

Abstract
Boronic acids have proved to be promising selective inhibitors of β-lactamases, acting as transition state analogues. Starting from a previously described nanomolar inhibitor of AmpC β-lactamase, three new inhibitors were designed to gain interactions with highly conserved residues, such as Asn343, and to bind more tightly to the enzyme. Among these, one was obtained by stereoselective synthesis and succeeded in placing its anionic group into the carboxylate binding site of the enzyme, as revealed by X-ray crystallography of the complex inhibitor/AmpC. Nevertheless, it failed at improving affinity, when compared to the lead from which it was derived. The origins of this structural and energetic discrepancy are discussed.
Introduction
Structure-based design is widely used to discover new enzyme inhibitors, often by mimicking interactions observed between the target enzyme and its natural substrates.1 Once a lead compound is identified, structure-based optimization can improve affinity on the basis of the structures of inhibition-enzyme complexes. When leads are substrate-analogues, these structures can also provide insight to enzyme mechanism and molecular recognition.
Great interest is focused on inhibition of serine-amidases, a class of enzymes that mediate several pathological conditions, such as thrombosis (thrombin, factor Xa, factor VIIa), inflammation and emphysema (elastase), hepatitis C (proteases involved in replication), and bacterial resistance against β-lactam antibiotics (β-lactamases).2 The structure of many of these enzymes has been exhaustively mapped and their mechanism of action carefully investigated. These studies have been advanced by inhibitors bearing an electrophilic centre (phosphonates, aldehydes, trifluoromethylketones and boronic acids)3 that can covalently modify the nucleophilic catalytic serine. Substrate-analogues bearing such electrophilic “warheads” have found extensive use as mechanistic probes4 and as leads for drug design.1
Among these classes, the boronic acids5 and the phosphonates6 have proven to be the most effective inhibitors of β-lactamases, with the former arguably having greater in-cell efficacies. β-lactamases are the most widespread resistance mechanism to the β-lactam class of antibiotics, such as the penicillins and cephalosporins. These enzymes catalyze the hydrolysis of the β-lactam bond in these antibiotics, rendering them inactive. In boronic acid inhibitors, the boron atom acts as an electrophile that mimics the carbonyl carbon of the β-lactam ring, and forms with the catalytic serine a tetrahedral adduct that closely resembles one of the transition states of the hydrolytic mechanism (Scheme 1, a–b).7
Scheme 1.
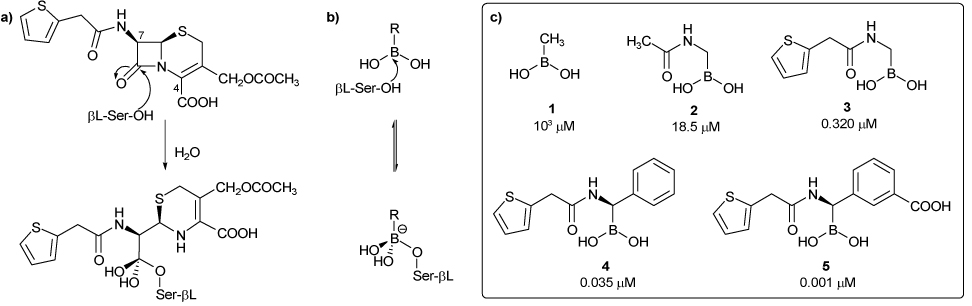
Mechanism of action of a β-lactamase (a) with a β-lactam (the antibiotic Cephalothin) and (b) with a boronic acid. (c) Growing mimesis of Cephalothin by boronic acids and their activity against the β-lactamase AmpC.
Our previous works in inhibition of the class C β-lactamase AmpC by boronic acids, based on a biomimetic approach, highlighted that the closer the boronic acid resembles the natural substrate in its interaction with the enzyme, the better its inhibition (Scheme 1c). Thus, as one moves from 1 to 5, a growing mimesis of the β-lactam cephalothin is displayed and higher inhibition towards the β-lactamase measured. In fact, if methaneboronic acid 1 (Ki 1000 µM) offers to the β-lactamase the sole interaction of the boron with the serine residue, compound 2 (Ki 30 µM), characterized by the presence of the acetamide moiety, gains the additional hydrogen bond with Asn152, Gln120 and Ala318, as also displayed by the amide at C7 of the natural substrate. A further improvement in inhibition is observed by insertion of more complex amide side chains on the boronic acid, and among these we selected the cephalothin one as a model, being a good compromise between complexity and inhibition (compound 3, Ki 0,32 µM).8 In addition, the stereo-controlled introduction of a phenyl group, mimicking the dihydrothiazine ring as well as the configuration at the C7 of cephalosporins led us to identify a hydrophobic binding pocket in the active site of AmpC β-lactamase, formed by Leu119 and Leu293, which accounts for 10 fold improvement in affinity (inhibitor 4, Ki 0.035 µM). Finally, the insertion of a m-carboxyphenyl moiety meant to gain the interaction of the carboxy group at C4 and further improved affinity led to the discovery of the most potent boronic inhibitor of AmpC β-lactamase ever tested (inhibitor 5, Ki 0,001 µM).9
A surprising feature of the crystal structure of 5 in complex with AmpC β-lactamase was the observation of a hydrogen bond between the carboxylate group of the inhibitor and that of the amide of Asn289. This was unexpected, because this Asparagine is not conserved among other members of the class C enzymes. In designing 5, we had hoped that the carboxylate would interact with the conserved site that in β-lactam complexes interacts with the C4 carboxylates of these substrates (Asn343, Asn346 and Thr316).10 Instead,11 in the AmpC/5 complex, these residues make no direct hydrogen bond with the inhibitor—the closest of them, Asn343, is 3.43 Å away from the inhibitor in the complexed structure. Thermodynamic cycle experiments confirmed that the Asn289 was critical to the recognition of compound 5—substitution of this residue with an Ala reduced affinity by 17-fold, and overall this interaction was found to contribute 2.1 kcal/mol to the overall binding affinity of 5. For many design projects, such accidental successes—interaction with a non-conserved and unintentional residues—would have occasioned little worry. Since β-lactamases are resistance enzymes, subject to rapid molecular evolution in the face of inhibitor pressure,12 we were concerned that this interaction could easily be overcome by mutation at Asn289, which is not necessary for AmpC function. Indeed, experiments with other members of the class C β-lactamase family that lack Asn289, such as the P99 β-lactamase from Enterobacter cloacae, suggested that such a substitution would reduce inhibitor affinity.
We wanted to find an analogue of 5 to exploit the carboxylate binding site implicated in substrate recognition, improving affinity and diminishing the ease of resistance substitutions. We reasoned that increasing the length of the anionic side chain would allow us to reach this sub-site. Here we describe the design, synthesis, biological and crystallographic evaluation of such inhibitors, resulting in the development of a new inhibitor of AmpC. This inhibitor succeeds in placing its anionic group into the carboxylate binding site of the enzyme, largely avoiding interaction with the mutable Asn289. Nevertheless, it fails at improved affinity compared to the lead from which it was derived. The origins of this structural and energetic discrepancy will be considered.
Results
Design
In the design of new boronic acids as β-lactamase inhibitors we chose to modify the lead structure of compound 5 following two main strategies. The first was to explore the effect on affinity of a different anionic group on the phenyl ring, replacing the carboxy moiety with a bioisosteric group such as the tetrazole (compound 6, Scheme 2), which should enhance the lipophilicity of the molecule and increase the distance of the anionic charge from the phenyl ring, allowing it to interact with more canonical anion recognition residues.
Scheme 2.
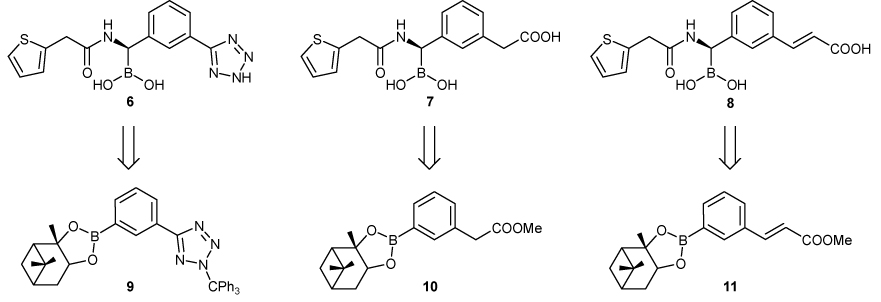
Newly designed potential inhibitors of β-lactamases and (+)-Pinanediol arylboronates respectively needed as their synthetic precursors.
The second was to insert different spacers between the carboxy group and the phenyl ring; this too would place the anionic moiety closer to the carboxylate sub-binding site, targeting its more conserved residues. To do so, we explored a methylene (compound 7) and a vinyl spacer (compound 8), distancing the anionic group from the phenyl ring by 2 and 4 Å respectively.
Molecular Modelling
Compounds 6–8 were designed and built in the active site of AmpC using Sybyl13 and Moloc.14 Each molecule was minimized by using the MAB force field allowing for flexibility only to protein residues within 7Å distance to the active site catalytic serine.15 During minimization the boron and Oγ of serine 64 were covalently linked. The process was driven by shape and polar complementarity between ligand and protein surface and the absence of unfavourable Van der Waals interactions. As shown in figure 1, these efforts suggested that compound 6, bearing the tetrazole moiety, would interact with Asn343, whereas the spacer-bearing compounds 7 and 8 would reach both the non-conserved residue Asn289 and the well conserved Asn343.
Figure 1.
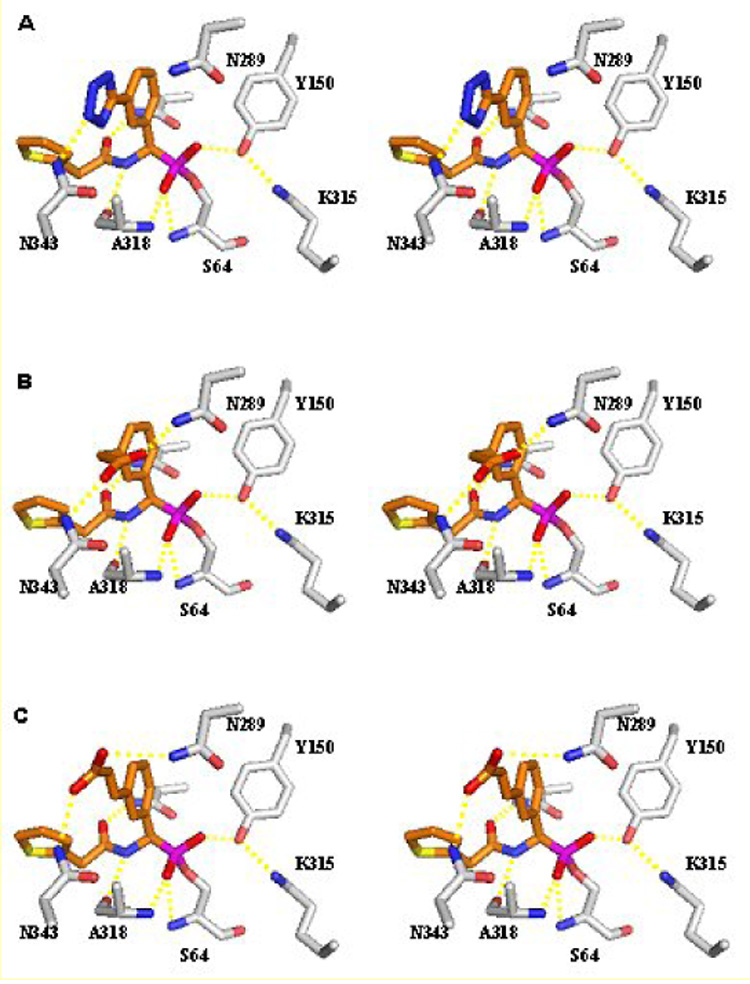
Stereo view of the interactions observed between AmpC and compounds 6–8 in the modelled conformation. (A) Compound 6. (B) Compound 7. (C) Compound 8. Carbon atoms are coloured grey for the protein and orange for the ligands; oxygen atoms, red; nitrogen atoms, blue; sulphur atoms, yellow; boron atoms, magenta. Atomic interactions within hydrogen bonding distance are shown as dashed yellow lines. This figure was generated with Pymol (htpp://pymol.sourceforge.net/).
Synthesis
As with inhibitor 5, all these molecules display a stereogenic carbon atom, which duplicates the stereochemistry on the correspondent C7 of cephalothin. In view of the general strategy developed by Matteson for the introduction of stereocentres α to the boron atom through highly stereoselective homologation of boronic esters, suitable arylboronates 9–11 were prepared from commercially available starting material as key intermediates for the synthesis of 6–8. (+)-Pinanediol was chosen as the chiral auxiliary as it is well known to induce the desired S configuration at the newly inserted chlorine-bearing carbon atom with very high diastereoselectivity (> 98%).16
(+)-Pinanediol 3-(2-trityltetrazol-5-yl)phenylboronate (9) was synthesized in two steps from 5-(3-bromophenyl)-2H-tetrazole. Protection of the tetrazole ring as a tritylate followed by boronation with n-butyllithium and trimethylborate at −78 °C and successive in situ transesterification with (+)-pinanediol afforded 9 in 48% overall yield.
(+)-Pinanediol (3-methoxycarbonylmethyl)phenylboronate (10) was obtained from pinacol (3-cyanomethylphenyl)boronate by treatment with gaseous hydrochloric acid at 0 °C in anhydrous diethyl ether followed by alcoholysis of the intermediate iminoester with methanol; final transesterification with (+)-pinanediol afforded 10 in 57% overall yield.17
(+)-Pinanediol (E)-3-(2-methoxycarbonylvinyl)phenylboronate (11) was synthesized by cross-coupling reaction of methyl trans-3-bromocinnamate with bispinacolate diboron, using (1,1’Bis-(diphenylphosphino)-ferrocene)-palladium dichloride [Pd(dppf)Cl2] as the catalyst in dry dimethylsulfoxide at 80 °C (59% yield, 65% conversion).18 The resulting pinacol ester was quantitatively converted to pinanediol ester by simple transesterification with (+)-pinanediol at rt.
Compounds 9–11 were then subjected to Matteson’s homologation protocol (Scheme 3).16 Pinanediol esters I were treated with in situ generated dichloromethyllithium from dichloromethane and n-butyllithium at −100 °C and the reaction mixture was warmed gradually to 0 °C. The presence of the α-chloroboronate II was investigated by means of 1H NMR analysis. Homologation resulted feasible for compounds 9 and 11: in fact, successful insertion of a chlorine-bearing carbon atom into the C–B bond was confirmed in the 1H NMR spectrum by the presence of a broad singlet at about 4.6 ppm. Compound 10, on the other hand, ended up in almost complete decomposition, probably due to the incompatibility of the benzylic enolizable protons of the methylcarboxylate moiety with strong nucleophiles such as dichloromethyllithium.
Scheme 3.
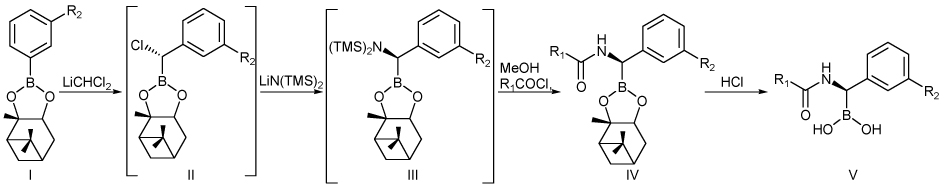
Stereoselective synthesis of chiral boronic acids.
It is worth to mention that although the homologation reaction generally proceeds with very high diastereoselection (> 98%), the benzylic stereocentre is prone to epimerization even for short exposure of the reaction mixture at rt.19 This annoying problem can be overcame never allowing the α-chloroboronate to reach temperatures over 0 °C,16c but treating in situ with lithiumhexamethyldisilylamide [LiN(TMS)2] at −78 °C. Substituted intermediate III was not isolated, but directly desilylated by treatment with methanol at 0 °C and immediately acylated at −78 °C with 2-thiophenacetyl chloride.
Compound 11 afforded the desired amide IV, whereas several attempts to pursue the same one-pot procedure with 9 only produced variable quantities of oxidation by-products, as the nucleophilic displacement at the halogenated carbon atom probably was most likely hampered by the steric hindrance of the trityl protective group, as also suggested by studies of molecular modelling. Final hydrolysis of both the chiral auxiliary and the methoxy protective group was accomplished by refluxing in 3N degassed hydrochloric acid to afford the free boronic inhibitor 8 in 25% yield
Enzymology and binding affinities
Almost all boronic acid inhibitors of AmpC β-lactamase previously studied are reversible, fast-on, fast-off, competitive inhibitors.20 The one important exception to this rule is compound 5, which has a time dependent component to it; this can, at least partially, be attributed to its low (1 nM) Kd value.21 Like compound 5, compound 8 had a time dependent component to its inhibition—we presume the origins of such were similar, reflecting the very high affinity of this inhibitor. Nevertheless, compound 8 remained reversible, and the inhibitor could be competed off by increasing substrate concentration. We have accounted for this time-dependent effect in the Ki value reported for this inhibitor (see Methods).
To investigate the effect of adding a spacer between the carboxylate group and the phenyl ring we determined the potency of compound 8. Compound 8 inhibited AmpC with a Ki of 1nM. While this inhibition is potent, it is no greater than that of the analogue compound 5 from which it was derived (Table 1). Comparison of 8 with compound 4 suggests that the m-vinylcarboxylate improves the binding energy by 2.1 kcal/mol, as did the m-carboxylic acid moiety of compound 5.23
Table 1.
Ki values of the cephalothin analogues boronic acids versus AmpC.
| Compound | Ki (µM) | ΔΔGb vs compound 3 (kcal/mol) |
|---|---|---|
| Cephalothin | 0.320a | 0 |
| 4 | 0.035 | 1.31 |
| 5 | 0.001 | 3.41 |
| 8 | 0.001 | 3.41 |
These Ki were determined in 50 mM KPi pH 7.0.7
The values in TRIS are typically two fold lower.
Differential free energy of binding relative to compound 1, calculated at 298 K. Positive values indicate improved affinity.
A key question was whether the affinity of compound 8 derived from the hydrogen bond to Asn289, as was the case for 5 (based on thermodynamic double-perturbation analyses),22 or whether the affinity derived through interactions with the conserved residues Asn343 or Asn346, as had been designed. To investigate this question, we measured the affinity of 8 versus the AmpC N289A mutant (Table 2). Whereas this substitution reduces affinity of compound 5 by 17-fold (1.7 kcal/mol), it led to only a 2.8-fold (0.6 kcal/mol) decrease in affinity for compound 8, consistent with the idea that this compound, even though it retains the affinity of the parent structure, no longer does so via an important hydrogen-bond with the non-conserved Asn289.
Table 2.
Ki values for compound 5 and 8 against various β-lactamases.
| Compound | Ki (nM) | |||
|---|---|---|---|---|
| AmpC | N289A | TEM-1 | TEM-30 | |
| 4 | 1 | 17 | 64 | 7800 |
| 7 | 1 | 2.8 | 106 | 3800 |
X-ray crystallographic structure determination
The crystal structure of AmpC in complex with 8 was determined at 2.0 Å resolution (Table 3, Fig. 1A, B). The inhibitor was unambiguously identified in the initial Fo - Fc difference map contoured at 3σ. Electron density connects the Oγ of the catalytic Ser64 to the boron atom of the inhibitor. The inhibitor was modelled at full occupancy in molecule B of the asymmetric unit, and at 0.6 occupancy in molecule A of the asymmetric unit. Excluding proline and glycine residues, 91.4% of the amino acids are in the most favoured regions of the Ramachandran plot (8.6% in the additionally allowed regions).23
Table 3.
Crystallographic summary for the complex AmpC
| AmpC/8 complex | |
|---|---|
| Cell constants (Å; °) | a=118.38 b=76.94 c=97.25 β=115.87 |
| Space group | C2 |
| Resolution (Å) | 2.00 |
| Unique reflections | 53061 |
| Total observation | 740048 |
| Rmerge (%) | 6.9 (49.2)a |
| Completeness (%) | 99.9 (99.4)a |
| <I>/<σI> | 18.5 (2.0)a |
| Resolution range for refinement (Å) | 20.0-2.00 (2.07-2.00) |
| Number of protein molecules | 716 |
| Number of water molecules | 482 |
| RMSD bond lengths (Å) | 0.013 |
| RMSD bond lengths (°) | 1.482 |
| Rcryst (%) | 17.6 |
| Rfree (%) | 21.9 |
| Average B-factor, protein atoms (Å2) | 27.2b |
| Average B-factor, inhibitor atoms (Å2) | 38.3b |
| Average B-factor, water atoms (Å2) | 34.5 |
Values in parentheses are for the highest resolution shell.
Values cited were calculated for both molecules in the asymmetric unit.
Overall, the AmpC/8 complex resembles that of AmpC/5 (Table 4). The boron geometry is tetrahedral, as expected, key hydrogen bond interactions are maintained and closely resemble those typically observed in β-lactamase structures with transition state analogues and with β-lactams. The major differences emerge in the point of substitution of this molecule, i.e. the vinylcarboxylate: as designed, the carboxylic acid group is observed to interact only with Nδ2 of Asn343, and is 3.3 Å from the side chain of Asn289.
Table 4.
Interactions in inhibitor bound and native AmpC β-lactamase
 | |||
|---|---|---|---|
| Interaction | Distance (Å) | ||
| AmpC/5a | AmpC/8b | Apob,c | |
| S64N-O12 | 3.1 | 3.1 | N.P.d |
| A318N-O12 | 2.7 | 2.7 | N.P. |
| A318O-O12 | 3.3 | 3.2 | N.P. |
| Y150OH-O13 | 2.7 | 2.5 | N.P. |
| Wat402-O12 | 2.8 | 2.7 | N.P. |
| Wat402-O13 | 3.0 | 2.8 | N.P. |
| Y150OH-K315Nξ | 2.9 | 2.9 | 2.5 |
| Y150OH-S64Oγ | 3.0 | 3.1 | 3.2 |
| Y150OH-K67Nξ | 3.3 | 3.2 | 3.1 |
| K67Nξ-A220O | 2.8 | 2.8 | 3.5 |
| K67Nξ-S64Oγ | 2.6 | 3.2 | 3.5 |
| Wat402-T316Oγ1 | 3.4 | 3.7 | 3.8 |
| Wat402-Wat403 | 2.6 | 2.9 | 2.9 |
| Wat403-N346Oδ1 | 2.7 | 2.6 | 2.7 |
| Wat403-R349Nη1 | 3.0 | 3.0 | 2.9 |
| A318O-N9 | 3.1 | 3.1 | N.P. |
| N152Nδ2-O8 | 2.8 | 2.9 | N.P. |
| Q120Nε2-O8 | 6.5 | 4.1 | N.P. |
| N152Oδ1-K67Nξ | 2.6 | 2.6 | 2.7 |
| N152Nδ2-Q120Oε1 | 7.1 | 3.2 | 3.0 |
| Wat181-O21 | 3.0 | N.P. | N.P. |
| Wat469-O22 | 3.1 | N.P. | N.P. |
| N343Nδ2-O22 | N.P. | 3.0 | N.P. |
| N289Nδ2-O21 | 2.9 | 3.3 | N.P. |
Distances are for monomer 1 of the asymmetric unit.
Distances are for monomer 2 of the asymmetric unit.
The Apo structures used as reference is PDB code 1KE420b
Not Present.
Discussion
Our goal was to obtain an inhibitor of AmpC β-lactamase that could interact with the carboxylate subsite formed by Asn343, Ans346 and Thr316, replacing the interaction with Asn289 experienced by compound 5. Our hope was that such a molecule would bind more tightly than the lead inhibitor 5, and would be less subject to resistance substitutions of the non-conserved Asn289. Compound 8 was the synthesized product of this design strategy. A key result of this study is that the design strategy was structurally successful–the x-ray crystal structure of the AmpC/8 complex reveals the new inhibitor to hydrogen-bond with Asn343, as called for by design. It is clear from studies of the mutant N289A enzyme that, in contrast with the lead inhibitor 5, the carboxylate of 8 no longer interacts significantly with Asn289, but instead is likely to owe its interaction energy to the new hydrogen bond with Asn343, also as designed. On the other hand, the affinity of 8 was no better than the lead compound 5. How can this apparent contradiction between a structurally successful design and lack of energetic improvement be reconciled? What lessons can we draw for future inhibitors of this antibiotic resistance enzyme?
The discrepancy between apparent structural “success” and energetic failure has two possible origins. First, the structure-based design may not have succeeded in every detail, and small discrepancies can foil energetic improvement. Second, the design may have been misplaced, with the canonical carboxylate site offering no real advantage over the fortuitous Asn289 interaction. We cannot discount either of these possibilities at this time. Comparing the overlap between the predicted and experimental structures (Figure 2C), it seems clear that the inhibitor is placed largely as designed. In the predicted structure, the carboxylate group interacts with Asn343, as well as Asn289. The distance between the carboxylate and Asn343 is 2.8 Å, and the distance between the carboxylate and Asn289 is 3.0 Å. In the experimental structure, conversely, the interaction between the carboxylate of 8 and Asn343 is 3.0 Å, and no interaction with Asn289 is observed. Thus, whereas overall the predicted and observed structures for the AmpC/8 complex correspond fairly well, the differences between them may be sufficient to erase any improvement in affinity.
Figure 2.
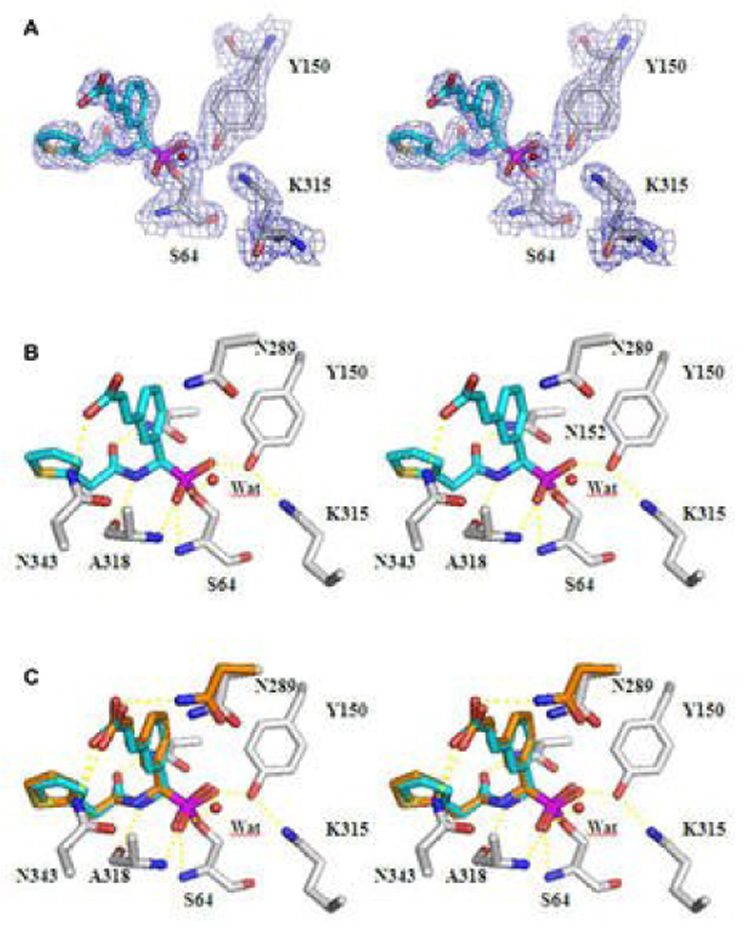
Stereo view of the Active Site Region of the AmpC/8 complex determined to 2.0 Å resolution. (A) The 2Fo – Fc electron density map is shown in blue, contoured at 1.0 σ. (B) Interactions observed between AmpC and compound 8 in the crystallographic complex. Carbon atoms are coloured grey for the protein and cyan for the ligand; oxygen atoms, red; nitrogen atoms, blue; sulphur atoms, yellow; boron atoms, magenta. Dashed yellow lines represent key hydrogen bonds. Red spheres represent water molecules. (C) Overlay of the modelled and crystallographic conformations of compound 8 in the AmpC site. Carbon atoms of compound 8 in the modeled conformation, orange; carbon atoms of compound 8 in the crystal structure, cyan. This figure was generated with Pymol (htpp://pymol.sourceforge.net/).
The possibility that the hydrogen-bond with Asn289 offers as much to the carboxylate as the canonical carboxylate site of the enzyme is also possible, though it seems harder to credit. Not only is Asn289 not conserved, but its interaction with the carboxylate of 5 occurs in a surface-exposed region. Asn343 is, by comparison, much more buried, forming a clear pocket along with Thr316 and Asn346 that typically accommodates substrate carboxylates. Of course, one might argue that as the ion-dipole interaction is buried, it will lose in solvation what it gains in interaction energy, and in some sense this must be true. Nevertheless, it seems surprising that one might gain so much from this interaction on the surface (the carboxylate with Asn289), and no more from an analogous interaction occurring in a pocket pre-formed over evolutionary time to accommodate it. Finally, it must be admitted that the vinyl carboxylate of 8 is not exactly equivalent to the direct carboxylate substituent of 5, and the former may pay entropic or strain energy costs in binding.24
Definitive answers to these questions are possible, but they must await further thermodynamic cycle analyses,22,25b involving site-directed mutagenesis and ligand derivatization. We remain convinced that the affinity in this series of inhibitors can be substantially improved by judicious choice of substitution, and the results of this study provide useful guidance in that direction. They also provide a cautionary lesson: even when computational design is structurally successful, with the appropriate ligand groups binding as predicted in subsequent experimental structures, it does not guarantee that affinity will improve. Predicting binding affinity changes remains challenging, with increases in interaction energies balanced by losses in solvation, entropic and strain energies. In this case, the balance was exact.
Experimental
Molecular modelling
The modelling was performed with SYBYL 6.8 and MAB/Moloc13–15 on a Linux workstation. Ligands were minimized in the pocket by using the MAB force field. During minimization the default parameters were applied and flexibility was allowed only for residues in 7 Å distance from the active serine.
Synthesis and Analysis
General
All reactions were performed under argon using oven-dried glassware and dry solvents. Anhydrous tetrahydrofuran (THF) and diethyl ether were prepared by standard methods and freshly distilled under argon from sodium benzophenone ketyl prior to use. Methanol was dried by storage upon 4 Ǻ molecular sieves. The −100 °C bath was prepared by addition of liquid nitrogen to a pre-cooled (−78 °C) mixture of ethanol/methanol (1:1). Degassed 3 N hydrochloric acid was prepared by diluting 37% hydrochloric acid with degassed water. All reactions were monitored by TLC on preloaded (0.25 mm) glass supported silica gel plates (Merk, Kieselgel 60, F254) and compounds were visualized by exposure to UV light and by the Hanessian’s cerium molybdate stain. Chromatographic purification of compounds was performed on silica gel (particle size 0.05–0.20 mm). Melting points were measured on a Büchi 510 apparatus. Optical rotations were recorded at 20 °C on a Perkin-Elmer 241 polarimeter and are expressed in 10−1 deg cm2 g−1. 1H and 13C NMR spectra were recorded on a Bruker DPX-200 spectrometer at 200 and 50 MHz, respectively; chemical shifts (δ) are reported in ppm downfield from TMS as internal standard. When peak multiplicities are given the following abbreviations are used: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broadened signal. Two-dimensional NMR techniques (Heteronuclear Single Quantum Coherence and Heteronuclear Multiple Bond Correlation) were used to aid signal assignment and were recorded on a Bruker Advance-400. Mass spectra were determined on a Finnigan MAT SSQ A mass spectrometer (EI, 70 eV). Elemental analyses were performed on a Carlo Erba Elemental Analyzer 1110.
The catalyst (1,1’Bis-(diphenylphosphino)-ferrocene)-palladiumdichloride [Pd(dppf)Cl2] was purchased from Sigma-Aldrich.
Synthesis of (+)-pinanediol 3-(2-trityltetrazol-5-yl)phenylboronate (9)
A solution of commercially available 5-(3-bromophenyl)-2H-tetrazole (1.07 g, 4.75 mmol) and Et3N (0.70 mL, 4.99 mmol) in THF (15 mL) was heated at 40 °C and trityl chloride (1.391 g, 4.99 mmol) in THF (6 mL) was added dropwise. A white abundant precipitate of ammonium salt formed immediately. After 30 min the solid was filtered off, the solvent was removed under reduced pressure and the resulting oil was crystallized from diethyl ether (20 mL) to afford 5-(3-bromophenyl)-2-trityltetrazole as a white solid (2.214 g, 100% yield). Mp 128–130 °C. 1H NMR (CDCl3): δ 7.10–7.50 (16H, m, H5, CPh3), 7.58 (1H, ddd, J = 8.0, 1.9, 1.1 Hz, H6), 8.10 (1H, ddd, J = 7.7, 1.7, 1.1 Hz, H4), 8.29 (1H, dt, J = 1.7, 0.3 Hz, H2),. 13C NMR (CDCl3): δ 122.8, 125.6, 127.8, 128.4, 129.9, 126.7, 130.3, 130.32, 133.2, 141.3. EI-MS: m/z 468 (M++2, <1%), 466 (M+, <1), 412 (<1), 410 (<1), 334 (<1), 332 (<1), 253 (1), 243 (100), 228 (5), 135 (38). Anal. Calcd for C26H19BrN4: C 66.82; H 4.10; N 11.99; Found: C 66.80; H 4.07; N 12.02.
In a 50 mL 3 necked flask 5-(3-bromophenyl)-2-trityltetrazole (0.972 g, 2.08 mmol) was dissolved in THF (20 mL) and cooled at −78 °C. n-Butyllithium (0.92 mL of a 2.5 M solution in hexanes, 2.29 mmol) was added dropwise over 10 min whereupon the solution turned red. The mixture was gradually allowed to reach −20 °C and stirred for 1 h, and a yellow solution with an abundant precipitate was obtained. The reaction was then re-cooled at −78 °C and a solution of freshly distilled trimethyl borate (0.234 mL, 2.08 mmol) in THF (1 mL) was added. After 1 h at −78 °C, the reaction mixture was treated with chlorotrimethylsilane (0.264 mL, 2.08 mmol) and the resulting clear solution was warmed to rt and stirred overnight. Finally, (+)-pinanediol (0.354 g, 2.08 mmol) was added at rt and the solution stirred for 1 h. The crude mixture was partitioned between water (20 mL) and ethyl acetate (100 mL) and the aqueous phase was extracted with ethyl acetate (2 × 50 mL). Combined organic layers were dried over MgSO4, filtered and concentrated in vacuo to give a white residue. Column chromatography using light petroleum/ethyl acetate (9:1) as the eluant afforded 9 as a white crystalline solid (0.560 g, 48% yield). Mp 185 °C. [α]D= +1.75 (αD = +0.031, c = 1.8%, CHCl3). 1H NMR (CDCl3): δ 0.91 (3H, s, pinanyl CH3), 1.23 (1H, d, J = 10.4 Hz, pinanyl Hendo), 1.33 (3H, s, pinanyl CH3), 1.51 (3H, s, pinanyl CH3), 1.80–2.60 (5H, m, pinanyl protons), 4.48 (1H, dd, J 8.5, 2.0 Hz, CHOB), 7.10–7.45 (15H, m, CPh3), 7.48 (1H, t, J = 7.6 Hz, H5), 7.91 (1H, dt, J = 7.6, 1.1 Hz, H6), 8.24 (1H, dt, J = 7.6, 1.1 Hz, H4), 8.59 (1H, s, H2). 13C NMR (CDCl3): δ 25.4, 27.9, 28.5, 30.1, 36.9, 39.6, 40.9, 52.8, 79.8, 87.8, 128.6, 129.1, 129.6, 131.2, 131.7, 134.7, 138.0, 142.8, 148.3, 165.5. C–B not seen. EI-MS: m/z 510 (M+−2N2, 100%), 132 (8), 357 (11), 330 (26), 280 (19), 253 (47), 243 (72), 178 (25), 165 (41), 135 (28), 107 (14), 93 (39). Anal. Calcd for C19H25BO4: C 69.53; H 7.68; Found: C 69.55; H 7.71.
Synthesis of (+)-pinanediol 3-(methoxycarbonylethyl)phenylboronate (10)
Commercially available pinacol 3-(cyanomethyl)phenylboronate (1.460 g, 6.0 mmol) was dissolved in anhydrous diethyl ether (4 mL) and dry methanol (0.29 mL, 7.18 mmol) was added. The solution was cooled at 0 °C and gaseous hydrochloric acid was bubbled. Upon storage in the refrigerator for 48 h the intermediate iminoester precipitated as a white solid and was recovered by quick filtration, washing with cooled diethyl ether (1.680 g, mp 92–96 °C). The crude solid was then stirred with methanol (2.5 mL) at rt for 24 h and subsequently diluted with diethyl ether, thus causing the precipitation of ammonium chloride. After filtration, the clear solution was concentrated in vacuo affording pinacol 3-(methoxycarbonylethyl)phenylboronate as a pale yellow oil which was used as such for the following step (1.242 g, 75% overall yield). 1H NMR (CDCl3): δ 1.35 (12H, s, CH3), 3.64 (2H, s, PhCH2), 3.69 (3H, s, OCH3), 7.25–7.47 (2H, m, H5,6), 7.66–7.78 (2H, m, H2,4). 13C NMR (CDCl3): δ 26.2, 42.4, 53.3, 85.2, 129.3, 133.5, 134.7, 134.9, 137.0, 173.4. C–B not seen. EI-MS: m/z 275 (M+−1, 14%), 260 (25), 215 (100), 200 (11), 190 (5), 176 (23), 131 (7), 117 (35), 91 (5), 59 (7). Anal. Calcd for C15H21BO4: C 65.24; H 7.67; Found: C 65.23; H 7.69.
The pinacol ester (1.242 g, 4.5 mmol) was dissolved in anhydrous THF (5 mL) and (+)-pinanediol (0.766 g, 4.5 mmol) was added. After stirring for 3 h at rt, the crude mixture was concentrated in vacuo and purified by column chromatography using light petroleum/ethyl acetate (7:3) as the eluant, affording 10 as a viscous oil (1.123 g, 76% yield). [α]D = +2.27 (αD = +0.031, c = 1.4%, CHCl3). 1H NMR (CDCl3): δ 0.89 (3H, s, pinanyl CH3), 1.22 (1H, d, J = 1.06 Hz, pinanyl Hendo), 1.30 (3H, s, pinanyl CH3), 1.48 (3H, s, pinanyl CH3), 1.80–2.61 (5H, m, pinanyl protons), 3.64 (2H, s, PhCH2), 3.68 (3H, s, OCH3), 4.45 (1H, dd, J = 8.8, 1.9 Hz, CHOB), 7.25–7.45 (2H, m, H5, H6), 7.65–7.80 (2H, m, H2, H4). 13C NMR (CDCl3): δ 25.4, 27.9, 28.5, 30.1, 36.9, 39.6, 40.9, 42.4, 52.8, 53.3, 79.7, 87.7, 129.4, 133.4, 134.7, 134.9, 137.0, 173.3. C–B not seen. EI-MS: m/z 328 (M+, 31%), 313 (28), 287 (20), 269 (33), 259 (100), 245 (30), 232 (82), 173 (20), 134 (19), 117 (57), 91 (41), 83 (52), 67 (48). Anal. Calcd for C19H25BO4: C 69.53; H 7.68; Found: C 69.55; H 7.71.
Synthesis of (+)-pinanediol 3-(2-methoxycarbonylvinyl)phenylboronate (11)
In a 3-necked flask, oven-dried potassium acetate (0.488 g, 4.98 mmol), bispinacolate diboron (0.463 g, 1.82 mmol) and commercially available trans-3-bromocinnamic acid (0.400 g, 1.66 mmol) were dissolved in anhydrous DMSO (5 mL). Pd(dppf)Cl2 catalyst (0.048 g, 0.066 mmol, 4%) was added to the solution, which turned red, and the mixture was heated at 80 °C for 1 h, thus leading to a black solution. As TLC analysis (light petroleum/diethyl ether 9:1) showed only incomplete conversion, additional catalyst (4%) was added and the mixture was heated for 1 h. The crude mixture was partitioned between water (50 mL) and ethyl acetate (200 mL) and the aqueous phase was extracted with ethyl acetate (2 × 100 mL). The organic layers were washed with brine, dried (MgSO4), filtered and concentrated in vacuo. The brown solid residue was purified by gradient column chromatography using light petroleum/diethyl ether (100% to 96:4) as the eluant and affording the desired coupling product pinacol 3-(2-methoxycarbonylvinyl)phenylboronate (0.280 g, 59% yield, 65% conversion). Attempts to scale-up the reaction, always resulted in poorer conversions. Mp 86 °C. 1H NMR (CDCl3): δ 1.36 (12H, s, CCH3), 3.80 (3H, s, COOCH3), 6.50 (1H, d, J = 16.0 Hz, Ph–CH=CH), 7.38 (1H, t, J = 7.4 Hz, H5 arom), 7.60 (1H, dt, J = 7.4, 1.2 Hz, H6 arom), 7.72 (1H, d, J = 16.0 Hz, Ph–CH=CH), 7.83 (1H, dt, J = 7.4, 1.2 Hz, H4 arom), 8.00 (1H, br, H2 arom). 13C NMR (CDCl3): δ 25.0, 51.5, 83.4, 117.9, 128.2, 130.7, 133.7, 134.4, 136.5, 144.8, 167.3. C–B not seen. EI-MS: m/z 288 (M+, 4%), 273 (2), 254 (2), 239 (17), 213 (2), 171 (4), 155 (6), 129 (5), 113 (6), 84 (100), 69 (20). Anal. Calcd for C16H21BO4: C 66.69; H 7.35; Found: C 66.71; H 7.38.
The pinacol ester (0.940 g, 3.26 mmol) was dissolved in anhydrous THF (3 mL), (+)-pinanediol (0.555 g, 3.26 mmol) was added and the mixture was stirred at rt. After 24 h the solvent was removed under reduced pressure and the crude yellow residue was purified on silica using light petroleum/ethyl acetate (8:2) as the eluant, thus affording 11 as a pale yellow oil which crystallizes on standing (1.111 g, 100% yield). Mp 113 °C, [α]D = −2.51 (αD = −0.024, c = 0.95%, CHCl3). 1H NMR (CDCl3): δ 0.91 (3H, s, pinanyl CH3), 1.22 (1H, d, J = 10.6 Hz, pinanyl Hendo), 1.33 (3H, s, pinanyl CH3), 1.50 (3H, s, pinanyl CH3), 1.90–2.55 (5H, m, pinanyl protons), 3.81 (3H, s, COOCH3), 4.48 (1H, dd, J = 8.6, 1.9 Hz, CHOB), 6.50 (1H, d, J = 16.0 Hz, Ph–CH=CH), 7.41 (1H, t, J = 7.4 Hz, H5 arom), 7.62 (1H, dt, J = 7.4, 1.2 Hz, H6 arom), 7.73 (1H, t, J = 16.1 Hz, Ph–CH=CH–COOCH3), 7.84 (1H, dt, J = 7.4, 1.2 Hz, H4 arom), 8.00 (1H, br, H2 arom). 13C NMR (CDCl3): δ 24.0, 26.5, 27.1, 28.7, 35.5, 38.2, 39.5, 51.4, 51.6, 78.4, 86.5, 117.9, 128.3, 130.7, 133.8, 134.4, 136.6, 144.8, 167.4. C–B not seen. EI-MS: m/z 340 (M+, 82%), 325 (30), 309 (14), 299 (22), 284 (26), 271 (100), 257 (27), 244 (67), 213 (16), 189 (15), 157 (32), 134 (45), 109 (21), 93 (22), 83 (66). Anal. Calcd for C20H25BO4: C 70.61; H 7.41; Found: C 70.60; H 7.38.
Synthesis of (1R)-1-(2-thiophen-2-yl-acetylamino)-1-(3-(2-carboxyvinyl)phenyl)methyl boronic acid (8)
A solution of dichloromethane (0.209 mL, 3.26 mmol) in THF (4 mL) was cooled at −100 °C and was treated with n-butyllithium (0.98 mL of a 2.5 M solution in hexanes, 2.45 mmol) under argon flow and mechanical stirring. After 30 min, a solution of 11 (0.694 g, 2.04 mmol) in THF (3 mL) was slowly added and the temperature was allowed to gradually reach 0 °C during 5 h. The clear solution became yellow and darkened upon warming. After stirring for 1 h at 0 °C, TLC analysis (light petroleum/ethyl acetate 8:2) showed complete conversion of the starting material to the α-chloro boronate. Hence the mixture was cooled at −78 °C and treated with lithium (hexamethyldisilyl)amide (2.24 mL of an 1 M solution in THF, 2.24 mmol) and allowed to warm to rt overnight. Dry methanol (0.980 mL of a 2.5 M solution in dry THF, 2.45 mmol) was then added at 0 °C and the resulting mixture was stirred for 1 h at 0 °C and at rt for one additional hour. Final acylation was accomplished at −78 °C by adding a solution of 2-thiophenacetylchloride (0.302 mL, 2.45 mmol) in THF (2 mL) and stirring at rt overnight. The crude mixture was partitioned between ethyl acetate (100 mL) and water (25 mL) and the acidic aqueous layer was extracted with ethyl acetate (2 × 50 mL). Combined organic phases were washed with satd. NaHCO3 (50 mL), dried over MgSO4, filtered and concentrated in vacuo. The resulting a brown oil was purified by column chromatography using diethyl ether as the eluant to afford (+)-pinanediol (1R)-1-(2-thiophen-2-ylacetylamino)-1-(3-(2-methoxycarbonylvinyl)phenyl)methylboronate as a pale yellow solid which was used for the final step (0.113 g, 11% yield). Mp 56 °C, [α]D = −1.27 (αD = −0.030, c = 1.18%, CHCl3). 1H NMR (CDCl3): δ 0.81 (3H, s, pinanyl CH3), 1.15 (1H, d, J = 10.1 Hz, pinanyl Hendo), 1.24 (3H, s, pinanyl CH3), 1.35 (3H, s, pinanyl CH3), 1.50–2.30 (5H, m; pinanyl protons), 3.81 (3H, s, COOCH3), 3.99 (2H, br, CH2CONH), 4.08 (1H, br, CHB), 4.21 (1H, dd, J = 8.5, 2.0 Hz, CHOB), 6.38 (1H, d, J = 16.0 Hz, Ph–CH=CH), 6.75 (1H, br, CONH), 6.96–7.08 (2H, m, C=CH–CH), 7.17 (1H, dt, J = 6.5, 1.9 Hz, H6 arom), 7.23–7.39 (4H, m, H2,4,5 arom, S–CH=CH), 7.63 (1H, d, J = 16.0 Hz, Ph–CH=CH). 13C NMR (CDCl3): δ 25.4, 27.7, 28.6, 30.0, 35.8, 37.4, 39.5, 41.1, 48.7 (br, C–B), 53.0, 53.4, 78.7, 86.3, 119.2, 127.0, 127.5, 127.7, 129.1, 129.6, 129.7, 130.3, 135.2, 135.8, 142.5, 146.3, 168.8, 175.4. EI-MS: m/z 493 (M+, 59%), 462 (2), 424 (2), 395 (2), 358 (4), 341 (13), 314 (7), 297 (8), 257 (40), 234 (12), 216 (8), 190 (19), 175 (13), 158 (7), 135 (15), 124 (24), 97 (100), 93 (36). Anal. Calcd for C27H32BNO5S: C 65.72; H 6.54; N 2.84; S 6.50; Found: C 65.70; H 6.56; N 2.87; S 6.55.
The (+)-pinanediol boronic ester (0.113 g, 0.23 mmol) was refluxed for 1 h in 3 N degassed hydrochloric acid (5 mL). The solution was extracted with diethyl ether (20 mL) and the aqueous layer was concentrated under reduced pressure to afford 0.030 g of a whitish solid residue. Insoluble impurities were precipitated with methanol and filtered off. The resulting clear solution was evaporated to dryness to give a pale yellow film which solidified into a white solid (25 mg) upon treatment with a single drop of water. 1H NMR analysis confirmed the presence of the desired product (25% yield) along with a deboronated by-product as impurity (22%), which displayed a doublet at 3.8 ppm corresponding to benzylic protons. The two products were chromatographically inseparable under all conditions tested. Under milder conditions and shorter reaction times removal of both protective groups failed. Mp 240 °C (dec.), [α]D = −62.7 (αD = −0.320, c = 0.51%, MeOH, corrected). 1H NMR (CD3OD): δ 3.84 (1H, s, CHB), 4.16 (2H, s, CH2CONH), 6.39 (1H, d, J = 16.0 Hz, Ph–CH=CH), 6.85–7.80 (8H, m, Ph–CH=CH, H2,4–6 arom, S–CH=CH–CH–C). 13C NMR (CD3OD): δ 32.1, 52.2 (br, C–B), 119.3, 126.1, 126.7, 127.1, 128.4, 129.1, 129.2, 129.8, 134.6, 135.6, 143.2, 146.4, 176.7, 179.2.
Enzymology
Inhibitors were initially dissolved in DMSO at a concentration of 50 mM; more dilute stocks (10 mM to 1 µM) were subsequently prepared as necessary. Kinetic measurements were performed in 50 mM Tris buffer, pH 7.0 and monitored in an HP8453 UV/Vis spectrophotometer. The concentration of each enzyme was determined spectrophotometrically; the enzyme was expressed and purified as described.26 For AmpC, 0.5 nM enzyme was used with 200 µM nitrocefin (Km 127 µM) or 50 µM centa (Km 19 µM) as initial substrate concentrations. For N289A, 1 nM enzyme was used with 400 µM nitrocefin (Km 27 µM) as initial substrate concentration. The hydrolysis of nitrocefin was monitored at 480 nm, and that of centa at 405 nm. To determine Ki values, inhibitor and enzyme were incubated together at their final concentration for five minutes before the reaction was initiated by the addition of substrate. The Ki values for compound 8 were obtained by comparison of progress curves in the presence and absence of inhibitor, using the method described by Waley27 which has been widely used for boronic acid inhibitors of β-lactamases. In these analyses, sufficient inhibitor was used to give at least 50% inhibition; the Ki values reported are the average calculated from reactions at five different inhibitor concentrations, each of which was repeated three times. In all reactions, rates were measured after reactions had overcome their initial lag phase and had reached a steady state.
Crystal growth and structure determination
Co-crystals of AmpC in complex with compounds 8 were grown by vapour diffusion in hanging drops equilibrated over 1.7 M potassium phosphate buffer (pH 8.7) using micro-seeding techniques. The initial concentration of the protein in the drop was 2.8 mg/ml, and the concentration of compounds 8 was 588 µM. The compound was added to the crystallization drops in a 1.2% DMSO, 1 M potassium phosphate buffer (pH 8.7) solution. Crystals appeared in 7–10 days after equilibration at 21 °C. Before data collection, crystals were immersed in a cryoprotectant solution of 20% sucrose, 1.7 M potassium phosphate, pH 8.7, for about 30 seconds, and flash cooled in liquid nitrogen. Diffraction data were collected on frozen crystals at the Advance Light Source (ALS, Lawrence Berkley Lab, CA). The data set was measured from single crystal. Reflections were indexed, integrated, and scaled using the HKL software package.28 The space group was C2, with two molecules in the asymmetric unit, each containing 358 residues. The initial phasing model was an AmpC apo structure (PDB entry 1KE4), with water molecules and ions removed. The model was positioned by rigid body refinement and its position further refined using the maximum likelihood target in CNS29 including simulated annealing, positional minimization, and individual B-factor refinement, with a bulk solvent correction. Sigma A-weighted electron density maps were calculated using CNS, with the program Coot used for manual model rebuilding and placement of water molecules.30 The inhibitors were built into the 2|Fo|–|Fc| and |Fo|–|Fc| difference density maps in each active site of the asymmetric unit. Subsequent refinement consisted of positional minimization and B-factor refinement in CNS.
Data Deposition
Atomic coordinates and structure factors have been deposited in the Protein Data Bank under PDB ID code 2RCX for AmpC in complex with compound 8.
Acknowledgements
Supported by GM63815 (to BKS and FP). We thank Dr. Paolo Davoli for reading this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Babine RE, Bender SL. Chem. Rev. 1997;97:1359–1472. doi: 10.1021/cr960370z. [DOI] [PubMed] [Google Scholar]
- 2.Yang W, Gao X, Wang B. Cap 13. In: Hall DG, editor. Boronic Acids. Preparation, applications in organic synthesis and medicine. Wiley-VCH Verlag GmbH & Co.; 2005. p. 481. [Google Scholar]
- 3.Pechenov A, Stefanova ME, Nicholas RA, Peddi S, Gutheil WG. Biochemistry. 2003;42:579–588. doi: 10.1021/bi026726k. [DOI] [PubMed] [Google Scholar]
- 4.a) Pongdee R, Liu H-W. Bioorganic Chemistry. 2004;32:393–437. doi: 10.1016/j.bioorg.2004.06.012. [DOI] [PubMed] [Google Scholar]; b) Fromm HJ. Methods in Enzymology. 1995;249:123–143. doi: 10.1016/0076-6879(95)49033-7. (Enzyme Kinetics and Mechanism, Part D) [DOI] [PubMed] [Google Scholar]
- 5.Ness S, Martin R, Kindler AM, Paetzel M, Gold M, Jensen SE, Jones JB, Strynadka NJC. Biochemistry. 2000;39:5312–5321. doi: 10.1021/bi992505b. [DOI] [PubMed] [Google Scholar]
- 6.Silvaggi NR, Anderson JW, Brinsmade SR, Pratt RF, Kelly JA. Biochemistry. 2003;42:1199–1208. doi: 10.1021/bi0268955. [DOI] [PubMed] [Google Scholar]
- 7.Powers RA, Caselli E, Focia PJ, Prati F, Shoichet BK. Biochemistry. 2001;40:9207–9014. doi: 10.1021/bi0109358. [DOI] [PubMed] [Google Scholar]
- 8.Caselli E, Powers RA, Blasczcak LC, Wu CY, Prati F, Shoichet BK. Chem. Biol. 2001;8:17–31. doi: 10.1016/s1074-5521(00)00052-1. [DOI] [PubMed] [Google Scholar]
- 9.Morandi F, Caselli E, Morandi S, Focia PJ, Blasquez J, Stoichet BK, Prati F. J. Am. Chem. Soc. 2003;125:685–695. doi: 10.1021/ja0288338. [DOI] [PubMed] [Google Scholar]
- 10.a) Beadle BM, Trehan I, Focia PJ, Shoichet BK. Structure. 2002;10:413–424. doi: 10.1016/s0969-2126(02)00725-6. [DOI] [PubMed] [Google Scholar]; b) Patera A, Blaszczak LC, Shoichet BK. J. Am. Chem. Soc. 2000;122:10504–10512. [Google Scholar]
- 11.Lobkovsky E, Billings EM, Moews PC, Rahil J, Pratt RF, Knox JR. Biochemistry. 1994;33:6762–6777. doi: 10.1021/bi00188a004. [DOI] [PubMed] [Google Scholar]
- 12.a) Rossolini GM, Docquier J-D. Future Microbiology. 2006;1:295–308. doi: 10.2217/17460913.1.3.295. [DOI] [PubMed] [Google Scholar]; b) Weinreich DM, Delaney NF, DePristo MA, Hartl DL. Science. 2006;312:111–114. doi: 10.1126/science.1123539. [DOI] [PubMed] [Google Scholar]
- 13.SYBYL. 6.8 Ed. St. Louis, MO: Tripos, Inc.; [Google Scholar]
- 14.Moloc. 01/05/08 Ed. Amden, Switzerland: Gerber Molecular Design; [Google Scholar]
- 15.Gerber PR, Muller K. J. Comput.-Aided Mol. Des. 1995;9:251–268. doi: 10.1007/BF00124456. [DOI] [PubMed] [Google Scholar]
- 16.a) Matteson DS, Ray R, Rocks RR, Tsai DJS. Organometallics. 1983;2:1536–1543. [Google Scholar]; b) Matteson DS. Acc. Chem. Res. 1988;21:294–300. [Google Scholar]; c) Matteson DS. Chem. Rev. 1989;89:1535–1551. [Google Scholar]; d) Matteson DS. J. Organomet. Chem. 1999;581:51–65. [Google Scholar]
- 17.a) Hunter MJ, Ludwig ML. J. Am. Chem. Soc. 1962;84:3491–3504. [Google Scholar]; b) McElvain SM, Stevens CL. J. Am. Chem. Soc. 1947;69:2663–2666. doi: 10.1021/ja01203a030. [DOI] [PubMed] [Google Scholar]; c) Guzman-Duran A, Guzman E, Pannell KH, Lloyd WD. Synth. Comm. 2003;33:3271–3283. [Google Scholar]
- 18.a) Ishiyama T, Murata M, Miyaura N. J. Org. Chem. 1995;60:7508–7510. [Google Scholar]; b) Zhu L, Duquette J, Zhang M. J. Org. Chem. 2003;68:3729–3732. doi: 10.1021/jo0269114. [DOI] [PubMed] [Google Scholar]; c) Nising CF, Shmid UK, Nieger M, Brase S. J. Org. Chem. 2004;69:6830–6833. doi: 10.1021/jo0490393. [DOI] [PubMed] [Google Scholar]
- 19.Matteson DS, Erdik E. Organometallics. 1983;2:1083–1088. [Google Scholar]
- 20.a) Weston GS, Blazquez J, Baquero F, Shoichet BK. J. Med. Chem. 1998;41:4577–4586. doi: 10.1021/jm980343w. [DOI] [PubMed] [Google Scholar]; b) Powers RA, Blazquez J, Weston GS, Morosini MI, Baquero F, Shoichet BK. Protein Sci. 1999;8:2330–2337. doi: 10.1110/ps.8.11.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrari S, Costi MP, Wade RC. Chem. & Biol. 2003;10:1183–1193. doi: 10.1016/j.chembiol.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 22.Roth TA, Minasov G, Morandi S, Prati F, Shoichet BK. Biochemistry. 2003;42:14483–14491. doi: 10.1021/bi035054a. [DOI] [PubMed] [Google Scholar]
- 23.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 24.Tirado-Rives J, Jorgensen WL. J. Med. Chem. 2006;49:5880–5884. doi: 10.1021/jm060763i. [DOI] [PubMed] [Google Scholar]
- 25.a) Fersht AR, Shi JP, Knill-Jones J, Lowe DM, Wilkinson AJ, Blow DM, Brick P, Carter P, Waye MMY, Winter G. Nature. 1985;314:235–238. doi: 10.1038/314235a0. [DOI] [PubMed] [Google Scholar]; b) Ranganathan R, Lewis JH, MacKinnon R. Neuron. 1996;16:131–139. doi: 10.1016/s0896-6273(00)80030-6. [DOI] [PubMed] [Google Scholar]
- 26.Usher KC, Blaszczak LC, Weston GS, Shoichet BK, Remington SJ. Biochemistry. 1998;37:16082–16092. doi: 10.1021/bi981210f. [DOI] [PubMed] [Google Scholar]
- 27.Waley SG. Biochem. J. 1982;205:631–633. doi: 10.1042/bj2050631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otwinowski Z, Minor W. Methods in Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 29.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallographica, Section D: Biological Crystallography. 1998 Sep 1;D54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 30.Emsley P, Cowtan K. Acta Crystallographica, Section D: Biological Crystallography. 2004;D60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]