

Catechol dioxygenases catalyze the opening of an aromatic ring and incorporation of both atoms of a molecule of dioxygen (reviewed by ref. 1; see Scheme 1). Initial deprotonation of a ring hydroxyl facilitates oxidation to a semiquinone-type state and loss of aromatic stabilization of the ring structure, but O2 also requires activation for the reaction to occur. Although the overall reaction is thermodynamically favorable, it is kinetically impeded by O2's spin triplet nature in the ground state, because this spin is not conserved in the reaction's singlet products (2). Thus, the catalyzed reaction must at least include mechanisms for recombination of electron spins, deprotonation of catechol, electron transfer from catecholate and to O2, and the chemical steps that follow. The early functions and proximate binding of O2 and catecholate can be performed by appropriate redox-active metal ions, so it is natural to focus on the metal ion when one is present. However, each of the early requirements can also be met by other means. In this issue of PNAS, Emerson et al. (3) use a pair of catechol dioxygenase enzymes to remind us of the versatility and variety of biochemical mechanisms. They show that despite the compelling elegance and many capabilities of redox-active metal ions as hammers, not all reactions are nails. Specifically, they argue that the Mn2+ of homoprotocatechuate 2,3-dioxygenase from Arthrobacter globiformis (MndD) and the Fe2+ of homoprotocatechuate 2,3-dioxygenase from Brevibacterium fuscum (Fe-HPCD) mediate reduction of O2 by bound catecholate without themselves undergoing formal oxidation.
Scheme 1.

Overall reaction catalyzed by MndD and Fe-HPCD. An H+ is also released.
The Fe2+-dependent catechol dioxygenases (CdOs) have Fe-binding sites that fall within a large and diverse host of so-called “two histidine, one carboxylate facial triad” (2H1E) enzymes in which Fe2+ is bound by one Glu/Asp and two His side chains on adjacent apices of the coordination octahedron, leaving three labile coordination sites for bidentate binding of a catecholate adjacent to dioxygen (4). This arrangement is very attractive from a chemical standpoint because it positions dioxygen close to both of the catecholate positions that are activated by binding to Fe (Fig. 1). The protein structural context of the active site varies, and it appears that 2H1E-triad sites have evolved on multiple independent occasions by convergent evolution (1).
Fig. 1.
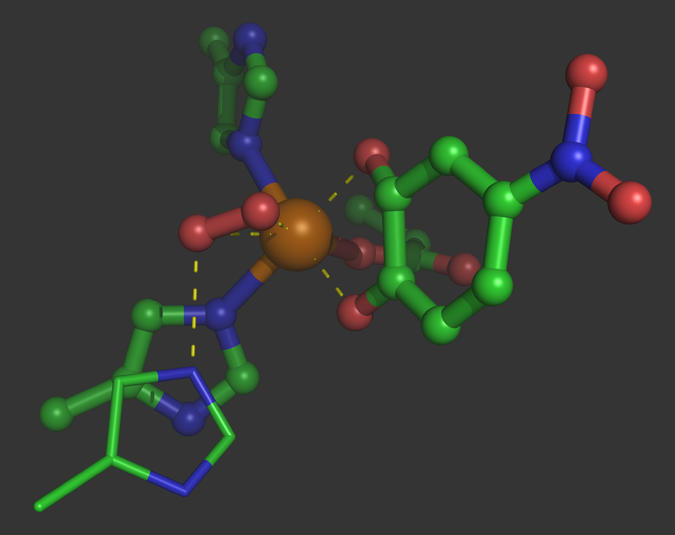
X-ray crystal structure of ternary complex between Fe-HPCD, O2, and 4-nitrocatechol, based on the structure of Kovaleva et al. (7), PDB ID code 2IGA chain C. O2 appears bound in a side-on conformation with an H bond from the conserved His200. 4-Nitrocatechol coordinates trans to the two His ligands. Also see ref. 11. This figure was generated by using PyMOL (12).
Catechol dioxygenation is thermodynamically favorable, and the catalyst's job may be simply to free the cork from the bottle. Once the reactive driving force of O2 is loosed, all manner of chemistry can follow spontaneously. The cork is the kinetic barrier posed by O2's spin triplet nature. Many enzymes overcome the spin barrier by reducing O2 to superoxide, whose single unpaired spin can combine with another unpaired spin or that of the metal ion (5, 6).
Fe-HPCD displays a superoxo intermediate, but in this case its formation is clearly accompanied by oxidation of the catecholate (7), arguing against oxidation of the metal ion. This has moreover been supported computationally (8). The Emerson article provides additional proof that redox activity on the part of the Fe is not required, by replacing Fe with Mn.
The teams of Lipscomb, Que, and Wackett over the years have built a body of evidence supporting the strong analogy between the Fe2+-containing CdO Fe-HPCD and a rogue CdO that employs Mn2+ instead. The two share strong structural homology (9) and very similar catalytic constants [see the work of Emerson et al. (3)]. So much has also been shown for the native Fe-SOD and Mn-SOD of Escherichia coli (reviewed in ref. 10). However, that is where the similarity ends. The two superoxide dismutases (SODs) apply very different redox tuning to the bound metal ion, thus compensating for the many hundreds of mV difference between the intrinsic reduction midpoint potential of Mn3+/2+ (adding one electron to an empty d-orbital) vs. that of high-spin Fe3+/2+ (adding an electron to an occupied d-orbital). Thus, the SOD proteins, with their greater number of protein ligands and their crucial coordinated solvent molecule that couples proton transfer to the redox event, achieve/ very similar reduction midpoint potentials in FeSOD and MnSOD. However, Fe-substituted-MnSOD and Mn-substituted-FeSOD are virtually inactive under standard conditions.
In the CdOs described by Emerson et al. (3), any differences in the redox tuning applied by the Fe-HPCD and MndT proteins does not appear to be crucial to activity, because both proteins achieve similar activity with either metal ion. The bound metal ions appear to retain much of their different redox characters, in each of the two protein contexts, as the authors find that the Fe is more prone to oxidation (lower Em) than the Mn (although the signature of some differential tuning is revealed by Fe-MndD's greater susceptibility than Fe-HPCD's). This is not unexpected. However, what is new is that even when Fe2+ is bound in the protein evolved to use Mn2+, it retains full activity, and similarly for Mn2+ bound to HPCD protein. Thus, even when any evolved protein-derived redox tuning should in fact reinforce intrinsic differences between Fe and Mn, activity is unaffected. Emerson et al. conclude that the Fe2+ and Mn2+ in MndD or HPCD do not need to assume the 3+ oxidation state in the course of catalysis and that they may convey a reducing equivalent between catechol and O2 without acquiring it. Nonetheless, it is interesting to ponder why the Fe3+ state of MndD is much less active than the Fe2+ version, if redox activity is not essential, and given that Fe3+ has the same d5 electronic configuration as Mn2+, which does support activity. This beautifully matched pair of enzymes with the same reaction but different native metal ions may have more to teach us still.
Emerson et al.'s (3) combination of spectroscopy, enzymology and x-ray crystallography shows that O2 activation need not depend on the metal ion's redox activity. This flies in the face of the metal-centered universe of chemistry many bioinorganic chemists imagine, but the metal is likely still important for spin reconciliation and transmission of electron density between catecholate and O2. Binding to the metal ion also stabilizes deprotonated states of the catechol and positions catechol and O2 adjacent to one another, but Emerson et al. present a strong and fascinating argument that a discrete oxidized Fe or Mn intermediate is not needed for reaction. It is the catecholate, not the metal ion, that reduces O2. The metal ion is not oxidized, but rather reduced, as it were, to a wire.
Footnotes
The author declares no conflict of interest.
See companion article on page 7347.
References
- 1.Vaillancourt FH, Bolin JT, Eltis LD. The ins and outs of ring-cleaving dioxygenases. Crit Rev Biochem Mol Biol. 2006;41:241–267. doi: 10.1080/10409230600817422. [DOI] [PubMed] [Google Scholar]
- 2.Pau MYM, Lipscomb JD, Solomon E. Substrate activation for O-2 reactions by oxidized metal centers in biology. Proc Natl Acad Sci USA. 2007;104:18355–18362. doi: 10.1073/pnas.0704191104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Emerson JP, Kovaleva EG, Farquhar ER, Lipscomb JD, Que L., Jr Swapping metals in Fe- and Mn-dependent dioxygenases. Evidence for oxygen activation without a change in metal redox state. Proc Natl Acad Sci USA. 2008;105:7347–7352. doi: 10.1073/pnas.0711179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koehntop KD, Emerson JP, Que L., Jr The 2-His-1-carboxylate facial triad: a versatile platform for dioxygen activation by mononuclear non-heme iron(II) enzymes. J Biol Inorg Chem. 2005;10:87–93. doi: 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- 5.Burger RN, Horwitz SB, Peisach J, Wittenberg JB. Oxygenated Bleomycin. A short-lived intermediate in the reaction of ferrous bleomycin with O2. J Biol Chem. 1979;254:12299–12302. [PubMed] [Google Scholar]
- 6.Xing G, et al. Evidence for C-H cleavage by an iron-superoxide complex in the glycol cleavage reaction catalyzed by myo-inositol oxygenase. Proc Natl Acad Sci USA. 2006;103:6130–6135. doi: 10.1073/pnas.0508473103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kovaleva EG, Lipscomb JD. Crystal structures of Fe2+ dioxygenase superoxo, alkylperoxo, and bound product intermediates. Science. 2007;316:453–457. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Georgiev V, Borowski T, Siegbahn PEM. Theoretical study of the catalytic reaction mechanism of MndD. J Biol Inorg Chem. 2006;11:571–585. doi: 10.1007/s00775-006-0106-9. [DOI] [PubMed] [Google Scholar]
- 9.Vetting MW, Wackett LP, Que L, Jr, Lipscomb JD, Ohlendorf DH. Crystallographic comparison of manga-nese- and iron-dependent homoprotocatechuate 2,3 dioxygenases. J Bacteriol. 2004;186:1945–1958. doi: 10.1128/JB.186.7.1945-1958.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller A-F. Redox tuning over almost 1 V in a structurally conserved active site: Lessons from Fe-containing superoxide dismutase. Acc Chem Res. 2008;41:501–510. doi: 10.1021/ar700237u. [DOI] [PubMed] [Google Scholar]
- 11.Kovaleva EG, Neibergall MB, Chakrabarty S, Lipscomb JD. Finding intermediates in the O2 activation pathways of non-heme iron oxygenases. Acc. Chem. Res. 2007;40:475–483. doi: 10.1021/ar700052v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeLano WL. Palo Alto, CA: DeLano Scientific; 2002. [Accessed April 28, 2008]. The PyMOL Molecular Graphics System. Available at: http://www.pymol.org. [Google Scholar]