronal sections of mouse brains harvested 2, 4, 6, or 8 dpi (Table 1). During later time-points of infection, spleens were severely depleted of lymphocytes and the peripheral lymph nodes had severe diffuse depletion of follicular mantle zones. Other tissues of infected mice were histologically normal and were comparable to sham-infected control tissues.
Table 1.
Time-course of pathology in the brains of mice infected i.n. with 106.1 CCID50 of TC-83 VEEV
| dpi | Normal Tissues | Perivascular cuffinga | Meningitisa | Encephalitisa | Lymphoid depletionb | Neuropil degenerationa | gliosisa |
|---|---|---|---|---|---|---|---|
| 2 | X | ||||||
| 4 | X | X | |||||
| 6 | X | X | X | X | |||
| 8 | X | X | X | X | X | X | |
| Cc | X |
Observed in the brain
Observed in spleen and lymph nodes
Sham-infected control mice
Cytokine profiles of brain samples were determined using a multiplex protein array. Significant elevations were seen with IL-1α, IL-1β, IL-6, IL-12, MCP-1, IFN-γ, TNF-α, MIP-1α, and RANTES (Fig. 3). Most of the peak elevations in these cytokine levels occurred at 6 or 7 dpi, with a reduction on subsequent days, although IFN-γ had peak levels on 8 dpi. In general, a decrease in cytokine levels would usually be associated with a recovery from viral infection, but in this model, a reduction in cytokine levels occurred shortly before death. Significant elevations were seen in levels of IL-2, IL-3, IL-4, IL-5, and IL-10 (Fig. 4), but these increases were below a 50% effective dose (ED50) required to achieve a physiologic response in vitro, suggesting that these elevations may not be biologically important. IL-9 levels were above the ED50 value, but were not significantly different than levels in uninfected mice (Fig. 4). There was no elevation in GM-CSF (data not shown).
Fig. 3.

Significantly elevated cytokine levels measured from brain homogenates taken from C3H/HeN mice on 3–8 days post-virus instillation with TC-83 VEEV. A multi-plex protein array was used Dashed line represents a 50% cell culture effective dose of each cytokine or chemokine. (***P<0.001, **P<0.01, *P<0.05 as compared with sham infection)
Fig. 4.
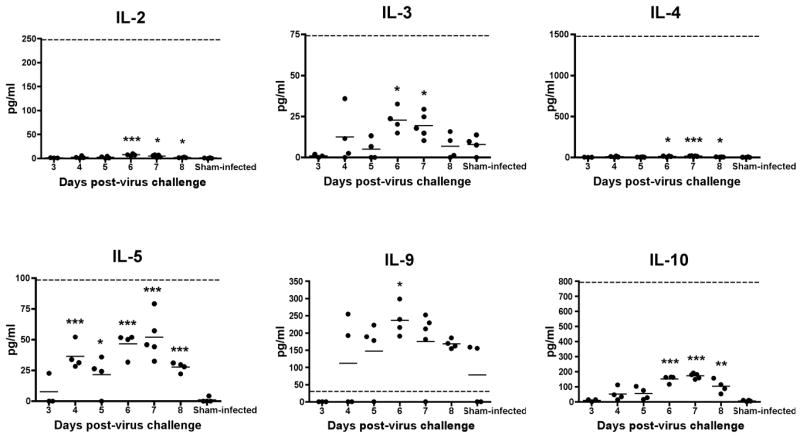
Levels of IL-2, -3, -4, -5, and -10 measured from brain homogenates taken from C3H/HeN mice on 3–8 dpi with TC-83 VEEV. These cytokines were significantly elevated as compared with levels from sham-infected animals, but were not higher than an average cell culture 50% effective dose (ED50) that is necessary to elicit a physiologic response in vitro. IL-9 levels were above the ED50, but were generally not significantly higher than corresponding amounts from sham-infected controls. Dashed line represents a 50% cell culture effective dose of each cytokine
Confocal microscopic double-staining with antibodies to VEE and neurospecific enolase (NSE) (Fig. 5 D-F) revealed that TC-83 VEE had infected neurons. However, considerable numbers of VEE-stained cells were not colocalized with NSE staining. Some VEE stained cells colocalized with glial fibrillary acid protein (GFAP) in the brains of infected mice (Fig. 5 J–L, arrows), but the colocalized staining was not as strong as was observed for VEE and neuron-specific staining with NSE. Many neuron-like cells were stained only with VEE and not with GFAP (Figure 5 L, arrowheads). No VEEV staining was observed in brains of uninfected control mice (Fig. 5 A, and I).
Fig. 5.

Confocal immunofluorescent staining of C3H/HeN mouse brain sections for VEEV and different cell marker proteins. Brain sections from uninfected (A, B, C, G, H, and I) or infected (D, E, F, J, K, and L) mice were double-stained for VEEV (A, D, G, and J) and for either neuron specific enolase (NSE) (B and E), a cell marker present on neurons, or with glial fibrillary acidic protein (GFAP) (H and K), a marker present on astrocytes. Double-staining of NSE with VEEV in uninfected (C) and infected (F) mouse brains is shown. Double-staining of GFAP and VEEV in uninfected (I) or infected (L) mouse brains is also shown. Double-staining of the cortex of infected mouse brains showed a colocalization of VEEV and NSE (F, arrows). A midbrain section of another infected mouse brain revealed colocalization of VEEV and GFAP (L, arrows) as well as further VEEV antigen staining of cells that appear to be neurons (L, arrowheads). Scale bar = 20μM.
Reversion of TC-83 to a more virulent phenotype may be occurring during replication in mice. This was monitored by determining plaque size of input (stock) virus and output virus obtained from brains of infected mice, based upon the report that virulent viruses produce much larger plaques than does the TC-83 virus (Kinney et al., 1993). Approximately 100 plaques isolated from brain samples of infected mice were measured, yielding an average diameter of 2.7 ± 0.4 mm, which was not significantly different from the average diameter of 2.7 ± 0.3 of plaques from stock virus. Plaque diameter ranged from 2.0 to 3.5 mm in plaques isolated from both brain samples and virus stock. Based on the work of Kinney et al, (1993) we would have expected a mean plaque size of at least 3.4 ± 0.5 mm to be indicative of virulent virus.
3.2. Treatment of VEEV disease
Mice were treated with 107, 106.5, or 106 IU/kg/d of IFN-α B/D, given i.p. for 8 days, beginning 4 h prior to virus challenge. A dose-response was observed with IFN-α B/D treatment (Fig. 6, Table 2). All but one of the mice treated with 107 IU/kg/d of IFN-αB/D survived through 21 dpi, and all but one mouse treated with placebo died, with a mean day-to-death of 9.6 ± 0.5. Survival was also significantly improved in mice treated with 106.5 IU/kg/d of IFN-α B/D, although 40% of mice in this group died. Weight change in infected animals was significantly improved in animals treated with 107 or 106.5 IU/kg/d IFN-α B/D as compared with weight change in placebo-treated controls (Table 2). Treatment with 106 IU/kg/d did not significantly improve survival or weight change, although a slight trend towards improvement of weight change was observed.
Fig. 6.
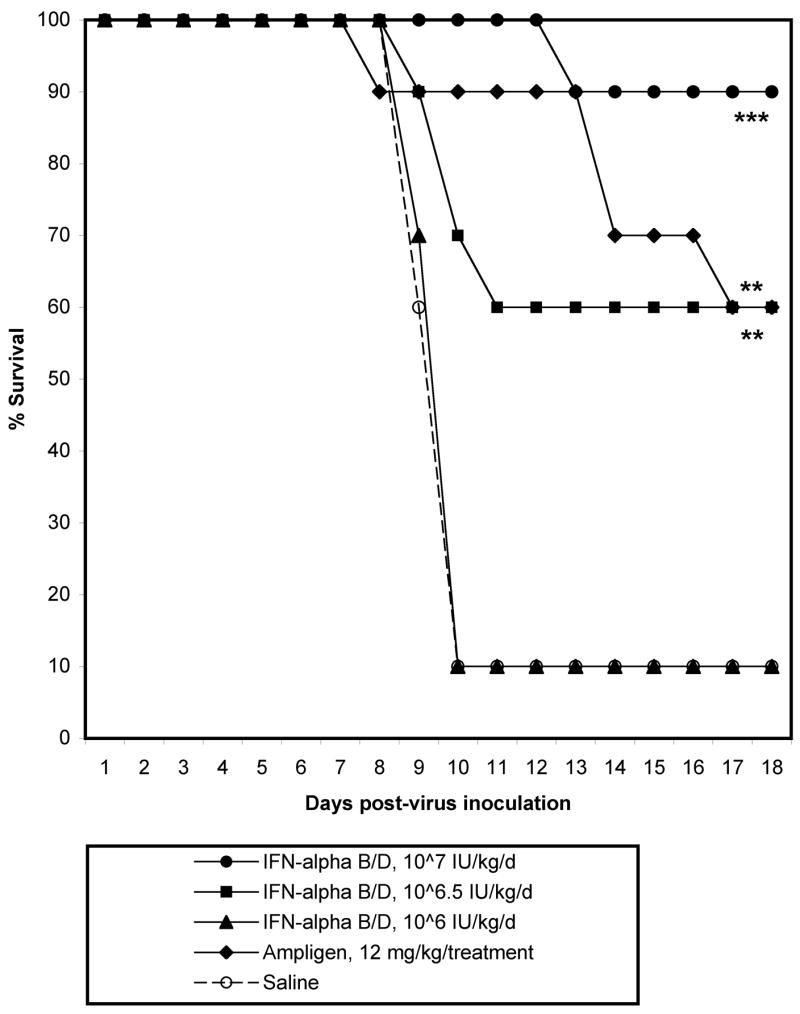
Survival of C3H/HeN mice infected with TC-83 VEEV and treated −4 h to 7 dpi with various concentrations of interferon alpha B/D or ampligen (***P<0.001, **P<0.01, as compared with saline treatment).
Table 2.
Effect of Interferon-α B/D or Ampligen Treatment on C3H/HeN Mice Infected with Venezuelan Equine Encephalitis Virusa
| Toxicity controls
|
Infected, treated
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Treatment | Dosage | Treatment Schedule | Alive/ Total | Mean wt.changeb(g) ± SD | Alive/Total | MDDc ± S D | Brain virus titerd(CCID50) | Mean wt.changeb (g) ± SD |
| Interferon-αB/D | 107 IU/kg/d | qd X 7 d beg. –1 | 3/3 | 2.0 ± 0.2 | 9/10*** | 13.0 ± 0.0 | 3.9 ± 0.4** | 1.2 ± 0.5*** |
| Interferon-αB/D | 106.5 IU/kg/d | qd X 7 d beg. –1 | -- | -- | 6/10** | 10.0 ± 0.6 | 5.4 ± 2.4* | 0.3 ± 1.6*** |
| Interferon-αB/D | 106 IU/kg/d | qd X 7 d beg. –1 | -- | -- | 1/10 | 9.7 ± 0.5 | 9.9 ± 0.9 | −2.2 ± 1.4 |
| Ampligen | 12 mg | −4 h and 2 dpi | -- | -- | 6/10** | 13.3 ± 3.8 | 5.0 ± 1.2** | 1.3 ± 1.9*** |
| Placebo | -- | qd X 7 d beg. –1 | -- | -- | 1/10 | 9.6 ± 0.5 | 9.5 ± 1.6 | −3.1 ± 1.5 |
| Normal Controls | -- | -- | 2/2 | 2.7 ± 0.4 | -- | -- | -- | -- |
TC-83 vaccine strain: 107.4 CCID50, administered via intranasal installation.
Difference between weight on 0 and 7 days post-virus injection (dpi).
Mean day to death of mice dying prior to 21 dpi.
Mean VEEV titer determined by infectious cell culture assay (± SD).
P 0.001,
P ≤ 0.01,
P ≤ 0.05, as compared with saline-treated controls.
Brain virus titers on 6 dpi were significantly (P<0.01) reduced by approximately 5.5 log10 after treatment with 107 IU/kg/d of IFN-α B/D. Titers were also reduced around 4 log10 with 106.5 IU/kg/d, which was also significant compared with placebo (P<0.05), but a further dilution of IFN-α B/D did not reduce viral titer in the brain (Table 2).
Cytokine levels in the brains of infected mice were quantified 6 dpi, after completion of the IFN-α B/D treatment regimen. All the cytokines found previously to be significantly upregulated in the brains of infected mice were reduced to background levels after treatment with IFN-α B/D at doses of 107 or 106.5 IU/kg/d (Table 3). Treatment with 106 IU/kg/d did result in a decrease in cytokine levels, but this decrease was only significant (P<0.05) in the case of IFN-1α (data not shown).
Table 3.
Select cytokine and chemokine levels (average ± SD) from the brains of infected mice, taken 6 dpi, after treatment with interferon-α B/D or ampligen. Cytokine and chemokine levels in the brains of treated mice were measured using a mouse cytokine array. P<0.01, P<0.05, as compared with saline-treated controls.
| Treatment (dose) | IL-1α | IL-1β | IL-6 | IL-12 | MCP-1 | IFN-γ | TNF-α | MIP-1α | RANTES |
|---|---|---|---|---|---|---|---|---|---|
| IFN-α B/D (107 IUa) | 27 ± 4.0 | 2.2 ± 1.8 | 1.3 ± 0.3 | 11 ± 2.1 | 120 ± 32 | 108 ± 20 | 4.4 ± 6.8 | 15 ± 13 | 4.4 ± 1.2 |
| IFN-α B/D (106.5 IU) | 36 ± 7.0 | 2.4 ± 2.9 | 1.1 ± 0.4 | 8.8 ± 3.5 | 107 ± 65 | 90 ± 42 | 2.7 ± 3.8 | 27 ± 16 | 4.2 ± 2.5 |
| IFN-α B/D (106 IU) | 257 ± 182 | 154 ± 137 | 2737 ± 2526 | 3592 ± 2310 | 18585 ± 13953 | 818 ± 680 | 256 ± 216 | 1247 ± 848 | 2620 ± 1826 |
| Ampligen (12 mg) | 20 ± 2.0 | 1.8 ± 1.4 | 0.9 ± 0.6 | 11 ± 2.3 | 101 ± 49 | 37 ± 16 | 0.6 ± 1.3 | 13 ± 12 | 18 ± 27 |
| Saline Placebo | 840 ± 472 | 208 ± 120 | 4962 ± 2853 | 4553 ± 2693 | 37041 ± 22238 | 1382 ± 871 | 701 ± 404 | 4267 ± 3064 | 8283 ± 5457 |
IFN-α B/D doses are in international units (IU)/kg/d.
Ampligen, an interferon inducer, was also effective in the treatment of VEEV when given i.p. at a dose of 12 mg/treatment at −4 h and 2 dpi. Survival was significantly improved in mice treated with ampligen as compared with placebo (Table 2). Weight change was also significantly improved in ampligen-treated mice. Ampligen treatment also significantly reduced viral titer and cytokine levels in the brains of mice infected with VEEV and harvested 6 dpi (Table 2).
Surviving mice from all treatment groups were re-challenged 21 dpi to determine if protective immunity was acquired after TC-83 infection. Mice that survived VEEV infection after IFN-α B/D or ampligen treatment were challenged i.n. with the same dose of virus given initially. Mice were not immune to this secondary challenge with 11 of 23 mice dying with a mean day to death of 8.6 ± 2.2 d after re-challenge. All naïve mice infected i.n. with the same inoculum died with a mean day to death of 10.3 ± 1.2 d after challenge, which was significantly (P<0.05) higher than the mean day-to-death in mice that survived previous VEEV infection. Although there was a 100% mortality rate observed in naïve mice, while mice that survived the previous infection had a 52% mortality rate, this difference in survival was not statistically significant.
4. Discussion
The main purpose of this study was to determine the utility of the C3H/HeN mouse model of TC-83 VEEV infection for antiviral and basic virological experiments. This model, as shown here and in previously published literature (Hart et al., 1997; Ludwig et al., 2001; Steele et al., 1998), is similar in many regards to the model of TrD strain infection in mice. Such similarities include high brain virus titer early in the course of disease continuing until death (Davis et al., 1994; Jackson et al., 1991; Jordan, 1973), cytokine induction (Grieder et al., 1997; Schoneboom et al., 2000), relative resistance (treatment was effective only at concentrations of 106.5 IU/kg/d and above) to interferon treatment (Lukaszewski and Brooks, 2000), and similar disease parameters including mean day-to-death (Grieder et al., 1997; Lukaszewski and Brooks, 2000), hunching (Jackson et al., 1991), weight loss, ruffled fur, and non-responsiveness (Ludwig et al., 2001). Immunohistochemical staining of brain samples showed co-localization of VEEV antigen with NSE and GFAP, indicating infection of neurons and astrocytes, which was similar to previously reported data (Jackson et al., 1991; Schoneboom et al., 2000; Weaver et al., 2004). Some differences observed in this study compared to those using other VEEV models include a reduced duration of viremia, lower frequency of tissue infection outside the CNS, and lack of susceptibility to peripheral inoculation (Ludwig et al., 2001). One shortcoming of the model is the fact that intranasal installation of virus does not properly replicate the natural course of infection in humans, which is generally through peripheral exposure after the bite of a mosquito carrying the virus. As mentioned previously, however, the time course and disease progression is similar to natural disease in many ways, thus providing a potentially useful model for the evaluation of antiviral compounds despite the difference in route of infection.
The course of VEEV infection in different animal hosts is biphasic, with an initial lymphocytic phase and viremia followed by a secondary phase of CNS infection (Gleiser et al., 1962). The infection with TC-83 in C3H/HeN mice seems to have a less pronounced initial lymphocytic phase, and viremia was low, similar to previously reported data (Ludwig et al., 2001). However, virus titer was detected by infectious cell culture assay in the spleens of infected mice, which was similar to spleen titers in mice infected with TrD (Lukaszewski and Brooks, 2000). Brain pathology included meningoencephalitis, gliosis, and multifocal neuropil vacuolation, which was similar to brain pathology seen in mouse infection with other VEEV strains (Charles et al., 2001; Jackson et al., 1991; Schoneboom et al., 2000). Compared with TrD infection in mice, infection with TC-83 results in a reduced viremia, both in terms of amount of circulating virus, as well as in duration (Ludwig et al., 2001). An important consideration in the development of antiviral agents for the treatment of VEEV is if the agent can inhibit virus after establishment of viral replication and disease in the brain, as clinical VEEV disease is usually diagnosed after the onset of neurological symptoms. The infection of C3H/HeN mice would allow for the testing of agents against VEEV CNS disease.
Intranasal instillation with TC-83 resulted in high morbidity and mortality when given at high dose (107.1 CCID50). The high virus inoculum was likely necessary because of the attenuated nature of this vaccine strain. Mortality is seen after inoculation with lower titers, but the rate is variable, making the detection of statistical significance more difficult. High titers of TC-83 are easily produced after one passage in Vero cells, so no concentration of virus was necessary to generate the large virus inoculum. A lower virus inoculum of TC-83 has been shown to cause lethal infection when injected intracranially into C3H/HeN (Ludwig et al., 2001). The large difference in virus stock needed for lethality between the present study and those data presented by Ludwig et al could be due to differences in stock preparation, method of stock quantification, or lower efficiency of i.n. infection.
Virus was detected in the brains of infected mice with peak titers 4 dpi, which continued at high levels until death. This is similar to the time-course of brain infection in mice inoculated through foot-pad injection with TrD strain VEEV (Davis et al., 1994). High virus titers in the brain have previously been observed for many different strains of VEEV, and brain titer is not dependent on the phenotype of the infecting strain. Many attenuated strains cause low or no mortality, but replicate efficiently in the brain (Jordan, 1973; Ludwig et al., 2001; Schoneboom et al., 2000). A comparison of TrD and TC-83 aerosol exposure in C3H/HeN mice showed a slightly reduced distribution of TC-83 in the brain as compared with the wild-type TrD (Steele et al., 1998). Death does not seem to correlate with peak brain virus titer in this model or in models of TrD infection, and is likely influenced by other factors, including the host immune response, rapidity of viral spread, or other unknown reasons (Charles et al., 2001; Jackson et al., 1991).
Reversion of TC-83 has been shown previously to occur after i.c. challenge with virus obtained after 5 brain passages in adult BALB/c mice (Ludwig et al., 2001). While it is possible that morbidity and mortality of C3H/Hen mice challenged i.n. with TC-83 VEEV was due to revertant viruses present in the inoculum, reversion to wild-type phenotype would likely involve at least two separate site-specific changes to restore wild-type virulence. Intranasal inoculation of C57BL/6 mice with TC-83 stock generated after one passage in Vero cells resulted in no observable morbidity or mortality while 80% of C3H/HeN mice infected in parallel exhibited disease signs and died. This indicates that stock virus is similar to TC-83 that has been used in previous studies (Hart et al., 1997; Ludwig et al., 2001; Steele et al., 1998). One indication of reversion to wild-type TrD phenotype may be plaque size of virus isolated from infected mice. An increase in plaque size was observed as various pathogenic elements of TrD VEEV were incorporated into a full-length molecular cDNA clone of TC-83, which also correlated with increased mortality in mice (Kinney et al., 1993). No mortality was observed in Swiss ICR mice after i.p. challenge with virus constructs that had average plaque sizes smaller than 3.4 mm (Kinney et al., 1993). Average plaque sizes of virus isolates we obtained from infected mouse brains taken 6 dpi and from stock TC-83 VEEV were identical to TC-83 plaque size previously reported (Kinney et al., 1993), suggesting that virus had undergone little if any adaptive mutations during replication cycles in the mice.
Death in C3H/HeN mice infected with TC-83 VEEV occurred around 8–10 dpi, which coincided with meningitis and encephalitis. This is similar to what has been seen in other models of VEEV infection (Grieder et al., 1997; Schoneboom et al., 2000). Cytokines that were significantly elevated generally peaked at 6 or 7 dpi with a decline on 8 dpi just prior to death, which gave the appearance of recovery from disease. Death did correlate with a rise in interferon-γ, which may indicate that this cytokine is important in mortality. Understanding the effects of the cytokine cascade in the brains of infected mice may be key in further options for therapeutic intervention. Weight change was also significantly reduced in infected animals and was indicative of disease severity, thus, can be used as a parameter when evaluating antiviral treatments. Very few mice survived past 8 dpi in this model (~10%). We had desired to obtain cytokine levels and other virological parameters at time-points beyond 8 dpi, but to do so would have required the infection of a prohibitive number of mice to obtain these data.
Treatment with IFN-alpha B/D improved disease parameters in a dose-responsive manner. Treatment with 107 IU/kg/d resulted in significant improvement in survival and weight change, while treatment with 106.5 IU/kg/d resulted in some mortality, although survival and weight change were significantly improved as compared with placebo treatment. Treatment with these concentrations also significantly reduced virus titer and levels of significantly elevated cytokines in the brains of infected mice 6 dpi. Cytokine levels were reduced somewhat with 106 IU/kg/d IFN-α B/D treatment, but these reductions were only significant in the case of IL-1α. Treatment with 106 IU/kg/d did not improve any of the other disease parameters analyzed, which is consistent with results seen in previous experiments with TrD strain of VEEV where treatment with 106 U/ml of IFN-α B/D did not improve disease in BALB/c mice infected with TrD VEEV (Lukaszewski and Brooks, 2000). The mutation in the 5′ UTR of TC-83 has been shown to be associated with increased sensitivity to interferon regulation in mice (White et al., 2001); however, in this study, treatment with 106 IU/kg/d of IFN-α B/D resulted in no increased survival, which was similar to previous results in the treatment of TrD VEEV (Lukaszewski and Brooks, 2000).
An interferon inducer, ampligen, was also effective in improving disease parameters. Early induction of the interferon response and other innate antiviral pathways is likely responsible for efficacy of ampligen pre-treatment in this model. Knockout mice lacking key components in the interferon response are more susceptible to VEEV-induced mortality, and infection of these mouse strains with attenuated strains results in high mortality and a reduced mean day-to-death (White et al., 2001).
Morbidity and mortality was observed after re-challenge of mice that survived initial infection with VEEV. This suggests that the initial significant increase in survival of infected mice was due to successful treatment with IFN-α B/D or ampligen. This may also suggest that infected mice treated with these compounds were not immunized after initial infection. However, infection of C3H/HeN mice with TC-83 has been shown to elicit a lower level of protection against aerosol challenge with TrD VEEV, despite a normal protection against peripheral inoculation with TrD, which was attributed to a suboptimal IgA response in the respiratory (including nasal) mucosa (Hart et al., 1997).
The C3H/HeN mouse model of TC-83 VEEV infection may be a useful model for the evaluation of antiviral compounds. Similarities between the CNS disease in this model as compared with other models of VEEV infection suggest that this model may be predictive for efficacy of an antiviral compound in the treatment of natural infection in humans. Studies are underway to compare the antiviral efficacy of other compounds in this model with the TrD model of VEEV disease.
Acknowledgments
We thank Luci Wandersee and Jeremy Strange for their work with treatments and data collection. This work was supported by contracts NO1-AI-15435 and NO1-AI-30048 from the Virology Branch, NIAID, NIH and grant 1-U54 AI-06357 from the Rocky Mountain Regional Centers of Excellence.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Justin G. Julander, Institute for Antiviral Research, Department of Animal, Dairy, and Veterinary Sciences, Utah State University, 5600 Old Main Hill, Logan, UT. 84322-5600. Ph (435)-797-7215, Fax (435)-797-3959, e-mail jgjulander@cc.usu.edu
Ramona Skirpstunas, Veterinary Diagnostic Laboratory, Department of Animal, Dairy, and Veterinary Sciences, Utah State University.
Venkatraman Siddharthan, Institute for Antiviral Research, Department of Animal, Dairy, and Veterinary Sciences, Utah State University.
Kristiina Shafer, Institute for Antiviral Research, Department of Animal, Dairy, and Veterinary Sciences, Utah State University.
Justin D. Hoopes, Department of Biology, Utah State University
Donald F. Smee, Institute for Antiviral Research, Department of Animal, Dairy, and Veterinary Sciences, Utah State University
John D. Morrey, Institute for Antiviral Research, Department of Animal, Dairy, and Veterinary Sciences, Utah State University
References
- Anonymous. Mismatched double-stranded RNA: polyI:polyC12U. Drugs R D. 2004;5:297–304. doi: 10.2165/00126839-200405050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronson JF, Grieder FB, Davis NL, Charles PC, Knott T, Brown K, Johnston RE. A single-site mutant and revertants arising in vivo define early steps in the pathogenesis of Venezuelan equine encephalitis virus. Virology. 2000;270:111–123. doi: 10.1006/viro.2000.0241. [DOI] [PubMed] [Google Scholar]
- Berge TO, Banks IS, Tigertt WD. Attenuation of Venezuelan equine encephalomyelitis virus by in vitro cultivation in guinea-pig heart cells. Am J Hyg. 1961;73:209–218. [Google Scholar]
- Charles PC, Trgovcich J, Davis NL, Johnston RE. Immunopathogenesis and immune modulation of Venezuelan equine encephalitis virus-induced disease in the mouse. Virology. 2001;284:190–202. doi: 10.1006/viro.2001.0878. [DOI] [PubMed] [Google Scholar]
- Charles PC, Walters E, Margolis F, Johnston RE. Mechanism of neuroinvasion of Venezuelan equine encephalitis virus in the mouse. Virology. 1995;208:662–671. doi: 10.1006/viro.1995.1197. [DOI] [PubMed] [Google Scholar]
- Davis NL, Grieder FB, Smith JF, Greenwald GF, Valenski ML, Sellon DC, Charles PC, Johnston RE. A molecular genetic approach to the study of Venezuelan equine encephalitis virus pathogenesis. Arch Virol Suppl. 1994;9:99–109. doi: 10.1007/978-3-7091-9326-6_11. [DOI] [PubMed] [Google Scholar]
- de la Monte S, Castro F, Bonilla NJ, Gaskin de Urdaneta A, Hutchins GM. The systemic pathology of Venezuelan equine encephalitis virus infection in humans. Am J Trop Med Hyg. 1985;34:194–202. doi: 10.4269/ajtmh.1985.34.194. [DOI] [PubMed] [Google Scholar]
- Gleiser CA, Gochenour WS, Jr, Berge TO, Tigertt WD. The comparative pathology of experimental Venezuelan equine encephalomyelitis infection in different animal hosts. J Infect Dis. 1962;110:80–97. doi: 10.1093/infdis/110.1.80. [DOI] [PubMed] [Google Scholar]
- Grieder FB, Davis BK, Zhou XD, Chen SJ, Finkelman FD, Gause WC. Kinetics of cytokine expression and regulation of host protection following infection with molecularly cloned Venezuelan equine encephalitis virus. Virology. 1997;233:302–312. doi: 10.1006/viro.1997.8617. [DOI] [PubMed] [Google Scholar]
- Grieder FB, Davis NL, Aronson JF, Charles PC, Sellon DC, Suzuki K, Johnston RE. Specific restrictions in the progression of Venezuelan equine encephalitis virus-induced disease resulting from single amino acid changes in the glycoproteins. Virology. 1995;206:994–1006. doi: 10.1006/viro.1995.1022. [DOI] [PubMed] [Google Scholar]
- Griffin D, Levine B, Tyor W, Ubol S, Despres P. The role of antibody in recovery from alphavirus encephalitis. Immunol Rev. 1997;159:155–161. doi: 10.1111/j.1600-065x.1997.tb01013.x. [DOI] [PubMed] [Google Scholar]
- Hart MK, Pratt W, Panelo F, Tammariello R, Dertzbaugh M. Venezuelan equine encephalitis virus vaccines induce mucosal IgA responses and protection from airborne infection in BALB/c, but not C3H/HeN mice. Vaccine. 1997;15:363–369. doi: 10.1016/s0264-410x(96)00204-6. [DOI] [PubMed] [Google Scholar]
- Jackson AC, SenGupta SK, Smith JF. Pathogenesis of Venezuelan equine encephalitis virus infection in mice and hamsters. Vet Pathol. 1991;28:410–418. doi: 10.1177/030098589102800509. [DOI] [PubMed] [Google Scholar]
- Johnson BJ, Kinney RM, Kost CL, Trent DW. Molecular determinants of alphavirus neurovirulence: nucleotide and deduced protein sequence changes during attenuation of Venezuelan equine encephalitis virus. J Gen Virol. 1986;67(Pt 9):1951–1960. doi: 10.1099/0022-1317-67-9-1951. [DOI] [PubMed] [Google Scholar]
- Jordan GW. Interferon sensitivity of Venezuelan equine encephalomyelitis virus. Infect Immun. 1973;7:911–917. doi: 10.1128/iai.7.6.911-917.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julander JG, Siddharthan V, Blatt LM, Schafer K, Sidwell RW, Morrey JD. Effect of exogenous interferon and an interferon inducer on western equine encephalitis virus disease in a hamster model. Virology. 2007;360:454–460. doi: 10.1016/j.virol.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney RM, Chang GJ, Tsuchiya KR, Sneider JM, Roehrig JT, Woodward TM, Trent DW. Attenuation of Venezuelan equine encephalitis virus strain TC–83 is encoded by the 5′-noncoding region and the E2 envelope glycoprotein. J Virol. 1993;67:1269–1277. doi: 10.1128/jvi.67.3.1269-1277.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehne RW, Pannier WL, Stephen EL. Indirect mouse model for the evaluation of potential antiviral compounds: results with Venezuelan equine encephalomyelitis virus. Antimicrob Agents Chemother. 1977;11:683–687. doi: 10.1128/aac.11.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyssen P, Drosten C, Paning M, Charlier N, Paeshuyse J, De Clercq E, Neyts J. Interferons, interferon inducers, and interferon-ribavirin in treatment of flavivirus-induced encephalitis in mice. Antimicrob Agents Chemother. 2003;47:777–782. doi: 10.1128/AAC.47.2.777-782.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig GV, Turell MJ, Vogel P, Kondig JP, Kell WK, Smith JF, Pratt WD. Comparative neurovirulence of attenuated and non-attenuated strains of Venezuelan equine encephalitis virus in mice. Am J Trop Med Hyg. 2001;64:49–55. doi: 10.4269/ajtmh.2001.64.49. [DOI] [PubMed] [Google Scholar]
- Lukaszewski RA, Brooks TJ. Pegylated alpha interferon is an effective treatment for virulent venezuelan equine encephalitis virus and has profound effects on the host immune response to infection. J Virol. 2000;74:5006–5015. doi: 10.1128/jvi.74.11.5006-5015.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muehlenbein MP, Cogswell FB, James MA, Koterski J, Ludwig GV. Testosterone correlates with Venezuelan equine encephalitis virus infection in macaques. Virol J. 2006;3:19. doi: 10.1186/1743-422X-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu J, Wang Y, Dixon R, Bowden S, Qiao M, Einck L, Locarnini S. The use of ampligen alone and in combination with ganciclovir and coumermycin A1 for the treatment of ducks congenitally-infected with duck hepatitis B virus. Antiviral Res. 1993;21:155–171. doi: 10.1016/0166-3542(93)90051-j. [DOI] [PubMed] [Google Scholar]
- Pinto AJ, Morahan PS, Brinton MA. Comparative study of various immunomodulators for macrophage and natural killer cell activation and antiviral efficacy against exotic RNA viruses. Int J Immunopharmacol. 1988;10:197–209. doi: 10.1016/0192-0561(88)90050-1. [DOI] [PubMed] [Google Scholar]
- Reed LJ, Muench CH. A simple method of estimating fifty percent endpoint. Am J Hyg. 1938;27:493–497. [Google Scholar]
- Schoneboom BA, Catlin KM, Marty AM, Grieder FB. Inflammation is a component of neurodegeneration in response to Venezuelan equine encephalitis virus infection in mice. J Neuroimmunol. 2000;109:132–146. doi: 10.1016/s0165-5728(00)00290-3. [DOI] [PubMed] [Google Scholar]
- Smee DF, Gilbert J, Leonhardt JA, Barnett BB, Huggins JH, Sidwell RW. Treatment of lethal Pichinde virus infections in weanling LVG/Lak hamsters with ribavirin, ribamidine, selenazofurin, and ampligen. Antiviral Res. 1993;20:57–70. doi: 10.1016/0166-3542(93)90059-r. [DOI] [PubMed] [Google Scholar]
- Spotts DR, Reich RM, Kalkhan MA, Kinney RM, Roehrig JT. Resistance to alpha/beta interferons correlates with the epizootic and virulence potential of Venezuelan equine encephalitis viruses and is determined by the 5′ noncoding region and glycoproteins. J Virol. 1998;72:10286–10291. doi: 10.1128/jvi.72.12.10286-10291.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele KE, Davis KJ, Stephan K, Kell W, Vogel P, Hart MK. Comparative neurovirulence and tissue tropism of wild-type and attenuated strains of Venezuelan equine encephalitis virus administered by aerosol in C3H/HeN and BALB/c mice. Vet Pathol. 1998;35:386–397. doi: 10.1177/030098589803500508. [DOI] [PubMed] [Google Scholar]
- Weaver SC, Ferro C, Barrera R, Boshell J, Navarro JC. Venezuelan equine encephalitis. Annu Rev Entomol. 2004;49:141–174. doi: 10.1146/annurev.ento.49.061802.123422. [DOI] [PubMed] [Google Scholar]
- Wesselingh SL, Levine B, Fox RJ, Choi S, Griffin DE. Intracerebral cytokine mRNA expression during fatal and nonfatal alphavirus encephalitis suggests a predominant type 2 T cell response. J Immunol. 1994;152:1289–1297. [PubMed] [Google Scholar]
- White LJ, Wang JG, Davis NL, Johnston RE. Role of alpha/beta interferon in Venezuelan equine encephalitis virus pathogenesis: effect of an attenuating mutation in the 5′ untranslated region. J Virol. 2001;75:3706–3718. doi: 10.1128/JVI.75.8.3706-3718.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]