

Abstract
To help understand the mechanism of pathogenesis of dengue virus (DV), we set out to create an infectious cDNA of the Brazilian prototype strain of DV serotype 1 (DV1-BR/90). PCR-amplified fragments of DV1-BR/90 cDNA were readily assembled into a sub-genomic cDNA that could be used to produce replicating RNAs (replicons), lacking the structural protein-encoding regions of the genome. However, assembly of a cDNA capable of producing infectious virus was only possible using a bacterial artificial chromosome plasmid, indicating that DV1 sequences were especially difficult to propagate in E. coli. While characterizing our cDNA we discovered a fortuitous temperature-sensitive mutation in the NS1 encoding region. Using our infectious cDNA and a renilla luciferase - expressing replicon we were able to demonstrate that this mutation produced a defect in RNA replication at 37°C, demonstrating that the DV1 NS1 protein plays an essential role in RNA replication.
Introduction
Dengue viruses (DV) are the etiologic agents of dengue fever (DF), dengue hemorrhagic fever and dengue shock syndrome. The viruses are transmitted to humans by Aedes mosquitoes. DV infections are a serious cause of morbidity and mortality in most tropical and subtropical areas of the world. Dengue cases are estimated to occur in up to 100 million individuals annually. DV belongs to the Flavivirus genus in the family Flaviviridae and there are four serotypes (DV1 to 4) (Burke and Monath, 2001). The four serotypes of DV do not confer cross-protective immunity, and epidemiological evidence indicates that immunity to one serotype of DV increases the chance of a more severe diseases upon infection with a second serotype by about ten-fold (Kurane and Ennis, 1997).
Flaviviruses are single-stranded, positive-sense RNA viruses and has approximately 11-kb genome length consisting of a single open reading frame encoding three structural proteins (C, preM/M and E) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5), and non-translated regions at its 5′ and 3′ terminal (5′UTR and 3′UTR). Flavivirus RNA replication occurs in the cytoplasm via a negative-strand intermediate, leading to the accumulation of positive-strand RNAs. Several NS proteins have been implicated in the process. The NS2B/NS3 serine proteinase is required for processing at multiple sites in the NS polyprotein. NS3 also possesses RNA triphosphatase and RNA helicase activities. NS5 contains methyltransferases and RNA-dependent RNA polymerase (Lindenbach and Rice, 2001).
NS1 is a highly conserved glycoprotein containing 12 invariant C residues and two potential N-linked glycosylation sites. Although the functions of NS1 have not been fully elucidated, several lines of evidence suggest that NS1 is involved in viral RNA replication (Lindenbach and Rice, 1997; Mackenzie et al., 1996; Muylaert et al., 1997; Westaway et al., 1997).
Infectious clones have been obtained for multiple flaviviruses. The first systems, developed for yellow fever virus {YFV (Rice et al., 1989)} and DV4 (Lai et al., 1991) overcame the genetic instability of flaviviral genomes in standard E. coli plasmids by propagating the 5′ and 3′ halves of the genomes into separate fragments, followed by in vitro ligation of the fragments to produce a template for production of synthetic genome-length RNAs. Subsequent strategies that have been used to propagate genome-length cDNAs that could be used directly as templates for the production of infectious RNAs have included the use of yeast plasmids (Polo et al., 1997), low-copy plasmids (Gualano et al., 1998) or bacterial artificial chromosomes (BAC) plasmids (van der Most et al., 1999) to overcome these E. coli instability problems.
Based on our successful strategy of using low-copy plasmids to propagate cDNAs for flavivirus replicons that were readily converted into genome-length clones for West Nile virus (WNV) (Rossi et al., 2005) and Japanese encephalitis virus (JEV) (Ishikawa, Konishi and Mason, unpublished), we designed a similar strategy to produce infectious cDNA for DV1. Although our low-copy plasmid was suitable for a DV1 replicon construction, we were unable to use this plasmid to produce an infectious genome-length DV1 cDNA. However, an infectious cDNA was readily obtained in a BAC plasmid. Furthermore, while characterizing this cDNA we discovered a fortuitous mutation in NS1 that produced a temperature-sensitive (ts) form of the protein that permitted us to demonstrate the importance of NS1 in DV genome replication
Results
Construction of cDNA encoding a replicon and an infectious DV1 RNA
Standard methods were used to create a subgenomic replicon from DV1-BR/90, the prototype strain of DV1 isolated in 1990 from a DF patient in Rio de Janeiro, Brazil. The viral RNA was extracted from infected C6/36 cells, reverse transcribed into cDNA, and amplified in individual dsDNA fragments as shown in the Fig.1. The five individual fragments required to produce a replicon-length cDNA were readily assembled into the low-copy plasmid pACDV1poly. Synthetic RNAs produced from this plasmid (designated pACDV1repNS1*NS3*; see below) replicated well when introduced into BHK cells (see supplemental data, Fig.S1). Comparison of the sequence of this replicon and the PCR amplicons used to generate it revealed two changes in encoded amino acids, one in NS1, and a second in NS3 (see Table 1, discussed in detail below). Nevertheless, cells transfected with this replicon produced high levels of antigen when detected with either a polyclonal sera (results not shown) or a MAb to NS1 (Supplemental data, Fig.S1).
Fig.1.

(A) Schematic representation of the DV1-BR/90 genome showing the position of the restriction enzymes sites (NotI, BspTI, NgoMIV, PshAI, PmlI, SapI, XhoI and MluI) and fragments used to assemble the replicon cDNA encoded by pACDV1repNS1*NS3*. Position of the T7 promoter is shown on the left, and positions of the SapI and the HDV-RZ/bacteriophage T7 terminator fragments (which included a downstream SwaI site for linearization and an XhoI site for insertion) are shown at right. (B) Schematic representation of the structural protein-encoding cDNA fragment present in pACDV1T7-C-NS1 and the cDNA present in BAC plasmid pBACDV1flcNS1*NS3*. (C) Structure and position of Rluc encoding cistron added to the 3′ UTR of DV1repNS1*. The “*”s above NS1 and NS3 are used to indicate the position of coding differences with the BR/90 amplicon (see Table 1 and text for details)
Table 1.
Summary of nucleotide and amino acid sequencea differences between the GenBank-deposited sequences of BR/90, the BR/90 amplicons used to generate our infectious cDNAs and various cloned cDNAs.
| Base position (codon)b | Coding region | BR/90 GenBankc | BR/90 amplicons | pACDV1rep NS1*NS3* | pACDV1 T7-C-NS1 | pBACDV1flc NS1*NS3* | pBACDV1flc |
|---|---|---|---|---|---|---|---|
| 325 | C | A | G | G | G | G | |
| 1213 | E | A | U | U | U | U | |
| 1258 | E | U | C | C | C | C | |
| 1291 | E | U | C | C | C | C | |
| 1378 | E | A | G | G | G | G | |
| 1639 | E | C | U | U | U | U | |
| 1648 | E | U | C | C | C | C | |
| 2719 | NS1 | G | A | A | A | A | |
| 3159 (247) | NS1 | A (Y)d | A (Y)d | G (C) | G (C) | A (Y)d | |
|
| |||||||
| 3265 | NS1 | C | U | U | U | U | |
| 3346 | NS1 | U | U | C | C | U | |
| 3424 | NS1 | C | U | U | U | U | |
| 3907 (144) | NS2A | C (H) | G (Q)d | G (Q)d | G (Q)d | G (Q)d | |
|
| |||||||
| 4627 | NS3 | G | A | A | A | A | |
| 5250 (211) | NS3 | A (Q)d | A (Q)d | G (R) | G (R) | A (Q)d | |
|
| |||||||
| 5413 | NS3 | A | A | A | A | G | |
| 6148 | NS3 | U | U | C | C | C | |
| 6598 | NS4A | A | A | G | G | G | |
| 10195 | NS5 | G | G | A | A | A | |
| 10258 | NS5 | A | A | G | G | G | |
Encoded amino acid sequences, when different, are shown in “()”.
Position of nucleotide change (within genome) and codon change (within the individual protein-encoding region).
To construct a genome-length, infectious cDNA from pACDV1repNS1*NS3*, we created a low-copy plasmid encoding the 5′UTR and C-NS1 polyprotein of DV1-BR/90 (designated pACDV1T7-C-NS1; Fig.1). Sequence analyses of pACDV1T7-C-NS1 confirmed that its sequence was identical to that in the DV1-BR/90 amplicons used to create it. Using multiple different restriction endonuclease assembly strategies, genome-length cDNAs were very difficult to assemble from pACDV1repNS1*NS3* and pACDV1T7-C-NS1. Furthermore, even when obtained, bacterial colonies harboring plasmids that had restriction maps consistent with full-length cDNAs grew very poorly. Moreover, when RNAs produced from the cDNA templates prepared from these bacteria were transfected into BHK cells, they failed to produce cells expressing any detectable DV1 antigen (results not shown). Since we successfully applied these same assembly methods to recover infectious cDNAs for WNV (Rossi et al., 2005) and JEV (Ishikawa, Konishi, and Mason, unpublished), these results suggested an especially severe genetic incompatibility/instability of full-length DV1 cDNAs in E. coli (see Introduction).
To overcome the apparent genetic instability/incompatibility of the DV1 cDNA sequences with our low-copy plasmid, we transferred the subgenomic replicon cDNA to a BAC plasmid, and then added back the 5′ end of the genome encoding the structural proteins from pACDV1T7-C-NS1 (see Fig.1). Initial analyses demonstrated that synthetic RNAs from all three full-length BAC plasmid clones isolated from the first assembly attempt produced large numbers of antigen-positive cells (supplemental data, Fig.S2). One of these BAC plasmids, designated pBACDV1flcNS1*NS3* was sequenced in its entirety (see Table 1) and used for all further studies.
Characterization of DV1flcNS1*NS3*
BHK cells transfected with synthetic RNA produced from pBACDV1flcNS1*NS3* produced large numbers of antigen-positive cells when stained with antibodies specific for either E or NS1 (up to 50% of the cells in these cultures were antigen-positive 2 days after electroporation). However, the number of immunopositive cells in these cultures did not increase upon further cultivation, and infectious titer of DV in the supernatant fluid recovered from cells at all days up to and including day 8 post-electroporation was undetectable (supplemental data Fig.S2 and results not shown). When C7/10 cells were electroporated with this RNA transcribed from this cDNA, a small number of immunopositive cells were detected on day 2, indicating that our electroporation methods was less efficient with mosquito cells than with BHK cells. However, by day 4, most of the cells in these transfected cultures were immunopositive, and by day 6, all cells stained strongly with DV-specific antibodies (Fig.2). These results suggest that DV1 infection spread in these C7/10 cells, furthermore, titration of the supernatant fluid recovered from these electroporated cells revealed viral replication (approximately 4×103 focus-forming units (ffu)/mL on day 5 when titrated on C7/10 cells), indicating that pBACDV1flcNS1*NS3* was an infectious cDNA. However, the foci size of this virus, designated DV1flcNS1*NS3*, appeared smaller than parental DV1BR/90 on C7/10, C6/36, and especially Vero cells (Supplemental data, Fig.S3).
Fig.2.

Micrograph of monolayers of C7/10 cells electroporated with the in vitro transcript of pBACDV1flcNS1*NS3* and then fixed at the indicated time post electroporation and stained with MAb D2-7E11.
Generation of repaired DV1 cDNA clones
To begin to dissect the reason of phenotypic difference between DV1flcNS1*NS3*, and DV1BR/90, a detailed comparison was made between the entire cDNA in pBACDV1flcNS1*NS3*, the RNA stock used to create it, and various DV1 sequences (Summarized in Table 1). These analyses confirmed that the assembly of the genomic fragments from the low-copy plasmids pACDV1repNS1*NS3* and pACDV1T7-C-NS1 into pBACDV1flcNS1*NS3* did not result in the selection of any nucleic acid differences, confirming the utility of the BAC plasmid in propagating the DV1 cDNAs. These analyses also confirmed that there were only 3 coding differences between the GenBank-deposited sequence of BR/90 (AF226685) and pBACDV1flcNS1*NS3*. Although a number of sequence differences appeared to be restricted to particular cloned fragments, it is unclear if the differences obtained in the E. coli-propagated cDNA fragments represented quasispecies in the viral RNA or a combination of PCR-introduced errors and E. coli selection (Table 1 and results not shown).
In the case of the three coding mutations, the NS2A position #144 difference between the GenBank-deposited sequence for DV1BR/90 and our PCR amplicon was discounted from consideration, since the Q encoded by our parental virus PCR amplicon and all of our cloned cDNAs (Fig.1, Table 1, and results not shown) was shared by multiple other GenBank sequences for DV1 (see Table 1). The NS1 Y at position 247 and the NS3 Q at aa position 211 were present in our BR/90 amplicon, and all GenBank-deposited DV1 sequences examined (see above, and Table 1), but were altered in pBACDV1flcNS1*NS3*, and the low-copy plasmid-propagated cDNAs used to create it (Table 1). Furthermore, sequencing of several other low-copy plasmid clones of DV1-BR/90 cDNA spanning these regions, demonstrated that it was possible to clone cDNA plasmids with the consensus sequence contained in the parental viral cDNA amplicon (results not shown), thus these two differences were considered mutations associated with PCR amplification and/or cloning in low-copy plasmids.
To examine the role of the NS1 and NS3 mutations on the DV1flcNS1*NS3* phenotype, several additional infectious cDNAs were engineered with wild-type (WT) codons at one or both of these positions (Fig.3). The infectivity of these full-length DV1 transcripts was tested in BHK and C7/10 cells. Cells were transfected with RNA transcripts by using Lipofectin, and cultured for 4 days under a semi-solid overlay, followed by immunostaining for DV1 antigen as shown in Fig.3B. When RNA were transfected into C7/10 cells, foci formation were observed in all clones, however, foci produced by RNAs obtained from pBACDV1flcNS1*NS3* and pBACDV1flNS3* were smaller than the foci produced from the cDNA plasmids encoding the WT NS3 Q, indicating that the R mutation in NS3 resulted in small foci size in C7/10 cells. In the case of transfected BHK cells, RNAs derived from clones with the WT NS1 sequences (pBACDV1flcNS3* and pBACDV1flc) produced foci (Fig.3B), whereas RNAs from pBACDV1flcNS1*NS3* did not produce foci, and very small foci were observed with RNAs produced from pBACDV1flcNS1* (Fig.3B). These results suggest that although mutations in either NS1 or NS3 affected viral production in BHK cells, the NS1 mutation was the primary cause of defect of DV1 growth in these cells. As expected, side-by-side plaque assays of DV1flc and the parental virus on C7/10 cells and Vero cells showed indistinguishable plaque morphologies (Fig.3C).
Fig.3.



Demonstration of the effect of NS1 and NS3 mutations on recombinant virus focus formation on monolayers of C7/10 and BHK cells. (A) Schematic representation of the genome of DV1 showing all of the amino acid coding differences between DV1-BR/90 and our BAC plasmid encoded genomes. The position of amino acid residues of NS1 and NS3 that differed between these genomes are shown at the top; bold type has been used to designate the amino acids of consensus sequence in DV1 (see text and Table 1). (B) Photographs of monolayers C7/10 and BHK cells transfected with Lipofectin using similar amounts of synthetic RNA (20 ngs) from the indicated BAC clones, overlaid with semi-solid media, and stained with anti-NS1 antibody 4 days after transfection. (C) Photographs of monolayers C7/10 and Vero infected with DV1-BR/90 or DV1flc, overlaid with semi-solid media, and fixed and immunostained with anti-NS1 antibody 4 days (C/710) or 5 days (Vero) after incubation at 30°C (C7/10) or 37°C (Vero).
Phenotypic characterization of NS1 mutant
To further investigate the effect of the NS1 mutation on viral production from our infectious cDNAs, we compared the production of virus from cells infected with WT and NS1 mutant viruses at 30°C or 37°C. To this end, BHK cells were electroporated with transcribed RNA from pBACDV1flcNS1* or pBACDV1flc, diluted with naïve BHK cells, and following attachment, the cell monolayers were cultured for 5 days under a semi-solid overlay, and then immunostained to reveal DV1 foci. As shown in Fig.4, foci morphology were similar for the progeny viruses derived from both pBACDV1flcNS1* or pBACDV1flc RNAs in BHK cells at 30°C. However, in contrast, foci produced by RNA from pBACDV1flcNS1* were smaller than those produced by RNA from pBACDV1flc when these viruses were propagated in the cells at 37°C (Fig.4). To further explore the effect of temperature on the replication of DV1flcNS1*, side-by-side growth curves were generated with this virus and DV1flc in infected Vero cells grown at 30°C and 37°C (Fig.5). These studies showed that at 30°C, DV1flcNS1* replicated at similar levels to DV1flc. However, at 37°C, DV1flcNS1* growth was delayed relative to DV1flc. Taken together, these results indicate that a C at codon 211 in NS1 produces a ts defect in NS1.
Fig.4.

Photographs of monolayers of cells transfected with synthetic RNA from pBACDV1flcNS1* and pBACDV1flc, and then grown at 30°C or 37C. BHK cells were electroporated with transcribed RNA from pBACDV1flcNS1* and pBACDV1flc, diluted with naïve BHK cells, incubated for 5 days at indicated temperature, then fixed and stained with MAb D2-7E11. In all cases, the same dilution (10-fold) of electroporated cells is shown.
Fig.5.

Growth curves of DV1flcNS1* and DV1flc on Vero cells at 30°C or 37°C. Monolayers of Vero cells were infected with the indicated virus at a multiplicity of infection (MOI) of 0.01 and incubated at the indicated temperature. At each time point, the media were removed and frozen for subsequent titration and fresh media were added. Virus titers in cell culture medium were determined by plaque assay in Vero cells incubated at 30°C.
Effect of NS1 mutation on virus entry
Since NS1 is retained in the same cellular compartment as immature flavivirus provirions (Lindenbach and Rice, 2001), we reasoned that mutation 211 in NS1 could alter virus maturation and/or NS1 could be associated with the virion, altering virion infectivity. To determine if the NS1 211 ts mutation altered virion maturation and/or its infectivity at the non-permissive temperature, we performed a temperature-shift experiment. In this assay, WT and mutant virus were incubated with Vero cells at 30°C or 37°C for 5 hrs (long enough to bind, penetrate, and uncoat the genome of DV), and then incubated for 5 days at either of these temperatures. The results of this experiment, shown in Fig.6, demonstrate that the NS1 mutation does not affect virus binding or establishment of replication, since the WT and mutant viruses produce similar numbers of foci and similar-sized foci if the infection is conducted at 30°C or 37°C and then followed by a 5-day incubation at 30°C. However, the number and size of foci produced by DV1flcNS1* were both drastically reduced when the post-entry phase of the experiment was performed at 37°C (Fig.6). These results suggest that this NS1 mutation has little (or no) effect on virus entry and uncoating, and is likely to effect RNA genome replication and/or packaging and virion release.
Fig.6.

Effect of temperature shift on infectious foci formation by cells infected with same numbers of ffu of DV1flcNS1* and DV1flc. The left side of the figure shows the incubation temperatures utilized for the 5-hr attachment/infection and 5-day growth/spread portion (performed under semi-solid overlay) of the experiment, and the right side of the figure shows the resulting monolayers, fixed, and stained with MAb D2-7E11 to reveal infectious foci (see Methods).
Effect of NS1 ts mutation on RNA replication
To evaluate the role of the NS1 mutation in RNA replication, we developed a replicon of DV1 containing the renilla luciferase (Rluc) reporter gene with or without the position-211 NS1 mutation (see Methods and Fig.1). These replicon RNAs were electroporated into BHK cells, and luciferase activities were measured at 6 hr and 4 days postelectroporation. The values obtained 6 hr after electroporation were nearly identical for both replicons at both temperatures, indicating that as expected the differences in NS1 position 211 had no effect on the initial translational activity of the reporter gene encoded by the transfected replicon RNA. For the later time point, these 6-hr activities were used to normalize for transfection efficiency (see legend for Fig. 7). These Rluc data show that the replicon harboring the NS1 mutation (DV1repNS1*) replicated much more poorly than the WT replicon at 37°C (Fig.7). In contrast, Rluc activities detected in lysates obtained from the same electroporations, and then incubated at 30°C contained indistinguishable levels of Rluc activity. These data indicate that the ts NS1 mutation reduces viral replication by reducing RNA replication.
Fig.7.
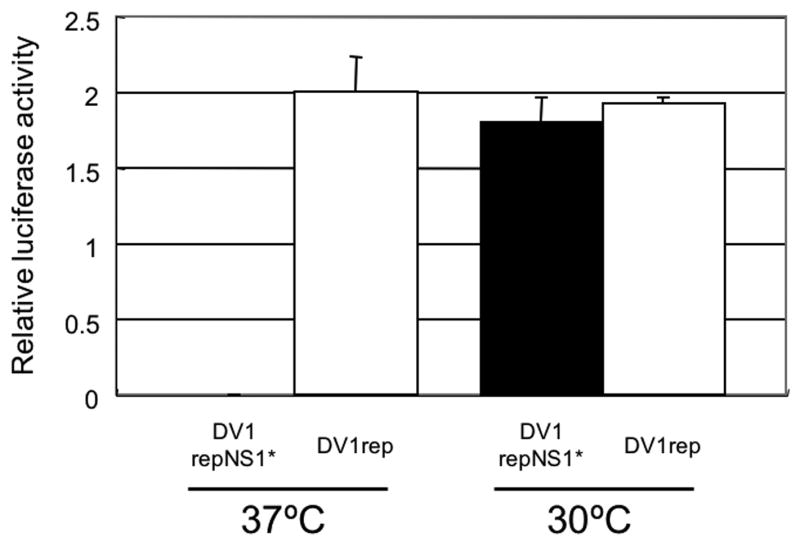
Effect of NS1 mutation on DV1 RNA replication. BHK cells were electroporated with identical amount of Rluc-expressing DV1 subgenomic replicon RNAs (with or without the NS1 position 211 mutation), seeded in multi-well plates, and incubated at 30°C or 37°C prior to harvesting lysates for the determination of Rluc activity (see Methods). Rluc activities were measured at 6 hr and 4 days postelectroporation. The Rluc activity obtained at 4 days are shown normalized to the activity obtained at 6 hr, to account for slight differences in transfection efficiency (see text). Data from each condition are shown as an average of triplicate values with error bars showing standard deviations.
Effect of NS1 mutation on polyprotein processing and NS1 secretion
To examine whether the ts mutation in NS1 affected processing and secretion of NS1 in infected cells, cells were infected with DV1flcNS1* or DV1flc for 5 days at 30°C, and then incubated for an additional 2 days at 37°C. As shown in Fig.8A, Western blot analyses revealed no obvious difference in the electrophoretic migration of NS1* versus NS1 or the amount of NS1 and NS1* present in cells at this time point. Furthermore, Western blot analyses of the NS1 present in the culture fluid harvested from these cells failed to reveal any gross defect in the dimerization or release of NS1* or NS1 from these cultures (Fig.8B). Specifically, a heat-labile NS1 form matching the predicted dimer MW of 90k was present in culture fluid harvested from cells infected with either DV1flcNS1* or DV1flc. Despite the lack of detectable differences in the quality of NS1 produced by DV1flcNS1* or DV1flc in cells grown at the non-permissive temperature for DV1flcNS1*, we did note a slight reduction in NS1* production from cells infected with the mutant virus, likely the result of the reduction in RNA replication detected with this virus when grown at this temperature (see above, and Fig.7). Since the ts mutation consisted of the addition of a C residue to NS1*, that could have altered disulfide bond formation in NS1*, we compared the electrophoretic migration of non-reduced forms of NS1 and NS1* in lysates of cells prepared at the non-permissive temperature. These analyses failed to reveal any altered migration (specifically aggregation, associated with improper disulfide formation; (Lorenz et al., 2002); results not shown) suggesting that the ts mutation was not exerting its influence on RNA replication through a gross alteration in NS1 structure or intracellular localization.
Fig.8.

Affect of NS1 mutation on NS1 synthesis and secretion. Western blots prepared from samples of cell lysates and supernatant fluids obtained from Vero cells infected at a MOI of 0.01 ffu with either DV1flcNS1* and DV1flc viruses (or mock-infected), incubated for 5 days at 30°C (to allow both viruses to infect the entire monolayer), and then shifted to 37°C for 2 days. The media were changed three times with serum free medium one day before harvest to insure that the NS1 protein analyzed represented protein produced at the final incubation temperature. (A) Replica of Western blot showing the NS1 protein (detected with MAb D2-7E11) present in cell lysates. (B) Replica of Western showing the NS1 protein released from the indicated cell monolayers at 37°C, following electrophoresis after denaturation in the presence or absence (to preserve dimers) of heating, and detection with MAb D2-7E11.
Discussion
Infectious cDNAs have been reported for a large number of flaviviruses, including most of the important human pathogens. In some cases, the apparent incompatibility of full-length viral cDNAs with commonly used E. coli recombinant DNA propagation systems has hampered development of these important reagents. Here we have demonstrated the utility of a BAC plasmid system for propagation of a highly infectious cDNA from a South American strain of DV1. Specifically, the cDNAs propagated in BAC plasmids could be used as template for RNA transcription that generated highly infectious RNAs with a specific infectivity of more than 104 ffu/ug of RNA. BAC plasmids have been used by others to produce infectious cDNA clones for YFV (van der Most et al., 1999), JEV (Yun et al., 2003) and DV2 (Pierro et al., 2006), but these investigators did not directly compare BAC plasmids to low-copy plasmids, which have been recently employed by several investigators to produce full-length clones of flaviviruses (see Introduction). In our hands, precisely the same low-copy plasmid methods that were used to successfully establish replicons and infectious cDNAs for WNV (Rossi et al., 2005) and JEV (Ishikawa, Konishi and Mason, unpublished), were successfully used to produce infectious DV1 replicons, but these methods were not able to be used to produce infectious full-length DV1 genomes. It is not clear the reason for the differences between these flaviviruses, but one possibility is that DV1 cDNAs or spontaneously produced translation products are more toxic to E. coli cells than the products produced from other flavivirus cDNAs.
The strain selected for our studies, DV1-BR/90 is a well-characterized South American prototype strain derived from an isolate obtained early after the introduction of DV1 to Brazil. We selected this strain due to the availability of animal-adapted derivatives (Duarte dos Santos et al., 2000), and the fact that DV1-BR/90 provides a useful starting point for comparisons to strains of DV1 that have evolved in Brazil since the introduction of DV1 to South America (Duarte dos Santos et al., 2002). Thus, this cDNA will be useful for understanding the involvement of animal-adapted and naturally evolved mutations in DV1 replication, transmission, and pathogenesis.
The DV NS1 protein contains two N-linked glycosylation sites and 12 conserved C residues that have been shown to form 6 disulfides (Wallis et al., 2004). NS1 is secreted from mammalian, but not mosquito cells (Mason, 1989), and exists as a heat labile homodimer (Winkler et al., 1988). Alteration of selected C residues, or the N residues that are glycosylated during NS1 processing result in the loss of dimerization and secretion of the NS1 protein, suggesting that dimerization is necessary for NS1 secretion (Pryor and Wright, 1993). However, cells infected with a Kunjin virus with a defect in NS1 dimer formation were able to secrete NS1, and the virus could replicate, suggesting that dimerization of NS1 is not an absolute requirement for NS1 secretion as well as NS1 function (Hall et al., 1999). Although a specific function of extracellular forms NS1 has not yet been demonstrated, NS1 has been shown to be important for replication of YFV. Specifically, mutations at the first or both N-linked glycosylation sites of YFV NS1 led to dramatic defects in RNA replication and virus production (Muylaert et al., 1996). Furthermore, a single amino acid substitution at position 299 in the YFV NS1 protein resulted in a defective for RNA accumulation and delayed viral production in a ts manner (Muylaert et al., 1997). Finally, trans-complementation studies revealed that NS1 has an important function at a very early stage in RNA replication (Lindenbach and Rice, 1997).
Our ts NS1 mutant virus, DV1flcNS1*, encodes an additional C residue between the naturally occurring C residues number 6 and 7. At the permissive temperature (30°C), this virus is indistinguishable from the WT virus (DV1flc) in terms of growth and infectious focus formation. In addition, DV1flcNS1* appears to infect and uncoat with equal efficiency as the WT virus. However, DV1flcNS1* is retarded with respect to virion production at 37°C. By using a subgenomic replicon encoding an Rluc reporter gene, we were able to demonstrate that the ts defect due to the additional C residue was manifest at the level of reduced RNA replication.
Examination of the processing of NS1* at the non-permissive temperature showed no obvious differences relative to WT NS1. Since the alteration in NS1* resulted in the substitution of an extra C residue for a Y residue, we specifically tested to see if this alteration was associated with changes in dimerization, secretion, or misfolding, which could accompany the addition of an unnatural C, resulting in impaired disulfide bond formation. However, we were unable to detect any difference in synthesis, secretion, or dimerization of NS1* when the ts mutant virus was grown at the non-permissive temperature (37°C). These results suggest that the Y residue at position 247 in NS1, which is highly conserved in all DV1 examined (Table 1) might be important for function of NS1. Interestingly, a P to L substitution at residue 250 in Kunjin virus NS1 also produced a mutant virus with delayed replication (Hall et al., 1999), and an R to A mutation at residue 299 in YFV NS1 was also responsible for a ts RNA synthesis phenotype (Muylaert et al., 1997), indicating that this C terminal region of NS1 plays a critical role in RNA replication for multiple flaviviruses.
The NS1* mutation resulted in a significant delay in virus production at the non-permissive temperature during first 5 days of DV1flcNS1* infection in Vero cells, consistent with a role for NS1 in the early phases of viral RNA replication. However, DV1flcNS1* titers were indistinguishable from DV1flc titers at 9-days post infection, suggesting that later stages of virus replication were less affected by this mutation. Interestingly, the ts mutation in NS1* appeared to be relatively stable, since we were unable to detect revertants following attempts to grow the virus at 37°C.
The mutation that we detected in DV1flcNS1* reduces virus yield at 37°C, suggesting that it could attenuate viremia in vivo, a property that is correlated with disease severity in man (Murgue et al., 2000; Vaughn et al., 2000). Thus, our NS1* mutation (or alternative mutations in the C-terminal region of NS1) might be a useful additions to live-attenuated vaccine candidates for dengue.
In summary, our work on an infectious cDNA system for DV1 has demonstrated the advantage of the BAC plasmid over a widely utilized low-copy plasmid, and has identified a novel ts mutation in NS1 that has been used to demonstrate a role for NS1 in DV RNA synthesis.
Materials and Methods
Plasmids, viruses and cell lines
A low-copy plasmid, designated pACDV1poly, was derived from pACNR (Ruggli et al., 1996) by adding a custom polylinker containing NotI, BspTI, BspEI, NgoMIV, PshAI, PmLI, and XhoI restriction endonuclease sites to facilitate cloning of DV1 cDNA fragments. A BAC plasmid designated pBACDV1poly was derived from pBeloBAC11 (a BAC plasmid containing the Ori2 origin of replication and F (fertility) factor of E. coli (Shizuya et al., 1992); obtained from I.Frolov), by removal of a non-essential NotI fragment and substitution of a directional polylinker containing a NotI, BamHI, XhoI and SwaI restriction endonuclease sites to facilitate directional insertion of DV1 cDNAs fragments cloned into low-copy plasmids.
DV1-BR/90, a Brazilian DV1 strain isolated from an adult male with DF in Rio de Janeiro, RJ, Brazil, in 1990 has been previously described (Despres et al., 1993; Nogueira et al., 1993) This isolate was obtained from P. Despres (Interactions Moleculaires Flavivirus-Hôtes, Institut Pasteur, 25 rue du Dr. Roux, 75724, Paris, cedex 15, France) and then was passaged an additional 4 times in C6/36 cells, prior to use. Virus stocks were maintained at −80°C.
BHK cells were maintained at 37°C in minimal essential medium (MEM – Invitrogen) supplemented with 10% fetal bovine serum (FBS – Gemini) and antibiotics. Vero cells were maintained at 37°C in MEM containing 6% FBS and antibiotics. C7/10 and C6/36 mosquito cell lines were maintained at 30°C in Leibovitz's L15 medium (Invitrogen) supplemented with 10% FBS, 10% tryptose phosphate broth (Sigma) and antibiotics.
cDNA synthesis, amplification and cloning
Total cellular RNA was extracted from BR/90-infected C6/36 cells using RNAqueous (Ambion). DV1 RNA present in this preparation was reverse-transcribed using random hex oligonucleotide primers and Improm-II reverse transcriptase (Promega). Fragments of DV1-BR/90 cDNA were amplified by the polymerase chain reaction (PCR) using specific oligonucleotide primers (see Fig.1) and a high-fidelity Taq polymerase (Sigma). The resulting double-stranded cDNA fragments were resolved by gel electrophoresis, purified by standard methods (Qiagen), digested with restriction endonucleases, and cloned into pACDV1poly (see Fig. 1). The oligonucleotide used to amplify the 5′UTR included a T7 promoter recognition site and an additional G preceding the first base of the viral genome. In addition, a synthetic antisense oligonucleotide was used to add a BspTI site at codons 30-31 of the C-coding region (following the cyclization sequence), and a sense oligonucleotide was designed that added the same site preceding the final transmembrane domain of the E protein coding sequence, to permit the ligation of C to NS1, permitting the construction of a sub-genomic replicon (Fig.1). Finally, two alternative strategies were designed to create synthetic run-off transcripts that contained a 3′ terminus identical to the viral RNA. The first consisted the introduction of a type IIS restriction endonuclease site (SapI) downstream of the 3′ end of the cDNA by using an antisense oligo containing an XhoI site and the SapI site fused to the complement of the 3′ end of the DV1 genome (CCGCTCGAGGCTCTTCGAGAACCTGTTGATTCAACAGC). The second strategy consisted of using oligonucleotides to fuse a hepatitis delta virus ribozyme (HDV-RZ)/bacteriophage T7 terminator fragment (Mason et al., 2002) to the 3′ end of the cDNA using an overlap PCR construction strategy (Higuchi et al., 1988). PCR-generated cDNA fragments were individually inserted into pACDV1poly (in some cases by a multiple step-wise addition process recreating the natural junctions) and the resulting molecularly cloned plasmids were isolated by standard techniques and sequences were checked by automatic nucleotide sequencing and compared to the sequence of PCR amplicons obtained from mosquito cells infected with BR/90 and the original BR/90 sequence deposited with GenBank (AF226685). Fragments were assembled into genome-length cDNAs in the pACDV1poly and pBACDV1poly by standard methods (see Fig.1).
To construct subgenomic DV1 replicons expressing a humanized Rluc reporter, overlap PCR (Higuchi et al., 1988) was used to fuse an EMCV IRES to a humanized Rluc gene derived from pGL4.73 (Promega), and this cassette was inserted into the variable region of the 3′-UTR (in place of bases −447 to −279 from the last base in the DV1 genome in pBACDV1rep and related plasmids (see Fig.1).
RNA synthesis using in vitro transcription reactions
Plasmids were purified by centrifugation in CsCl gradients using standard methods. Plasmids were prepared for run-off transcription by digestion with SapI restriction endonucleases or SwaI (in the case of HDV-RZ-encoding plasmids) and the resulting template DNAs were in vitro transcribed using MegaScript T7 synthesis kit (Ambion) supplemented with a 7mG(ppp)G cap analogue (NEB). This 7mG(ppG) cap analogue was added to permit efficient capping of this synthetic transcript, which was designed to encode an unnatural G in front of the normal DV A residue in order to facilitate high level synthesis with phage T7 polymerase (which produces much higher levels of RNA from transcripts beginning with a G). The yield and integrity of transcripts were analyzed by gel electrophoresis under nondenaturing condition, and aliquots of transcription reactions were used for transfection without additional purification.
RNA transfection
RNA was transfected into BHK or C7/10 monolayers using Lipofectin (Invitrogen) by a slight modification of the manufacturer's suggested protocol. Three hours after transfection, the RNA/Lipofectin/media was removed from the cell layer and the cells were re-fed with growth media and incubated at 30 or 37°C, as required. BHK or C7/10 cells were electroporated with T7 transcription reactions as previously described (Rossi et al., 2005).
Detection of DV-antigen in transfected cells by immunohistochemical staining
DV1-antigen expressing cells were labeled by a modification of previously described methods (Rossi et al., 2005). Briefly, cell monolayers were rinsed with PBS, fixed in cold acetone/methanol (1:1), air-dried, rehydrated with PBS containing 1% normal horse serum (NHS; Sigma) and incubated with DV-specific antibodies (polyclonal hyperimmune murine ascitic fluid (HIMAF) from DV1-infected mice (supplied by R.B.Tesh), an E-specific monoclonal antibody (MAb) D1-4G2 (Gentry et al., 1982) (supplied by R.B.Tesh), or the NS1-specific MAb D2-7E11 (Mason et al., 1990) (provided by R.J.Putnak, WRAIIR, Washington DC), followed by a peroxidase-conjugated anti-mouse secondary antibody (KPL), and peroxidase-labeled cells were stained with the VIP substrate (Vector Laboratories).
Western blot analysis
Proteins present in cell culture fluid or obtained from triton-lysed monolayers (lysed in 0.1% triton X-100, 300 mM NaCl, 50 mM Tris, pH 7.6) of infected cells were resolved by SDS-PAGE (NuPAGE; Invitrogen) and then electrophoretically transferred to polyvinylidene difluoride membranes (Immobilon). After blocking, these membranes were probed with MAb D2-7E11 (anti-NS1), and then washed and incubated with a peroxidase-conjugated anti-mouse antibody (KPL). The decorated NS1 proteins were then detected with the ECL Plus Western Blotting Detection System (GE Healthcare) and the image was captured on Xray film.
Viral titrations and growth curves
Cell monolayers prepared in multi-well plates were incubated with dilutions of virus and then overlaid with growth media (in some cases containing 0.8 % carboxymethyl cellulose (CMC; Sigma, medium viscosity), and incubated for the indicated times and then fixed and immunostained as described above, and foci were counted and used to calculate a titer of ffu/mL. For growth curves, Vero cells were infected and then incubated at the indicated temperature and at selected intervals the media was removed for storage at −80C (for subsequent titration), and replaced with fresh media.
Renilla luciferase assay
Monolayers of BHK cells electroporated with Rluc-expressing plasmids were lysed by addition of Reporter Lysis Buffer (Promega), and the lysates were stored at −20C for subsequent assay. Prior to assay, the extracts were thawed and clarified to remove insoluble debris, and a portion of each extract was mixed with 5 volumes of an Rluc reaction buffer (100 mM EDTA, 50 mM Tris, pH8.0, containing 5ug/ml Coelenterazine; Nanolight technology) in black-walled 96-well microtitration plates. Following a 1 min incubation period, luminescence was determined using a Microplate Luminometer (Applied Biosystems).
Supplementary Material
Fig.S1. Micrograph of an immunostained monolayer of BHK cells electroporated with in vitro synthesized DV1 subgenomic replicon RNA. Cells were electroporated with transcribed RNA from pACDV1repNS1*NS3*, seeded, and then fixed and immunostained with an anti-NS1 MAb 3 days post-electroporation.
Fig.S2. Micrographs of immunostained monolayers of BHK cells electroporated with full-length transcripts from three independently assembled full-length cDNAs assembled in a BAC plasmid (pBACDV1flcNS1*NS3*). The cells were fixed and immunostained with a HIMAF generated to DV1 (DV1poly), or MAbs D1-4G2 (Anti-E), or D2-7E11 (Anti-NS1) 2 days post-electroporation.
Fig.S3. Micrographs of immunostained foci of infection produced by DV1BR/90 (parental virus) or the recombinant DNA derived virus DV1flcNS1*NS3* on monolayers of C7/10, C6/36 and Vero cells. Monolayers of each cell type were infected with DV1-BR/90 or the virus derived from pBACDV1flcNS1*NS3*, overlaid with semi-solid media, fixed and stained with anti-NS1 antibody 4 days post-infection. The mosquito cell monolayers where grown at 30°C whereas the Vero cell monolayers were grown at 37°C. All micrographs were obtained using the same magnification.
Acknowledgments
We thank Dr. R.J.Putnak, WRAIIR, Washington, DC, for providing the anti NS1 MAb, and Dr. R.B.Tesh of UTMB for providing the polyclonal anti-DV1 HIMAF, and hybridoma cells producing the E-specific Mab, D1-4G2, and I.Frolov for supplying pBeloBAC11. We also thank I.Frolov and D. Beasley for helpful discussion and suggestions. This work was supported by a grant from NIAID to PWM through the Western Regional Center of Excellence for Biodefense and Emerging Infectious Disease Research (NIH grant number U54 AI057156). We also acknowledge Fundacao Oswaldo Cruz (FIOCRUZ) and Conselho de Desenvolvimento Cientifico e Tecnologico (CNPq, Brazil) for their support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Burke DS, Monath TP. Flaviviruses. In: Roizman B, editor. Fields Virology. Fourth. Vol. 1. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 1043–1125. 2 vols. [Google Scholar]
- Despres P, Frenkiel MP, Deubel V. Differences between cell membrane fusion activities of two dengue type-1 isolates reflect modifications of viral structure. Virology. 1993;196:209–19. doi: 10.1006/viro.1993.1469. [DOI] [PubMed] [Google Scholar]
- Duarte dos Santos CN, Frenkiel MP, Courageot MP, Rocha CFS, Vazeille FMC, Wien MW, Rey FA, Deubel V, Despres P. Determinants in the envelope E protein and viral RNA helicase NS3 that influence the induction of apoptosis in response to infection with dengue type 1 virus. Virology. 2000;274:292–308. doi: 10.1006/viro.2000.0457. [DOI] [PubMed] [Google Scholar]
- Duarte dos Santos CN, Rocha CF, Cordeiro M, Fragoso SP, Rey F, Deubel V, Despres P. Genome analysis of dengue type-1 virus isolated between 1990 and 2001 in Brazil reveals a remarkable conservation of the structural proteins but amino acid differences in the non-structural proteins. Virus Res. 2002;90:197–205. doi: 10.1016/s0168-1702(02)00180-6. [DOI] [PubMed] [Google Scholar]
- Gentry MK, Henchal EA, McCown JM, Brandt WE, Dalrymple JM. Identification of distinct antigenic determinants on dengue-2 virus using monoclonal antibodies. Am J Trop Med Hyg. 1982;31:548–55. doi: 10.4269/ajtmh.1982.31.548. [DOI] [PubMed] [Google Scholar]
- Gualano RC, Pryor MJ, Cauchi MR, Wright PJ, Davidson AD. Identification of a major determinant of mouse neurovirulence of dengue virus type 2 using stably cloned genomic-length cDNA. J Gen Virol. 1998;79(Pt 3):437–46. doi: 10.1099/0022-1317-79-3-437. [DOI] [PubMed] [Google Scholar]
- Hall RA, Khromykh AA, Mackenzie JM, Scherret JH, Khromykh TI, Mackenzie JS. Loss of dimerisation of the nonstructural protein NS1 of kunjin virus delays viral replication and reduces virulence in mice, but still allows secretion of NS1. Virology. 1999 Nov;264:66–75. doi: 10.1006/viro.1999.9956. [DOI] [PubMed] [Google Scholar]
- Higuchi R, Krummel B, Saiki RK. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16:7351–67. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurane I, Ennis FA. Immunopathogenesis of dengue virus infections. In: Gubler DJ, Kuno G, editors. Dengue and Dengue Hemorrhagic Fever. CABI Publishing; Oxon, UK: 1997. pp. 273–290. [Google Scholar]
- Lai CJ, Zhao BT, Hori H, Bray M. Infectious RNA transcribed from stably cloned full-length cDNA of dengue type 4 virus. Proc Natl Acad Sci U S A. 1991;88:5139–43. doi: 10.1073/pnas.88.12.5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach BD, Rice CM. trans-Complementation of yellow fever virus NS1 reveals a role in early RNA replication. J Virol. 1997;71:9608–17. doi: 10.1128/jvi.71.12.9608-9617.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach BD, Rice CM. Flaviviridae: The Viruses and Their Replication. In: Knipe DM, Howley PM, Griffin DG, Lamb RA, Martin MA, Roizman B, editors. Fields Virology. Fourth. Vol. 1. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 991–1041. 2 vols. [Google Scholar]
- Lorenz IC, Allison SL, Heinz FX, Helenius A. Folding and dimerization of tick-borne encephalitis virus envelope proteins prM and E in the endoplasmic reticulum. J Virol. 2002;76:5480–5491. doi: 10.1128/JVI.76.11.5480-5491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie JM, Jones MK, Young PR. Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology. 1996;220:232–40. doi: 10.1006/viro.1996.0307. [DOI] [PubMed] [Google Scholar]
- Mason PW. Maturation of Japanese encephalitis virus glycoproteins produced by infected mammalian and mosquito cells. Virology. 1989;169:354–64. doi: 10.1016/0042-6822(89)90161-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason PW, Bezborodova SV, Henry TM. Identification and characterization of a cis-acting replication element (cre) adjacent to the internal ribosome entry site of foot-and-mouth disease virus. J Virol. 2002;76:9686–94. doi: 10.1128/JVI.76.19.9686-9694.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason PW, Zugel MU, Semproni AR, Fournier MJ, Mason TL. The antigenic structure of dengue type 1 virus envelope and NS1 proteins expressed in Escherichia coli. J Gen Virol. 1990;71(Pt 9):2107–14. doi: 10.1099/0022-1317-71-9-2107. [DOI] [PubMed] [Google Scholar]
- Murgue B, Roche C, Chungue E, Deparis X. Prospective study of the duration and magnitude of viraemia in children hospitalised during the 1996-1997 dengue-2 outbreak in French Polynesia. J Med Virol. 2000;60:432–438. doi: 10.1002/(sici)1096-9071(200004)60:4<432::aid-jmv11>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Muylaert IR, Chambers TJ, Galler R, Rice CM. Mutagenesis of the N-linked glycosylation sites of the yellow fever virus NS1 protein: effects on virus replication and mouse neurovirulence. Virology. 1996;222:159–68. doi: 10.1006/viro.1996.0406. [DOI] [PubMed] [Google Scholar]
- Muylaert IR, Galler R, Rice CM. Genetic analysis of the yellow fever virus NS1 protein: identification of a temperature-sensitive mutation which blocks RNA accumulation. J Virol. 1997;71:291–8. doi: 10.1128/jvi.71.1.291-298.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira RM, Miagostovich MP, Lampe E, Souza RW, Zagne SM, Schatzmayr HG. Dengue epidemic in the stage of Rio de Janeiro, Brazil, 1990-1: co-circulation of dengue 1 and dengue 2 serotypes. Epidemiol Infect. 1993;111:163–70. doi: 10.1017/s0950268800056788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierro DJ, Salazar MI, Beaty BJ, Olson KE. Infectious clone construction of dengue virus type 2, strain Jamaican 1409, and characterization of a conditional E6 mutation. J Gen Virol. 2006;87:2263–8. doi: 10.1099/vir.0.81958-0. [DOI] [PubMed] [Google Scholar]
- Polo S, Ketner G, Levis R, Falgout B. Infectious RNA transcripts from full-length dengue virus type 2 cDNA clones made in yeast. J Virol. 1997;71:5366–74. doi: 10.1128/jvi.71.7.5366-5374.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor MJ, Wright PJ. The effects of site-directed mutagenesis on the dimerization and secretion of the NS1 protein specified by dengue virus. Virology. 1993;194:769–780. doi: 10.1006/viro.1993.1318. [DOI] [PubMed] [Google Scholar]
- Rice CM, Grakoui A, Galler R, Chambers TJ. Transcription of infectious yellow fever RNA from full-length cDNA templates produced by in vitro ligation. New Biol. 1989;1:285–96. [PubMed] [Google Scholar]
- Rossi SL, Zhao Q, O'Donnell VK, Mason PW. Adaptation of West Nile virus replicons to cells in culture and use of replicon-bearing cells to probe antiviral action. Virology. 2005;331:457–70. doi: 10.1016/j.virol.2004.10.046. [DOI] [PubMed] [Google Scholar]
- Ruggli N, Tratschin JD, Mittelholzer C, Hofmann MA. Nucleotide sequence of classical swine fever virus strain Alfort/187 and transcription of infectious RNA from stably cloned full-length cDNA. J Virol. 1996;70:3478–87. doi: 10.1128/jvi.70.6.3478-3487.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shizuya H, Birren B, Kim UJ, Mancino V, Slepak T, Tachiiri Y, Simon M. Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc Natl Acad Sci U S A. 1992;89:8794–7. doi: 10.1073/pnas.89.18.8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Most RG, Corver J, Strauss JH. Mutagenesis of the RGD motif in the yellow fever virus 17D envelope protein. Virology. 1999 Dec;265:83–95. doi: 10.1006/viro.1999.0026. [DOI] [PubMed] [Google Scholar]
- Vaughn DW, Green S, Kalayanarooj S, Innis BL, Nimmannitya S, Suntayakorn S, Endy TP, Raengsakulrach B, Rothman AL, Ennis FA, Nisalak A. Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J Infect Dis. 2000;181:2–9. doi: 10.1086/315215. [DOI] [PubMed] [Google Scholar]
- Westaway EG, Mackenzie JM, Kenney MT, Jones MK, Khromykh AA. Ultrastructure of Kunjin virus-infected cells: colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J Virol. 1997;71:6650–61. doi: 10.1128/jvi.71.9.6650-6661.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler G, Randolph VB, Cleaves GR, Ryan TE, Stollar V. Evidence that the mature form of the flavivirus nonstructural protein NS1 is a dimer. Virology. 1988;162:187–196. doi: 10.1016/0042-6822(88)90408-4. [DOI] [PubMed] [Google Scholar]
- Yun SI, Kim SY, Rice CM, Lee YM. Development and application of a reverse genetics system for Japanese encephalitis virus. J Virol. 2003;77:6450–65. doi: 10.1128/JVI.77.11.6450-6465.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig.S1. Micrograph of an immunostained monolayer of BHK cells electroporated with in vitro synthesized DV1 subgenomic replicon RNA. Cells were electroporated with transcribed RNA from pACDV1repNS1*NS3*, seeded, and then fixed and immunostained with an anti-NS1 MAb 3 days post-electroporation.
Fig.S2. Micrographs of immunostained monolayers of BHK cells electroporated with full-length transcripts from three independently assembled full-length cDNAs assembled in a BAC plasmid (pBACDV1flcNS1*NS3*). The cells were fixed and immunostained with a HIMAF generated to DV1 (DV1poly), or MAbs D1-4G2 (Anti-E), or D2-7E11 (Anti-NS1) 2 days post-electroporation.
Fig.S3. Micrographs of immunostained foci of infection produced by DV1BR/90 (parental virus) or the recombinant DNA derived virus DV1flcNS1*NS3* on monolayers of C7/10, C6/36 and Vero cells. Monolayers of each cell type were infected with DV1-BR/90 or the virus derived from pBACDV1flcNS1*NS3*, overlaid with semi-solid media, fixed and stained with anti-NS1 antibody 4 days post-infection. The mosquito cell monolayers where grown at 30°C whereas the Vero cell monolayers were grown at 37°C. All micrographs were obtained using the same magnification.