

Abstract
Oxidative stress-induced cell death plays a major role in the progression of ischemic acute renal failure. Using microarrays, we sought to identify a stress-induced gene that may be a therapeutic candidate. Human proximal tubule (HK2) cells were treated with hydrogen peroxide (H2O2) and RNA was applied to an Affymetrix gene chip. Five genes were markedly induced in a parallel time-dependent manner by cluster analysis, including activating transcription factor 3 (ATF3), p21WAF1/CiP1 (p21), CHOP/GADD153, dual-specificity protein phosphatase, and heme oxygenase-1. H2O2 rapidly induced ATF3 approximately 12-fold in HK2 cells and approximately 6.5-fold in a mouse model of renal ischemia-reperfusion injury. Adenovirus-mediated expression of ATF3 protected HK2 cells against H2O2-induced cell death, and this was associated with a decrease of p53 mRNA and an increase of p21 mRNA. Moreover, when ATF3 was overexpressed in mice via adenovirus-mediated gene transfer, ischemia-reperfusion injury was reduced. In conclusion, ATF3 plays a protective role in renal ischemia-reperfusion injury and the mechanism of the protection may involve suppression of p53 and induction of p21.
Acute renal failure (ARF) is associated with high morbidity and mortality rates, although it was recently reported that in-hospital mortality has declined.1,2 Ischemia reperfusion injury (IRI), a common cause of ARF, occurs during kidney transplantation,2 cardiopulmonary bypass,3 aortic bypass surgery, and sepsis.4,5 There are a number of potential mediators of renal IRI, which are characterized as abnormal vascular reactivity, tubular obstruction, epithelial cell dysfunction, necrosis, and apoptosis. It is generally believed that reactive oxygen species are the key mediators of the renal IRI.
Many researchers are keenly interested in identifying key genes involved in renal IRI. To achieve this goal, we used DNA microarrays to elucidate candidate IRI genes that could have therapeutic potential. DNA microarray technology is a powerful tool for therapeutic discovery, optimization, and clinical validation.6 Several studies have exploited the high-throughput capability of DNA microarrays to obtain a molecular profile of oxidative stress in a variety of organs. We previously reported that the renal ischemia reperfusion animal model induces many sets of genes including activating transcription factor 3 (ATF3),7,8 which could represent a therapeutic target.
ATF3 is a member of the mammalian activation transcription factor/cAMP responsive element-binding protein family of transcription factors.9,10 ATF3 represses rather than activates transcription from promoters with ATF binding elements. In animal models, ATF3 is induced by myocardial, renal, and liver ischemia reperfusion, by chemicals, and in the skin by wounding. In cultured cells, ATF3 is induced by a variety of signals including cytokines, genotoxic agents, and cytotoxic agents. It has been reported that the induction of ATF3 was almost completely inhibited by N-acetyl-L-cysteine, an oxidant scavenger, suggesting that ATF3 is induced by oxidative stress.11 However the role of ATF3 in renal ischemia reperfusion injury remains unclear.
Here we report that ATF3 is rapidly induced in human kidney cells in response to oxidative stress and may protect the cells. Overexpression of ATF3 in mice via adenovirus-mediated gene transfer ameliorated IRI, significantly decreasing creatinine and improving the pathologic score for acute tubular injury.
Results
Microarray Analysis
Human kidney-2 cells (HK2 cells: human renal proximal tubular epithelial cells) were stimulated with 100 μM H2O2 for 15 min, 1, 4, and 16 h. After the indicated time, mRNAs were extracted and DNA microarray analysis was performed. Differential gene expression profiles after H2O2 stimulation were clustered into groups of coregulated genes as shown in Figure 1A. The changes were analyzed using the manufacture's software and fold changes were indicated by “affy-fold”. Among the groups clustered by expression, one group contained seven highly induced genes (Figure 2B) that included p21WAF1/CiP1 (p21), ATF3, CHOP/GADD153 (CHOP), dual-specificity protein phosphatase (DSP), heme oxygenase-1 (HO1), ID1, and Period. Using real-time reverse transcription (RT)-PCR we confirmed that five of these genes, namely p21, ATF3, CHOP, DSP, and HO1, were significantly upregulated in a time-dependent (Figure 2A) and dose-dependent (Figure 2B) manner. The time courses and dose-dependence for induction of these five genes were comparable (Figure 2B). Other gene clusters of up- and downregulated genes, including expressed sequence tags (ESTs) (AA149307, AA522537, etc.) or orphan genes (AB020713, etc.), were unremarkable in terms of either fold changes or known roles in cell injury and repair, and thus were not pursued further.
Figure 1.

Expression profile of H2O2 induced alteration of gene expression. (A) A tree of the clustering analysis is represented. (B) Expression profile of genes upregulated by H2O2. The markedly upregulated genes at 1 and 4 h include p21, ATF3, CHOP, DSP, HO1, ID1, and Period from the top at 4 h.
Figure 2.

H2O2-Induced alterations of gene expression were confirmed by real-time RT-PCR. (A) Time course of gene expression in HK2 cells treated with 100 μM H2O2 at the indicated period were confirmed. ATF3, CHOP, DSP, HO1 and p21 were significantly upregulated. (B) Dose-response of genes in cells treated with the indicated concentrations of H2O2 for 4 h. Each experiment was performed in triplicate.
Confirmation of Upregulation of ATF3 Expression
All five upregulated genes are known to have roles in oxidative stress. However, of these genes, ATF3 was most markedly changed by H2O2 treatment and is a known transcriptional regulator. For these reasons ATF3 was selected as a key regulatory gene that might mediate injury events associated with oxidative stress. To confirm that H2O2 induced ATF3 expression at the protein level, Western blot analysis was performed. Induction was clearly observed at 1, 4, and 16 h (Figure 3A). Immunohistological examination showed that, under control, nonstress conditions, ATF3 is present in the cytosolic regions of HK2 cells (Figure 3B). The staining of ATF3 shifted to the nucleus after 4 h of H2O2 treatment (100 μM) (Figure 3C). These observations are consistent with ATF3 functioning as an activated transcription factor in response to oxidant stress.
Figure 3.

Induction of ATF3 by 100 μM H2O2 treatment. (A) Induction of ATF3 was clearly observed by Western blot analysis. Immunohistochemistry of ATF3 in the presence (B) and absence (C) of 100 μM H2O2 stimulation in HK2 cells for 4 h. Magnification: ×400.
Interaction between ATF3 and Other Upregulated Genes
To analyze the interaction between ATF3 and other upregulated genes, ATF3 was silenced using small RNA interference (siRNA) methodology. H2O2-Induced ATF3 expression was suppressed by ATF3-siRNA transfection (Figure 4) of HK2 cells. Interestingly, suppression of ATF3 with ATF3-siRNA blocked induction of p21 but did not influence the increased expression of CHOP, DSP, or HO1. Thus, p21 appears to be a downstream target of ATF3.
Figure 4.

siRNA silencing of ATF3 suppresses p21 induction, but not other genes in HK2 cells in real-time PCR (n = 4). Values are mean ± SEM. *P < 0.05 versus ATF3RNAi (−).
ATF3 Prevented H2O2-Induced Cell Death in HK2 Cells
Next, we examined whether overexpression of ATF3 could protect cells from oxidant stress. For this purpose, HK2 cells were infected with AdATF3 or AdLacZ, treated with H2O2, and cell viability was examined with an assay using acetomethoxy ester of calcein (calcein-AM; Molecular Probes, Eugene, OR). As shown Figure 5A, AdATF3-treated HK2 cells were significantly protected from cell death. Thus our in vitro experiment indicated that ATF3 could diminish H2O2-induced cell death. To examine a whether this protective effect of ATF3 could involve a decreased expression of p53 as previously reported,12 we investigated the p53 mRNA expression in the HK2 cells with or without AdATF3. Real-time RT-PCR revealed that p53 mRNA was significantly downregulated in the AdATF3-treated HK2 cells by approximately 0.7-fold compared with AdLacZ-overexpressed HK2 cells (Figure 5B). Notably, p21 mRNA was induced by AdATF3 by approximately 1.8-fold.
Figure 5.

Effects of adenovirus-mediated gene transfer and overexpression of ATF3 (A). HK2 cells were significantly protected from H2O2 by AdATF3 at various doses of treatment with H2O2. A cell viability assay was performed using calcein AM (n = 6). (B) p53 mRNA was significantly decreased and p21 mRNA was significantly increased by treating with AdATF3 using real-time RT-PCR. (n = 4). Values are mean ± SEM. *P < 0.05 versus AdLacZ.
ATF3 Overexpression by Adenovirus Gene Transfer Protected against Renal IRI In Vivo
In an in vivo study we next examined whether ATF3 plays a role in IRI. First we characterized the changes in ATF3 levels in the kidney of a mouse model of IRI. In the kidney, at 12 and 24 h after ischemia there was a significant increase in ATF3 mRNA levels (Figure 6A) and corresponding increases in ATF3 protein levels (Figure 6B). Immunofluorescent staining of kidney (Figure 6C) showed that, with IRI, ATF3 protein was localized principally in the nuclei of the distal tubules that were not stained for NHE3.
Figure 6.

ATF3 was markedly induced by renal IRI in the mouse model. (A) Real-time PCR showed that ATF3 mRNA was significantly induced by IRI at 12 and 24 h after ischemia reperfusion compared with sham. (B) Representative Western blot analysis showed that ATF3 was significantly induced by IRI at 12 to 48 h after ischemia reperfusion compared with sham. (C-a) Immunofluorescent staining showed that ATF3 (green) was not detected in sham. (C-b) However, ATF3 (green) was induced by IRI for 24 h and was localized mainly in the nuclei of tubules (arrows). (C-c) NHE3 (red), the marker of proximal tubular cells (arrow), stained by immunofluorescent study. (C-d) Overlay picture of ATF3, NHE3, and DAPI (blue) is shown. ATF3 and DAPI were costained in the nuclei (arrow) of the distal tubule cells that were not stained for NHE3. For (A) and (B), values are mean ± SEM (n = 4). *P < 0.05 versus sham. Magnification for (C), ×200.
Because ATF3 overexpression inhibited oxidative stress induced cell death in HK2 cells, we hypothesized that ATF3 might protect the kidney from IRI. Therefore, we examined the protective effect of overexpression of ATF3 in our model of IRI in the mouse kidney. First, ATF3 gene transfer was performed by hydrodynamic injection of AdATF3 into the mouse tail vein. Overexpression of ATF3 mRNA and protein in the kidney was confirmed by RT-PCR and Western blot analysis (Figure 7, A and B). Immunofluorescence revealed that ATF3 was prominently overexpressed in all nephron segments of mice injected with AdATF3 (Figure 7D) but not with AdLacZ (Figure 7C). In the kidneys of mice injected with AdLacZ, marked damage (i.e., exfoliation of epithelial cells, tubular cast formation, and tubular dilation) was observed (Figure 7E). In contrast, the injury observed in the kidneys of mice injected with AdATF3 was attenuated compared with control mice injected with AdLacZ (Figure 7, E and F). An associated improvement in renal function was observed in AdATF3-treated mice as serum creatinine levels were significantly ameliorated compared with control mice injected with AdLacZ (Figure 8A). An improvement in the acute tubular necrosis (ATN) score was also observed in the kidneys of IRI mice with ATF3 overexpression (Figure 8B).
Figure 7.

ATF3 protected tubular injury from ischemia reperfusion in mouse kidney. (A) RT-PCR showed that ATF3 mRNA was markedly induced by injection of AdATF3 in the kidney. (B) ATF3 induction by AdATF3 injection was confirmed by Western blotting. (C and D) Representative immunohistochemical stain for ATF3 is shown. ATF3 was upregulated mainly in the all-nephron segments in the mouse injected with AdATF3 (D). (E and F) Representative histochemical staining is shown. AdATF3 protected the tubular damage against IRI compared with AdLacZ (E). Magnification, ×200.
Figure 8.
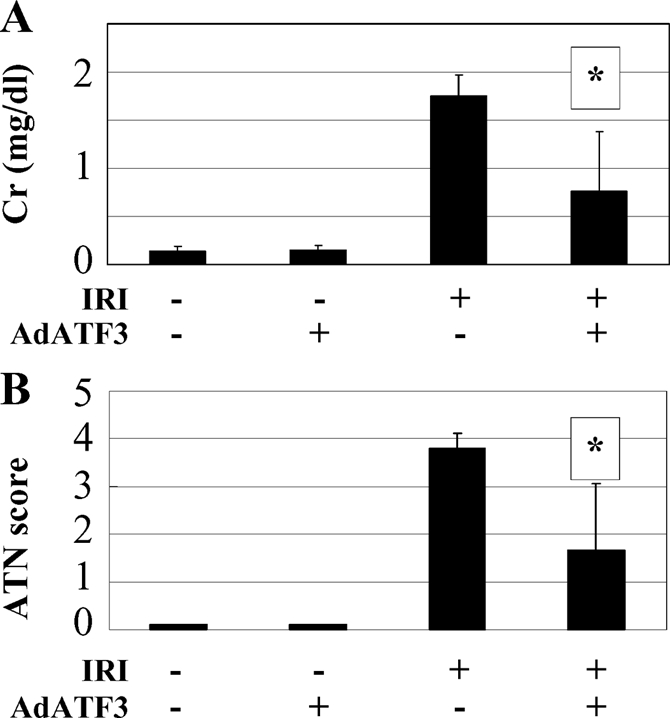
Protective effect of AdATF3 from renal IRI. Serum creatinine (A) and ATN score (B) in the sham (−) and IRI (+) treated mouse with AdATF3 or AdLacZ after 24 h reperfusion are presented. Both serum creatinine and ATN score were significantly ameliorated compared with treatment with AdLacZ. Values indicated by means ± SEM (n = 4 to 5). *P < 0.05 compared with AdLacZ.
Discussion
We have three major findings. First, five markedly upregulated genes, including ATF3, were identified in response to H2O2 treatment in HK2 cells. Second, ATF3 plays a protective role in the H2O2-induced cell death in the HK2 cells. Third, ATF3 also plays a protective role in mouse renal IRI.
A cluster analysis was used to search for key genes, because genes with similar patterns of expression can represent genes that are coregulated at the transcription level. Genes involved in a specific biochemical pathway or developmental process have been shown to have coordinated transcriptional regulation, and functionally unknown genes showing the same regulation can be consequently be implicated in the same pathway.
We found five H2O2-induced genes in HK2 cells using DNA microarrays. These genes included ATF3, p21, CHOP, DSP, and HO1. All of these genes have been reported to be involved in oxidative stress. Yuen et al. reported about microarray-identified genes, including ATF3, p21, and HO1.13 HO1 is known to be upregulated in response to an ischemic insult and to play an important role in protection against oxidative stress including renal IRI,14,15 which was supported by our result. DSP was reported to be induced by H2O2 stimulation and is implicated in ERK and JNK inactivation in the nucleus.16 Therefore this gene could also be related to the mechanism of IRI. Id1 was observed as increased in the microarray analysis, but this was not confirmed by real-time PCR for reasons that are unclear. Nonetheless, further work with Id1 may be warranted because Kang et al. suggested that ATF3 can mediate Id1 repression as a common target of TGF-β/Smad signals and stress signals via p38 kinase.17
In this article we demonstrated that ATF3 was dramatically induced by H2O2 in HK2 cells and IRI in the mouse kidney. ATF3 is a member of the ATF/cAMP-responsive element-binding protein family and is known to be activated under a variety of extracellular stresses.9 The gene products of this family have a basic region followed by the leucine-zipper motif and bind to the consensus DNA sequences TGACGT(C/A)(G/A) as transcription factors. Several reports have noted that ATF3 is rapidly and preferentially induced by various stimuli.9,18 It has been reported that the ATF3 gene is induced when organs such as the liver, heart, and kidney are subjected to oxidative stresses caused by ischemia reperfusion.11,19,20 Our result is consistent with these previous reports. Our previous report did not report the upregulation of ATF3 gene in the animal model of IRI because the probe of ATF3 was not on the microarray. A report by Supavekin et al. also did not show the induction of ATF3 for reasons that are not apparent.21
Herein we described the protective effect afforded by ATF3 against H2O2-induced cell death in a cultured cell line and in renal IRI in the mouse. The mechanism, however, is not clear. As shown in our results, the increase in p21 was suppressed by silencing the ATF3 induction by H2O2. Furthermore, AdATF3 induced p21. The cyclin-dependent kinase inhibitor p21 plays a pivotal role in apoptosis through regulation of the cell cycle. Therefore, the mechanism could be that the oxidative stress–mediated increase in p21 associated with overexpression of ATF3 contributed to the protection against oxidative stress through cell-cycle arrest (G1 arrest) and thus inhibited apoptosis. The experiment for further detail about apoptosis and proliferation should be achieved in the next project. Another possibility is that overexpression of ATF3 suppressed the downregulation of p53, which can cause apoptosis. Nobori and colleagues reported that overexpression of ATF3 inhibited doxorubicin-induced apoptotic cell death in cardiac myocytes.12 They suggested that the downregulation of p53 may function as a cytoprotective transcription factor in doxorubicin-treated cardiac myocytes. Kawauchi et al. also reported that ATF3 protects human umbilical-vein endothelial cells from TNFα through downregulation of p53.22 These reports are compatible with our results. Therefore, the downregulation of p53 also may play a protective role in renal tubules against IRI.
CHOP is a key gene in renal IRI that physically interacts with ATF3. Our results indicated that CHOP was elevated by H2O2 stimulation but unaffected by siRNA silencing of ATF3, which suggests that its induction is not mediated by ATF3. CHOP is a member of the CCAAT/enhancer binding protein (C/EBP), can function as a complex with ATF3, and is known to be an apoptosis-induced gene.23–25 ATF3 and CHOP transcripts were regulated in parallel, and ATF3/CHOP heterodimers may be formed with and related to necrotic cell death. Therefore, CHOP would play an important role with ATF3 in the caspase signaling pathway in oxidative stress–induced apoptosis in the human proximal tubule. Moreover, we would suggest that during oxidant stress the mechanism responsible for ATF3 induction may also be also responsible for CHOP induction as well.
Other mechanisms for ATF3 in the protection against cell death have also been reported. Nakagomi et al. hypothesized another mechanism in which ATF3 binds with c-Jun and the heterodimer of ATF3/c-Jun induces heat shock protein 27 (Hsp27), which contains a heat shock element in the promoter.26 Hsp27 is known to inhibit cell death in neuronal cells.26–29 These authors further suggested that Hsp27 activated the Akt pathway and inhibited MEKK1-c-Jun-JNK, thereby inhibiting apoptosis in neuronal cells. Therefore, Hsp27 may play an important role with ATF3 in IRI. In another study, Alam et al. reported that ATF3 as well as the transcription factors Nrf2 and MafG bind to stress-response elements and contribute to the cytoprotective function of HO1 activity during heme-mediated cellular injury.30
In conclusion, in this study we have shown that there is a marked induction of ATF3 by oxidative stress and renal ischemic injury. Furthermore, ATF3 promotes a protective effect against renal IRI, potentially through regulation of p53 and p21. Thus, overexpression of ATF3, either by gene therapy or by infusion of recombinant ATF3 protein, may provide a new therapeutic intervention in ischemia-mediated acute renal failure. However, further study may be needed to clarify the role of ATF3 because alternative mechanisms for the protective effect of ATF3 have been reported. In addition, an appropriate ATF3 gene delivery system needs to be developed to put this approach into clinical practice.
Concise Methods
Cell Culture
HK2 cells were cultured in RPMI1640 (Life Technology, Tokyo, Japan) supplemented with 10% FBS (JRH Biosciences, Lexena, KS), 100 units/ml penicillin, and 100 μg/ml streptomycin (Life Technology). Cells were grown at 37°C in 5% CO2 and 95% air.
Adenovirus and Gene Transfer
An adenovirus vector encoding the human ATF3 gene (AdATF3) and β-galactosidase (AdLacZ) using an Adenovirus Expression Vector kit was a kind gift from Dr. Nobori of Tokyo Medical and Dental University (Tokyo, Japan). For adenovirus-mediated gene transfer, HK2 cells were exposed to the adenoviral vectors and cultured for the indicated time on the basis of the DNA-terminal protein complex method as described elsewhere.10 Purified recombinant adenoviruses were added to HK2 cells at a dose of 3.3 × 108 plaque-forming units. For hydrodynamic injection, AdATF3 or AdLacZ in 1 ml of PBS was injected within 10 s into the mouse tail vein.
Animals and Sample Preparation
C57BL/6j mice (20 to 25 g) were housed in a 12-h light/dark cycle and allowed free access to food and water. AdATF3, human recombinant ATF3, or AdLacZ was injected 24 h before treatment to mice. Nobori reported that human recombinant ATF3 functioned in mice.12 IRI was carried out as described previously.8 Briefly, animals were anesthetized intraperitoneally with pentobarbital sodium (65 mg/kg). Both renal arteries were exposed with a midline incision, then clamped for 30 min and followed by reperfusion on the heat pad. Sham animals had an incision plus 30 min of waiting time without clamping. Animals were then followed for 12 h, 1 d, or 2 d (n ≥ 6 per group per time point). The animals were killed humanely at the end of the experiment. The kidneys were quartered, then two of the kidney specimens were snap-frozen in liquid nitrogen, and the remaining specimens were fixed with either 15% neutral buffered formalin or 4% paraformaldehyde in PBS. Total RNA was isolated from the kidneys with Trizol (Invitrogen, Tokyo, Japan) extraction.
Measurement of Biochemical Parameters
Plasma concentrations of creatinine were measured with an autoanalyzer (SRL, Tokyo, Japan) as an indicator of impaired glomerular function.
RNA Isolation, Reverse Transcription, and Real-Time RT-PCR
Total RNA was extracted from the kidney using Trizol (Invitrogen).7 cDNA was prepared with the Super Script kit (Invitrogen) from 5 μg total RNA with Oligo (dT) for analysis with real-time PCR using Prism 7700 (Applied Biosystems, Tokyo, Japan) according to the manufacturer's instructions. Commercially available mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers (Rodent, Tokyo, Japan) were used; the PCR primers were as follows:
- Mouse ATF3 (product size 195)
- 5′ AACTGGCTTCCTGTGCACTT′ (forward)
- 5′ TGAGGCCAGCTAGGTCATCT′ (reverse)
- Human ATF3 (product size 115)
- 5′CCTCGGAAG TGAGTGCTTCT3′ (forward)
- 5′ ATGGCAAACCTCAGCTCTTC3′ (reverse)
- Human p53 (product size 173)
- 5′AGCCACCTGAAG TCCAAAAA3′ (forward)
- 5′ CAAGGG TTCAAAGACCCAAA 3′ (reverse)
- Human GAPDH primers (product size 127)
- 5′CCACCCAGAAGACTGTGGAT3′ (forward)
- 5′TTCAGCTCAGGGATGACCTT3′ (reverse)
Western Blot Analysis
For whole-cell extract preparation, HK2 cells treated as indicated were washed in PBS, resuspended in 50 μl of lysis buffer (50 mM Hepes-KOH (pH 7.5), 150 mM NaCl, 1% Triton X-100, 1.5 mM MgCl2, 1 mM EGTA, 0.1 mM PMSF, 10 μg/ml each leupeptin and aprotinin, 200 μM Na vanadate, and 100 mM NaF, and 10% glycerol), and incubated on ice for 10 min. The cells were then centrifuged at 10,000 rpm for 10 min, and the supernatant was taken as the whole-cell extract. The amounts of protein were measured with the Lowry method using BSA as a standard. Whole-cell extracts (20 μg of protein) were separated on an SDS-PAGE, transferred onto a nitrocellulose membrane, and subjected to a Western blot antibody against mouse ATF3 at a 1:4000 dilution. The ECL Western Blotting System (Amersham Life Sciences) was used for detection. Bands were visualized and their intensity was calculated with Image J (an image processing and analysis program; National Institutes of Health, Bethesda, MD).
Cell Viability Assay
To measure cell viability, the cell culture medium containing H2O2 was removed and replaced with D-PBS containing 10 μM calcein AM (Molecular, Tokyo, Japan) and cells were then incubated at 25°C for 30 min. Fluorescence was measured with a multilabel fluorescence plate reader (PerkinElmer Life Sciences, Tokyo, Japan).
DNA Microarray Analysis
Affymetrix GeneChip technology was used as described previously.31 Briefly, cDNA was synthesized from total RNA using a Superscript Choice kit (Gibco BRL, Tokyo, Japan) and a T7 polymerase (Mega Script T7 kit; Ambion, Tokyo, Japan). Total RNA (8.0 μg) was annealed to T7-(dt)24 primer (100 pmol/μl) at 70°C for 10 min. Reverse transcription was carried out at 42°C for 1 h in a mixture with final concentrations of 1 × First Strand Buffer, 10 mM dithiothreitol, 500 μM dNTP, and 20,000 U of Superscript II reverse transcriptase per ml. Second-strand synthesis was carried out in 150 μl, incorporating the entire 20-μl first-strand reaction mixture and a 130-μl second-strand reaction mixture containing final concentrations of 1 × Second Strand Buffer, 250 μM dNTP, 1.2 mM dithiothreitol, 65 U of DNA ligase per ml, 250 U of DNA polymerase I per ml, and 13 U of RNase H per ml. The mixture was incubated at 16°C for 2 h, whereupon 2 μl of T4 DNA polymerase at 5 U/μl were added and incubation at 16°C was prolonged for 5 min. After purification, the cDNA was precipitated with 5 M ammonium acetate and absolute ethanol at −20°C for 20 min. The pellet was resuspended in 1.5 μl of water. Synthesis of biotin-labeled cRNA was carried out using the MEGA script T7 In Vitro Transcription Kit (Ambion). The amplified cRNA was purified with an affinity resin column (RNeasy, Qiagen, Tokyo, Japan). The cRNA was fragmented by incubation at 94°C for 35 min in the presence of 40 mM Tris-acetate (pH 8.1), 100 mM potassium acetate, and 30 mM magnesium acetate. Twenty micrograms of the fragmented cRNA was hybridized to the GeneChip U74A Array Set (Affymetrix, Santa Clara, CA), which allowed us to monitor the abundance of about 12,000 mRNA transcripts. A 220-μl hybridization solution of 1 M NaCl, 10 mM Tris (pH 7.6), 0.005% Triton X-100, 50 pM control oligonucleotide B2, control cRNA, 0.1 mg of herring sperm DNA per ml, and 0.05 μg of the fragmented cRNA per μl was heated to 95°C and then cooled to 40°C. Hybridization occurred at 45°C in a rotisserie hybridization oven at 60 rpm for 16 h. Subsequent washing and staining of the arrays was carried out using the GeneChip fluidics station protocol EukGE-WS2. After washing and staining, probe arrays were scanned twice at 3 μm resolution using the GeneChip System confocal scanner (Hewlett-Packard, Santa Clara, CA). Data analysis was performed using Affymetrix GeneChip 3.1 software (Affymetrix).
siRNA Design and Transfection
Control siRNA (scramble) and siRNA for ATF3 were designed as follows: Control siRNA, 5′CTGAAGGCTCAGATTGAGGAGC 3′; ATF3 siRNA, 5′CTGTGAGATAAGCGGGACTCAG 3′. One picomole of the siRNA dissolved in 500 μl of culture medium was transfected into HK2 cells using 20 μl Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.
Histological Examinations
Kidneys were fixed with 15% neutral buffered formalin and then embedded in paraffin. For morphological evaluations, paraffin sections (3 μm thick) were routinely stained with periodic acid-Schiff stain. Histological examinations were performed by three renal pathologists in a blinded fashion. Histological changes caused by tubular necrosis were quantified by calculation of the percent of tubules that displayed cell necrosis, loss of the brush border, cast formation, and tubule dilation as follows: 0, none; 1, 10%; 2, 11% to 25%; 3, 26% to 45%; 4, 46% to 75%; and 5, >76%. At least 15 fields (×200) were reviewed for each slide in the HE stained specimens (ATN score).
Frozen Immunohistochemical Analysis for ATF3, NHE3, and Nuclear DNA
Kidneys were fixed with 4% paraformaldehyde in PBS for 16 h at 4°C, were immersed in 10%, 15%, and 20% sucrose in PBS for 12 h at 4°C, embedded in optimal cutting temperature compound, and immediately frozen in liquid nitrogen. Triple labeling for ATF3, NHE3, and 4′,6-diamidine- 2′-phenylindole,dihydrochloride (DAPI) were performed. For immunohistochemical analysis, a primary antibody against recombinant human ATF3 and a secondary detection antibody (Alexa Fluor 488 goat anti-mouse IgG (H+L), Molecular Probes, Tokyo, Japan) were used. To counterstain for NHE3, chicken anti-Na+/H+ exchanger-3 polyclonal antibody was used. Nonspecific binding sites were blocked in sections (3 μm thick) with goat serum (Life Technologies, Tokyo, Japan) for 1 h at room temperature, blocked with anti-mouse IgG (Vector Laboratories, Tokyo, Japan) for 3 h with TBS (20 mM Tris, 225 mM NaCl), and then incubated with the primary antibody that was used at a 1:300 dilution for 16 h at 4°C. After three 5-min washing steps with TBS, the sections were incubated with the secondary antibody that was used at a 1:200 dilution for 1 h at room temperature in the dark. After three 5-min washing steps with TBS, the sections were mounted using VectaShield (Vector Laboratories). Staining for nuclear DNA was performed using DAPI dye (1 μg/ml, Kirkegaard Perry Laboratories, Tokyo, Japan).
Statistical Analyses
All data are presented as means ± SEM. Differences between groups were analyzed with the t test. P < 0.05 was considered significant.
Disclosures
None.
Acknowledgments
We especially wish to thank to Mayuko Tsuboi, Ai Munekawa, and Atsuko Teraoka for their technical assistance.
Published online ahead of print. Publication date available online at www.jasn.org.
References
- 1.Waikar SS, Curhan GC, Wald R, McCarthy EP, Chertow GM: Declining mortality in patients with acute renal failure, 1988–2002. J Am Soc Nephrol 17: 1143–1150, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Star RA: Treatment of acute renal failure. Kidney Int 54: 1817–1831, 1998 [DOI] [PubMed] [Google Scholar]
- 3.Mangano CM, Diamondstone LS, Ramsay JG, Aggarwal A, Herskowitz A, Mangano DT: Renal dysfunction after myocardial revascularization: Risk factors, adverse outcomes, and hospital resource utilization. The Multicenter Study of Perioperative Ischemia Research Group. Ann Intern Med 128: 194–203, 1998 [DOI] [PubMed] [Google Scholar]
- 4.Kazmers A, Jacobs L, Perkins A: The impact of complications after vascular surgery in Veterans Affairs Medical Centers. J Surg Res 67: 62–66, 1997 [DOI] [PubMed] [Google Scholar]
- 5.Tracey KJ, Beutler B, Lowry SF, Merryweather J, Wolpe S, Milsark IW, Hariri RJ, Fahey TJ 3rd, Zentella A, Albert JD, Shires GT, Cerami A. Shock and tissue injury induced by recombinant human cachectin. Science 234: 470–474, 1986 [DOI] [PubMed] [Google Scholar]
- 6.Gerhold DL, Jensen RV, Gullans SR. Better therapeutics through microarrays. Nat Genet 32[Suppl]: 547–551, 2002 [DOI] [PubMed] [Google Scholar]
- 7.Yoshida T, Kurella M, Beato F, Min H, Ingelfinger JR, Stears RL, Swinford RD, Gullans SR, Tang SS: Monitoring changes in gene expression in renal ischemia-reperfusion in the rat. Kidney Int 61: 1646–1654, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Yoshida T, Tang SS, Hsiao LL, Jensen RV, Ingelfinger JR, Gullans SR: Global analysis of gene expression in renal ischemia-reperfusion in the mouse. Biochem Biophys Res Commun 291: 787–794, 2002 [DOI] [PubMed] [Google Scholar]
- 9.Hai T, Hartman MG: The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: Activating transcription factor proteins and homeostasis. Gene 273: 1–11, 2001 [DOI] [PubMed] [Google Scholar]
- 10.Liang G, Wolfgang CD, Chen BP, Chen TH, Hai T: ATF3 gene. Genomic organization, promoter, and regulation. J Biol Chem 271: 1695–1701, 1996 [DOI] [PubMed] [Google Scholar]
- 11.Allen-Jennings AE, Hartman MG, Kociba GJ, Hai T. The roles of ATF3 in glucose homeostasis. A transgenic mouse model with liver dysfunction and defects in endocrine pancreas. J Biol Chem 276: 29507–29514, 2001 [DOI] [PubMed] [Google Scholar]
- 12.Nobori K, Ito H, Tamamori-Adachi M, Adachi S, Ono Y, Kawauchi J, Kitajima S, Marumo F, Isobe M. ATF3 inhibits doxorubicin-induced apoptosis in cardiac myocytes: A novel cardioprotective role of ATF3. J Mol Cell Cardiol 34: 1387–1397, 2002 [DOI] [PubMed] [Google Scholar]
- 13.Yuen PS, Jo SK, Holly MK, Hu X, Star RA: Ischemic and nephrotoxic acute renal failure are distinguished by their broad transcriptomic responses. Physiol Genomics 25: 375–386, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raju VS, Maines MD: Renal ischemia/reperfusion up-regulates heme oxygenase-1 (HSP32) expression and increases cGMP in rat heart. J Pharmacol Exp Ther 277: 1814–1822, 1996 [PubMed] [Google Scholar]
- 15.Blydt-Hansen TD, Katori M, Lassman C, Ke B, Coito AJ, Iyer S, Buelow R, Ettenger R, Busuttil RW, Kupiec-Weglinski JW. Gene transfer-induced local heme oxygenase-1 overexpression protects rat kidney transplants from ischemia/reperfusion injury. J Am Soc Nephrol 14: 745–754, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Tournier C, Thomas G, Pierre J, Jacquemin C, Pierre M, Saunier B. Mediation by arachidonic acid metabolites of the H2O2-induced stimulation of mitogen-activated protein kinases (extracellular-signal-regulated kinase and c-Jun NH2-terminal kinase). Eur J Biochem 244: 587–595, 1997 [DOI] [PubMed] [Google Scholar]
- 17.Kang Y, Chen CR, Massague J: A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell 11: 915–926, 2003 [DOI] [PubMed] [Google Scholar]
- 18.Hai T, Wolfgang CD, Marsee DK, Allen AE, Sivaprasad U: ATF3 and stress responses. Gene Expr 7: 321–335, 1999 [PMC free article] [PubMed] [Google Scholar]
- 19.Yin T, Sandhu G, Wolfgang CD, Burrier A, Webb RL, Rigel DF, Hai T, Whelan J: Tissue-specific pattern of stress kinase activation in ischemic/reperfused heart and kidney. J Biol Chem 272: 19943–19950, 1997 [DOI] [PubMed] [Google Scholar]
- 20.Okamoto Y, Chaves A, Chen J, Kelley R, Jones K, Weed HG, Gardner KL, Gangi L, Yamaguchi M, Klomkleaw W, Nakayama T, Hamlin RL, Carnes C, Altschuld R, Bauer J, Hai T: Transgenic mice with cardiac-specific expression of activating transcription factor 3, a stress-inducible gene, have conduction abnormalities and contractile dysfunction. Am J Pathol 159: 639–650, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Supavekin S, Zhang W, Kucherlapati R, Kaskel FJ, Moore LC, Devarajan P. Differential gene expression following early renal ischemia/reperfusion. Kidney Int 63: 1714–1724, 2003 [DOI] [PubMed] [Google Scholar]
- 22.Kawauchi J, Zhang C, Nobori K, Hashimoto Y, Adachi MT, Noda A, Sunamori M, Kitajima S: Transcriptional repressor activating transcription factor 3 protects human umbilical vein endothelial cells from tumor necrosis factor-alpha-induced apoptosis through down-regulation of p53 transcription. J Biol Chem 277: 39025–39034, 2002 [DOI] [PubMed] [Google Scholar]
- 23.Wolfgang CD, Chen BP, Martindale JL, Holbrook NJ, Hai T: Gadd153/Chop10, a potential target gene of the transcriptional repressor ATF3. Mol Cell Biol 17: 6700–6707, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen BP, Wolfgang CD, Hai T: Analysis of ATF3, a transcription factor induced by physiological stresses and modulated by gadd153/Chop10. Mol Cell Biol 16: 1157–1168, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR, Wek RC: Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol 24: 1365–1377, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakagomi S, Suzuki Y, Namikawa K, Kiryu-Seo S, Kiyama H: Expression of the activating transcription factor 3 prevents c-Jun N-terminal kinase-induced neuronal death by promoting heat shock protein 27 expression and Akt activation. J Neurosci 23: 5187–5196, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Costigan M, Mannion RJ, Kendall G, Lewis SE, Campagna JA, Coggeshall RE, Meridith-Middleton J, Tate S, Woolf CJ: Heat shock protein 27: Developmental regulation and expression after peripheral nerve injury. J Neurosci 18: 5891–5900, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis SE, Mannion RJ, White FA, Coggeshall RE, Beggs S, Costigan M, Martin JL, Dillmann WH, Woolf CJ: A role for HSP27 in sensory neuron survival. J Neurosci 19: 8945–8953, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benn SC, Perrelet D, Kato AC, Scholz J, Decosterd I, Mannion RJ, Bakowska JC, Woolf CJ: Hsp27 upregulation and phosphorylation is required for injured sensory and motor neuron survival. Neuron 36: 45–56, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Alam J, Killeen E, Gong P, Naquin R, Hu B, Stewart D, Ingelfinger JR, Nath KA: Heme activates the heme oxygenase-1 gene in renal epithelial cells by stabilizing Nrf2. Am J Physiol Renal Physiol 284: F743–F752, 2003 [DOI] [PubMed] [Google Scholar]
- 31.Miyake S, Makimura M, Kanegae Y, Harada S, Sato Y, Takamori K, Tokuda C, Saito I: Efficient generation of recombinant adenoviruses using adenovirus DNA-terminal protein complex and a cosmid bearing the full-length virus genome. Proc Natl Acad Sci U S A 93: 1320–1324, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]