

Abstract
Hepatocellular carcinoma (HCC) is a leading cause of cancer-related death worldwide. HCC patients frequently present with disease that has metastasized to other regions of the liver, the portal vein, lymph nodes, or lungs, leading to poor prognoses. Therefore, model systems that allow exploration of the molecular mechanisms underlying metastasis in this disease are greatly needed. We describe here a metastatic HCC model generated after the somatic introduction of the mouse polyoma virus middle T antigen to mice with liver-specific deletion of the Trp53 tumor suppressor locus, and demonstrate the cell autonomous effect of p53 loss of function on HCC metastasis. We additionally find that cholangiocarcinoma (CC) also develops in these mice and some tumors display features of both HCC and CC, suggestive of origin from liver progenitor cells. Concomitant loss of the Ink4a/Arf tumor suppressor locus accelerates tumor formation and metastasis, suggesting potential roles for the p16 and p19 tumor suppressors in this process. Significantly, tumor cell lines isolated from tumors lacking both Trp53 and Ink4a/Arf display enhanced invasion activity in vitro relative to those lacking Trp53 alone. Thus, our data illustrate a new model system amenable for the analysis of HCC metastasis.
Keywords: HCC, mouse model, Trp53, Ink4a/Arf, metastasis
Introduction
Hepatocellular carcinoma (HCC) is a leading cause of cancer mortality worldwide. More than 500,000 people are diagnosed annually with this malignancy, with a 5-year survival rate below 5 percent (1). HCCs are resistant to current chemotherapeutic intervention strategies and therefore surgery remains the only curative option (2). However, surgical resection is not an option in patients with metastasis - most commonly observed in the extra-hepatic region of the portal vein, lymph nodes, and lungs - a common finding at the time of diagnosis (3). Therefore, understanding the mechanisms underlying the spread of HCC is of great importance.
HCC is commonly associated with chronic infection by the hepatitis B (HBV) and hepatitis C (HCV) viruses, and the mechanisms by which these viruses contribute to tumor initiation and progression remain critical areas of investigation (2, 4, 5). Previous studies have suggested that protein products of HBV and HCV - the HBx and NS5 proteins respectively - interact with the p53 tumor suppressor protein and inhibit its function (6, 7). In the cellular genome, mutations are commonly found in TP53, confirming the significance of loss of p53 function in HCC pathogenesis (8). Silencing of the INK4A/ARF locus also occurs in a majority of HCC cases (8-12). Additionally, while RB1 is seldom inactivated by mutation, the retinoblastoma tumor suppressor protein is frequently inactivated by overexpression of the oncoprotein gankyrin, which functionally targets both p53 and pRb (13). Thus, the Arf-p53 and p16-pRb tumor suppressor axes are targeted for inactivation in the majority of HCC cases.
Many previous studies have described mouse models for HCC (14-23). We have recently described a mouse model for HCC in which somatic delivery of avian retroviral vectors encoding the mouse polyoma virus middle T antigen (PyMT) to the livers of albumin-tv-a transgenic mice leads to the formation of hepatic tumors (24). While these tumors can exceed 1 cm in diameter, they rarely metastasize to the lungs. Significantly, we found that while induction of tumors in tv-a transgenic mice with germline Trp53 deletion did not lead to a higher tumor incidence, it led to a higher rate of lung metastases (24). These findings are consistent with the hypothesis that loss of TP53 is a late event associated with tumor progression in HCC, as well as with previously published findings on Trp53 loss and tumor progression in mouse models (25). However, mice with germline deletion of Trp53 are not suitable for studies of cancer progression because of the rapid occurrence of thymic lymphomas and sarcomas that lead to a shortened lifespan (26, 27). Furthermore, because all cells within the mouse lack the p53 tumor suppressor, it is not possible to identify cell autonomous requirements for tumor metastasis using these mice. Therefore, mouse models without these deficiencies will be of great benefit to the field.
We report here the development of mouse models for HCC after the somatic delivery of oncogene-bearing avian retroviruses into mice with liver-specific deletion of tumor suppressor genes. We find that PyMT is able to induce metastatic liver tumors in mice with liver-specific deletion of Trp53, suggesting that the effect of p53 on metastasis in our HCC model occurs in a cell autonomous fashion. Furthermore, we find that concomitant loss of Trp53 and Ink4a/Arf accelerates tumorigenesis and the development of metastatic lung lesions. Consistent with this finding, cell lines generated from tumors lacking both Trp53 and Ink4a/Arf exhibited strong migration and invasion capabilities, whereas a cell line generated from a Trp53 null tumor did not. Significantly, despite their poor migration and invasion capabilities, Trp53 null tumor cells, as well as cells lacking Trp53 and Ink4a/Arf, efficiently colonized the lungs of nude mice after intravenous injection, suggesting that the ability to colonize distant sites can occur before the acquisition of invasive capability. Thus, our data collectively demonstrate the cell autonomous role for p53 in the metastatic phenotype of HCC cells, and illustrate the utility of our model system for the elucidation of the molecular mechanisms underlying HCC metastasis.
Materials and Methods
Mouse lines
Albumin-tv-a and albumin-cre transgenic mice, and Trp53 and Ink4a/Arf conditional mutant mice have been previously described (24, 28-30). All animals were kept in specific pathogen-free housing at the University of Massachusetts Medical School with abundant food and water. All experiments were reviewed and approved by the University of Massachusetts Institutional Animal Care and Use Committee.
Virus delivery
The RCAS-GFP and RCAS-PyMT vectors have been previously described (31, 32). DF1 chicken fibroblasts (33, 34) transfected with RCAS vectors were maintained in DMEM supplemented with 10% fetal bovine serum in humidified 37°C incubators under 5% CO2. Cells to be injected were harvested, washed once with PBS and resuspended in PBS at a final concentration of 2 x 105 cells/μl. 5μl of the cell suspension was delivered by injection into the liver parenchyma of 2- or 3-day old animals using Hamilton syringes attached with 26 gauge needles.
Tumor harvest and histology
Animals were sacrificed with a lethal dose of CO2 as per institutional guidelines. Tumors, liver tissue, and lungs were removed and either fixed in 10% buffered formalin overnight at room temperature or snap frozen in liquid nitrogen. Fixed tissues were paraffin embedded and 5μm sections placed on sialynated slides.
Immunohistochemistry
Paraffin sections were deparaffinized and rehydrated by passage through Clear-Rite 3 and a graded alcohol series. Endogenous peroxidase activity was inactivated by treatment with 3% hydrogen peroxide after which antigen retrieval was performed utilizing heated citric buffer. Slides were blocked for 1 hour, and then incubated with appropriate primary and secondary antibodies according to manufacturers' instructions. Substrate incubation and color development were performed according to the manufacturer's instructions (Vector Labs). Primary antibodies: mouse anti-E-cadherin 1:50 (BD); mouse anti-β-catenin 1:100 (BD); TROMA-1 (anti-keratin 8) 1:50 (University of Iowa Developmental Studies Hybridoma Bank); and rat anti-polyoma virus T antigens 1:500 (gift of Michelle Fluck, Michigan State University).
Immunoblotting
Cell lysates were isolated in RIPA buffer after incubation on ice for 30 minutes. Protein concentration was determined using BCA reagent (Pierce) and 20 or 30μg of protein loaded per well. After transfer to PVDF or nitrocellulose membranes, primary antibodies were incubated for 1 hour at room temperature in Tris buffered saline, 0.1% Tween-20 (TBS-T), and appropriate blocking reagent, according to the manufacturer's instructions. After washing with TBS-T, blots were incubated with appropriate secondary antibodies (Jackson Immunochemicals) at room temperature for 45 minutes. Chemiluminescence was performed using either ECL reagent (Amersham) or Super Signal reagent (Pierce, Rockford, IL). Primary antibodies: rabbit anti-p-Akt 1:1000 (Cell Signaling); rabbit anti-p-Erk 1:1000 (Cell Signaling); rabbit anti-Akt 1:1000 (Cell Signaling); and rabbit anti-Erk 1:1000 (Cell Signaling).
Reverse-transcription polymerase chain reaction (RT-PCR)
RNA was isolated from frozen tumor tissue and tumor cell lines as previously described (24). 0.5μg of purified total RNA was utilized for RT-PCR. All RT-PCR reactions were performed in 50μl total reaction volume using the Superscript® III One-Step RT-PCR kit (Invitrogen). Primers: Cxcr4: 5′-ACCTCCTCTTTGTCATCACACTCC-3′ and 5′-AACACCACCATCCACAGGCTATCG-3′; p16/p19 (exon 2): 5′-ATGATGATGGGCAACGTTC-3′ and 5′-CAAATATCGCACGATGGTC-3′ Primers for tv-a, IGF2, snail, and β-actin have been previously described (24, 35). All reactions were performed for 35 cycles except for β-actin, which was performed for 25 cycles. Bands were quantified using a densitometer.
Isolation and culture of tumor cell lines
Immediately after harvest, tumor tissue was minced and dissociated by pipetting in DMEM. Cells were washed once in sterile PBS and plated in a 10 cm collagen-coated culture dish with DMEM supplemented with 10% fetal bovine serum (FBS) and antibiotics. After 48 hours, non-adherent cells were removed, adherent cells washed with PBS and fed with fresh medium containing FBS.
Cell proliferation assays
103 cells were seeded in quadruplicate onto 96-well plate with collagen coated and incubated at 37°C under 5% CO2. After 24 hours, viable cell numbers were measured in quadruplicate every period for 4 days using CellTiter 96 Aqueous One Solution Cell proliferation assay (Promega, Madison, WI) according to the manufacturer's instructions. The proliferation curves were constructed by calculating the mean value of optical density measurement at 490 nm using a 96-well plate reader.
Soft agar assays
Soft agar colony formation assays were performed as previously described (36).
Migration and invasion assays
2.5 x 104 cells in 0.5 ml of serum-free DMEM were plated into either control or matrigel-coated invasion inserts (BD). Inserts were then placed in wells with 0.75 ml of DMEM containing 10% FBS as a chemoattractant. After culture for 20-24 hours at 37°C, cells were fixed with methanol for 8 minutes at room temperature (RT) and stained with Giemsa reagent diluted 5X in H2O. Cells on the upper sides of the inserts were removed with a cotton swab, and the insert membranes removed and mounted on glass slides. Cell numbers for migration and invasion were then determined by counting the number of cells present in 5 microscope fields per insert at 50X magnification. The percent invasion was calculated as the number of invading cells divided by the number of migrating cells. All experiments were performed in duplicate and repeated a minimum of 3 times. Data are shown for representative experiments.
Tail vein injections
8-10-week old male nude mice were placed in a tail vein injector and restrainer, the tail swabbed with ethanol, and 105 cells resuspended in 300μl sterile PBS injected into the tail vein. Pressure was applied to the injection site for several seconds to prevent bleeding.
Results
Liver-specific Trp53 deletion induces tumors with metastatic potential
We have previously observed that liver tumors induced after somatic introduction of PyMT to albumin-tv-a mice with germline deletion of Trp53 metastasized to the lung in approximately 40% of cases (24). However, these animals have a reduced lifespan due to the development of thymic lymphomas and sarcomas, and therefore the analysis of tumor progression in these mice is compromised. We hypothesized that delivery of PyMT to mice bearing liver-specific deletion of Trp53 would lead to tumors with a similar phenotype, without the complications of additional non-hepatic tumors, thus providing a system to pursue studies related to tumor progression. We therefore crossed albumin-tv-a transgenic mice to animals bearing conditional mutant alleles (floxed alleles) of the p53 tumor suppressor, in which loxP sites are located in intron 1 and intron 10 (28), and to the albumin-cre transgenic mouse line (30), to generate triple transgenic mice with liver-specific p53 loss of function. To induce liver tumors in these mice, DF1 chicken fibroblasts producing RCAS-PyMT were injected into the livers of newborn mice as previously described (24). The animals were sacrificed at 6 or 8 months of age and analyzed for tumor development.
Consistent with our published findings (24), we found that 10/18 injected animals sacrificed at 6 months of age had liver tumors, with 7 of these animals bearing tumors greater than 6mm in diameter (Table 1). The primary tumors displayed a range of histologies, from aggregates of architecturally normal but cytologically atypical hepatocytes reminiscent of large cell change in humans, to frankly anaplastic and poorly differentiated carcinomas with regions of necrosis (Figure 1A, and data not shown). In addition, cholangiocarcinomas (CC) consisting of irregularly shaped glandular and micropapillary structures were observed in 2 tumor-bearing mice, either as a separate lesion (Figure 1A, right panel) or as a region within larger a HCC (combined hepatocellular-cholangiocarcinoma) (37). These histologic features resemble those of tumors found in animals with germline Trp53 deletion, suggesting that the histopathologic features are determined by the tumor cells themselves, and are not appreciably influenced by interactions with p53-deficient stromal and immune cells.
Table 1.
Tumor Incidence in Albumin-tv-a mice
| TSG Genotype | RCAS Virus | Age (Months) | Tumor Incidence | Metastasis Incidence |
|---|---|---|---|---|
| Trp53 null | PyMT | 6 | 10/18 | 3/10 |
| Trp53 null | PyMT | 8 | 3/6 | 0/3 |
| Trp53 null | GFP | 12 | 1/10 | 0/1 |
| Trp53 null | GFP | 15 | 1/15 | 0/1 |
| Ink4a/Arf null* | PyMT | 6 | 5/15 | 1/5 |
| Trp53, Ink4a/Arf null | PyMT | 4 | 8/9 | 2/8 |
| Trp53, Ink4a/Arf null | PyMT | 6 | 8/9 | 5/8 |
| Trp53, Ink4a/Arf null | GFP | 9 | 0/17 | N/A |
TSG = tumor suppressor gene
germline deletion
Figure 1.

(A) Histologic features of liver tumors induced by RCAS-PyMT in Trp53 null livers. Tumors with features of large cell dysplasia (left panel), hepatocellular carcinoma (center panel), and cholangiocarcinoma (right panel) are present. (B) Metastatic lung lesions derived from a hepatocellular carcinoma (left) and a cholangiocarcinoma (right). (C) Histology of a liver from an age-matched animal injected with RCAS-GFP.
Of the 7 mice bearing tumors greater than 6mm in diameter, 3 also had pulmonary metastases, a frequency higher than that observed in wild type animals (p<0.05) (24), indicating that loss of p53 contributes to metastasis in a cell autonomous fashion (Figure 1B). By histology and staining with anti cytokeratin 19 (CK19) antibodies, we determined that both HCCs and CCs display metastatic potential in our mouse model (Figure 1B, and data not shown). Consistent with the human disease, the metastatic cells grew as discrete expansile parenchymal nodules without infiltrating into the surrounding lung parenchyma.
Three of six animals sacrificed at 8 months of age had tumors, with only two of these exceeding 6mm in diameter, and none displaying lung metastases. The absence of higher tumor incidence and metastasis rates in 8-month-old mice suggests that, in our model system, the aggressiveness of the tumor and its metastatic capability are manifested by 6 months of age. Only 2/25 animals infected with RCAS-GFP and sacrificed between 12 and 15 months of age developed liver tumors (Figure 1C and Table 1).
The loss of E-cadherin is proposed to play a role in metastatic progression in several tumor types, including HCC. We therefore analyzed the expression of E-cadherin and its interacting partner β-catenin in metastatic lung lesions. Of the two lung metastases able to be analyzed, we found retention of membrane-localized E-cadherin and β-catenin in one lesion (consisting of a cholangiocarcinoma) and reduced expression of E-cadherin and β-catenin in the other (consisting of a HCC) (Figure 2A). Nuclear localization of β-catenin, indicative of activation of the canonical Wnt signaling pathway, was observed only in rare cells, predominantly stromal in origin, in the metastatic lesions or the primary tumors (Figure 2A, and data not shown).
Figure 2.
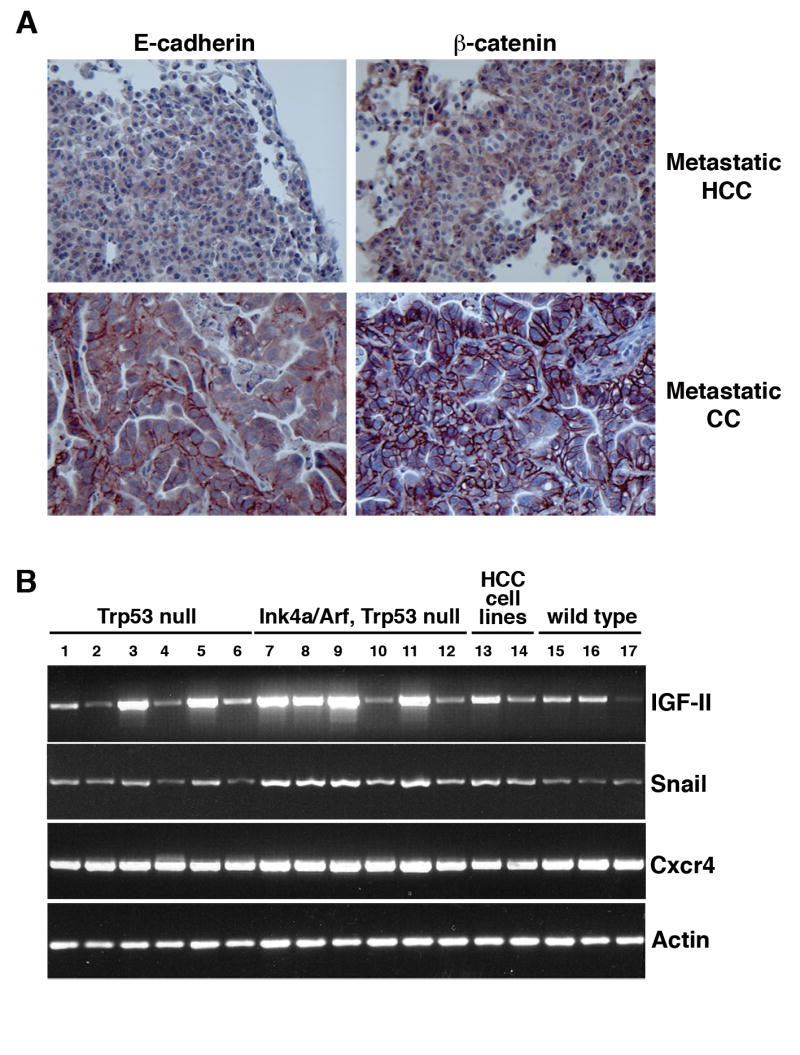
(A) Reduced expression of E-cadherin and β-catenin in an HCC-derived lung metastasis (middle panels) and retention of E-cadherin and β-catenin expression in a CC-derived lung metastasis (bottom panels) relative to a primary HCC (top panels). (B) RT-PCR analysis of wild-type, Trp53 null, and Trp53 plus Ink4a/Arf null primary liver tumors. Lanes 1-5, Trp53 null tumors; lane 6 Trp53 null GFP-infected non-tumorous liver; lanes 7-11, Trp53 plus Ink4a/Arf null tumors; lane 12, Trp53 plus Ink4a/Arf null GFP-infected non-tumorous liver; lanes 15-17, wild-type tumors; lane 13, Trp53 plus Ink4a/Arf null HCC cell line MM189; lane 14 Trp53 null cell line BL185.
Elevated mRNA levels for insulin-like growth factor 2 (Igf2) are observed in liver tumors induced by PyMT in animals with germline Trp53 deletion, and IGF-stimulated signaling correlates with HCC pathogenesis and tumor progression (24, 38, 39). To determine whether these changes occur in a cell autonomous fashion, we assayed by reverse transcriptase polymerase chain reaction (RT-PCR) the mRNA levels for Igf2 in tumors with liver-specific p53 loss. We observed a 1.6 fold increase in Igf2 mRNA levels in Trp53 null tumors relative to Trp53 wild type tumors (Figure 2B). These data suggest that the absence of p53 in PyMT-induced liver tumors enhances Igf2 expression, although the induction is not as robust we previously observed (24).
Recent reports have demonstrated that expression of the transcription factor snail in HCC samples correlates with metastasis and poor patient outcome (40, 41). Additional reports have suggested that the expression of the chemokine receptor Cxcr4 is associated with the development of lung metastasis in breast carcinoma cells and may regulate the migration of CC cells (42, 43). We therefore examined whether these genes were induced in tumors lacking p53. We found that snail mRNA levels are modestly (2-fold) induced in p53-deficient tumors relative to wild-type tumors (Figure 2B). We additionally observed that expression of Cxcr4 is constant in all tumors, even those with wild-type p53 alleles that display low metastatic potential (Figure 2B). These findings suggest that loss of p53 cooperates with PyMT to stimulate the expression of some pro-metastasis genes, such as Snail and Igf2, while the expression of other genes is not influenced by p53 status.
Concomitant deletion of Ink4a/Arf accelerates the metastatic phenotype
The Ink4a/Arf locus encodes two tumor suppressor proteins, both of which are commonly inactivated in HCC, and one of which, the p19Arf tumor suppressor protein, acts upstream of p53 (44). Therefore, to determine whether loss of the Ink4a/Arf locus stimulates the formation of lung metastases, we delivered RCAS-PyMT to albumin-tv-a mice with germline deletion of the Ink4a/Arf locus. 5/15 animals sacrificed at 6 months of age had grossly visible liver tumors (Table 1). Furthermore, of the 3 mice with tumors greater than 5mm in diameter, only one had metastasis to the lung. Thus, germline deletion of the Ink4a/Arf locus does not accelerate PyMT-induced liver tumorigenesis (24).
To identify whether the simultaneous loss of the Trp53 and Ink4a/Arf loci would cooperate in HCC tumor formation and metastasis, we generated compound mice transgenic for albumin-tv-a and albumin-cre, and bearing conditional mutant alleles at the Trp53 and Ink4a/Arf loci. Delivery of RCAS-PyMT producer cells led to the formation of tumors in 8/9 animals sacrificed and analyzed at 4 months of age (Table 1). All 8 tumor-bearing mice had tumors that exceeded 6mm in diameter, and metastatic lung lesions were observed in two of these animals. As was the case with Trp53 deficiency, liver-specific deletion of Trp53 and Ink4a/Arf combined with PyMT expression to induce a range of tumor histologies, including HCC (Figure 3A, left panel), cholangiocarcinoma (Figure 3A, 2nd panel), and combined hepatocellular-cholangiocarcinoma (Figure 3A, 3rd panel). In addition, we identified undifferentiated tumors with frankly anaplastic cells (Figure 3A, right panel). 8/9 animals sacrificed at 6 months of age also had tumors, with 7 of these animals bearing lesions larger than 6mm in diameter. Of these 7 mice, 5 displayed visible metastases to the lungs or diaphragm (Table 1 and Figure 3B). By histopathology, all of the metastatic lesions in this cohort appeared to be derived from HCCs.
Figure 3.

(A) Histologic findings in RCAS-PyMT injected Trp53 plus Ink4a/Arf null livers. Left panel - Hepatocellular carcinoma; 2nd panel - cholangiocarcinoma (arrow) growing adjacent to a hepatocellular carcinoma (asterisk); 3rd panel – aggressive cholangiocarcinoma (arrow) within a hepatocellular carcinoma; right panel – hepatocellular carcinoma cells with giant nuclei (arrowheads). (B) Metastatic lesions from Trp53 plus Ink4a/Arf null tumors found in the lung (left panel) and diaphragm (right panel). M denotes metastatic lesion, D denotes diaphragm. (C) Immunoblots demonstrating levels of phosphorylated and total Erk 1/2 and Akt in Trp53 null and Trp53 plus Ink4a/Arf null liver tumors. T= tumor and N= normal liver.
To determine whether the accelerated tumorigenesis reflected enhanced signaling by the PyMT oncoprotein, we examined the activation status of the MAP kinase and PI3 kinase signaling pathways by immunoblotting. Intriguingly, we found that the MAP kinase and PI3 kinase signaling pathways are more highly activated in p53 deficient tumors than in those lacking both p53 and Ink4a/Arf (Figure 3C). These data suggest that the accelerated tumor phenotype in the double null mice is not a consequence of the enhanced activation of these signaling pathways.
To identify whether metastasis of Trp53, Ink4a/Arf double null tumors required the loss of E-cadherin expression, we analyzed metastatic lesions for the presence of E-cadherin and β-catenin. As was observed for Trp53-deficient lesions, E-cadherin levels were reduced in 2/4 metastatic lesions (data not shown), indicating that loss of membrane-localized E-cadherin and β-catenin is associated with metastasis, but is not required for the metastatic phenotype. As was seen in Trp53 null tumors, double null tumors did not display nuclear accumulation of β-catenin (data not shown).
We next assayed whether these phenotypes were linked to elevated expression of Igf2, Snail, and Cxcr4. Consistent with our findings in p53-deficient tumors, we found that Cxcr4 mRNA levels were not increased relative to tumors with wild-type tumor suppressor gene loci (Figure 2B). However, double null tumors displayed further enhancement of Igf2 (2.5 fold) and Snail (3.8 fold) mRNA levels relative to wild-type tumors (Figure 2B). Given the suspected roles of Igf2 and snail in HCC progression, this finding may represent a potential explanation for the earlier onset of metastatic lesions in the double null animals (38, 40, 41).
Trp53-, Ink4a/Arf-deficient tumor cells invade in vitro
To further characterize the properties of tumor cells lacking Trp53 alone, or both Trp53 and Ink4a/Arf, we established cell lines from primary liver tumors. We were able to establish one cell line, BL185, lacking Trp53, and two cell lines, MM189 and BL322, lacking both Trp53 and Ink4a/Arf. Immunostaining of these cell lines with antibodies against PyMT and keratin 8 confirmed their origin from transformed hepatic epithelial cells (Figure 4A). Further, analysis by RT-PCR demonstrated the expression of tv-a, and the appropriate expression (or lack thereof) of p16 and p19 in these cell lines (Figure 4B).
Figure 4.
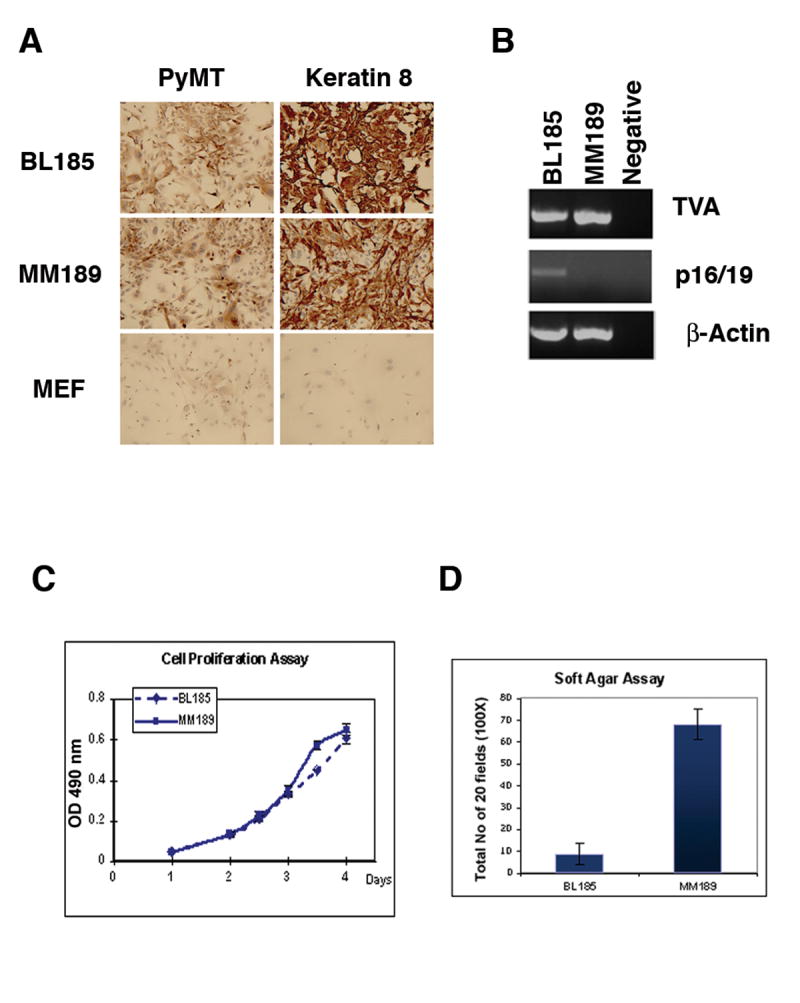
(A) Immunostaining demonstrating expression of PyMT and the epithelial marker keratin 8 in cell lines derived from PyMT-induced liver tumors. Mouse embryo fibroblasts (MEFs) are used as a negative control. (B) RT-PCR analysis for TVA and p16/19 in the HCC cell lines. β-actin serves as a control. Measurement of the proliferation (performed in quadruplicate) (C), and soft agar colony formation potential (performed in triplicate) (D) of these cell lines. Data are from representative experiments. Error bars represent standard deviation.
We next characterized the phenotypic features of the MM189 and BL185 cell lines in further detail. We found that both cell lines displayed similar growth rates when cultured in the presence of 10% fetal bovine serum, suggesting that the loss of the Ink4a/Arf locus does not increase the inherent growth rate of p53-deficient tumor cells in culture (Figure 4C). Interestingly, both cell lines proliferated in medium containing 0.1% serum without undergoing apoptosis (data not shown). When we measured the ability of these cell lines to grow in soft agar, we found that MM189 cells, which lack both Trp53 and Ink4a/Arf, have a greatly enhanced ability to form colonies relative to the BL185 cell line lacking Trp53 alone (Figure 4D). Thus, the loss of the Ink4a/Arf locus may enhance the transformation of p53-deficient hepatocytes.
An essential property of metastatic tumor cells is the ability to migrate and invade (45). We therefore measured the ability of the MM189 and BL185 cell lines to migrate and invade using cell culture chamber assays. We found that BL185 cells do not migrate or invade in significant numbers in response to serum as a chemoattractant (Figure 5A). By contrast, MM189 cells, which lack both Trp53 and Ink4a/Arf, display robust migration and invasion in response to serum (Figure 5A). Similar results were obtained with the BL322 cell line that also lacks both Trp53 and Ink4a/Arf (data not shown). Thus, our results suggest that loss of both the Trp53 and Ink4a/Arf tumor suppressor loci is required to induce tumor cell migration and invasion in our model system.
Figure 5.
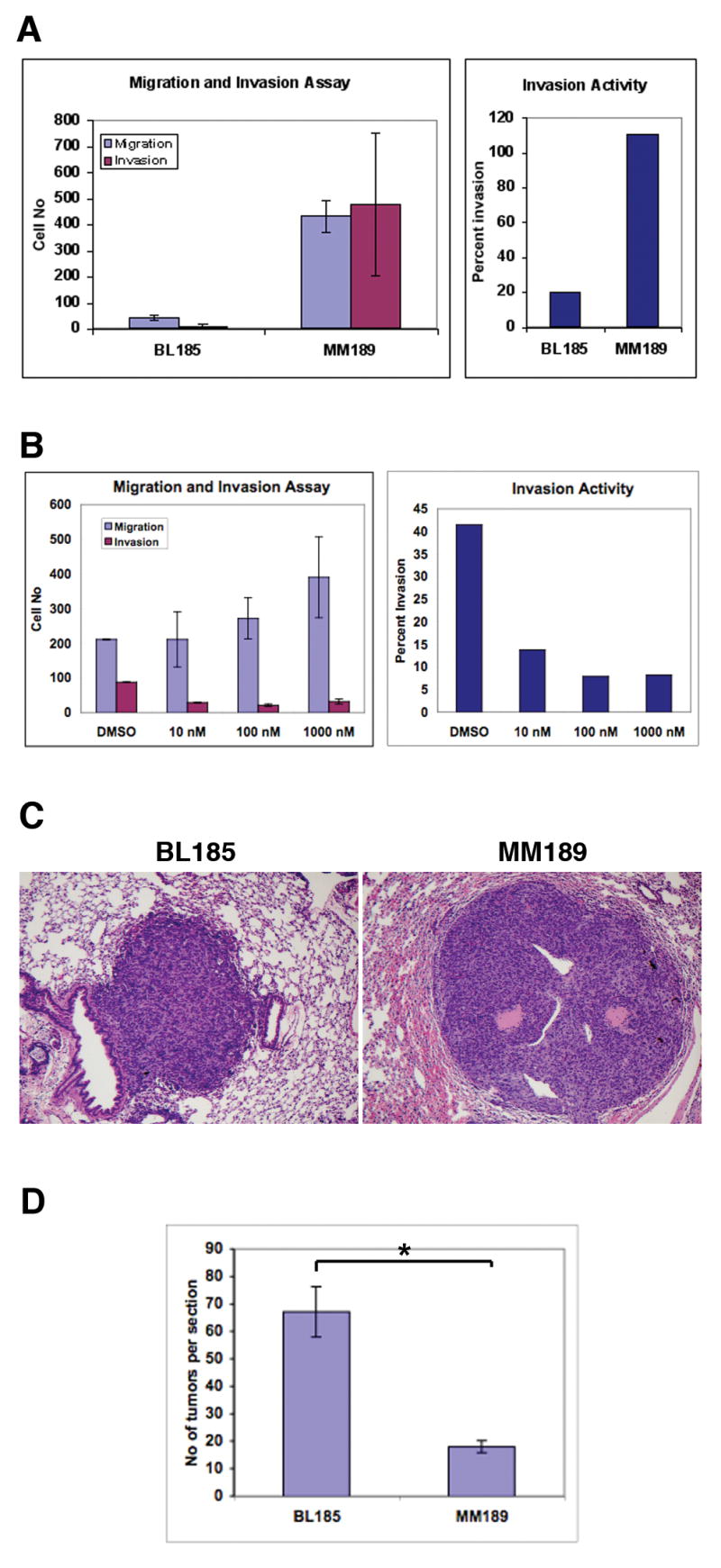
(A) Measurement of the migration (blue) and invasion (red) capabilities of the BL185 and MM189 cell lines (left panel). Error is standard deviation from the mean. The number of invading cells is plotted as a percentage of the number of migrating cells (right panel). (B) Migration and invasion activity of MM189 cells in response to increasing concentrations of the mTOR antagonist rapamycin (left panel). Error is standard deviation from the mean. The number of invading cells is plotted as a percentage of the number of migrating cells (right panel). (C) H&E stained images of lung lesions induced after tail vein injection of the BL185 (left panel) and MM189 (right panel) cell lines. (D) Quantification of lung colony formation in the tail vein injection assay. * p<0.001. Error bars are standard error of the mean.
We have previously demonstrated that attenuation of PyMT signaling through the PI3-kinase/Akt signaling pathway impairs lung metastasis in our HCC mouse model (24). To identify whether this occurs, in part, due to reduced cell migration and/or invasion, we measured the migration and invasion activities of MM189 cells treated with the compound rapamycin that inhibits mTOR, a key downstream effector of Akt signaling. We found that rapamycin inhibits tumor cell invasion at a concentration of 10nM (Figure 5B). Paradoxically, while rapamycin does not affect cell migration at this concentration, it progressively stimulates migration at higher concentrations (Figure 5B). Thus, in our cell culture model system, rapamycin blocks invasion and uncouples invasion from cell migration.
Trp53-deficient tumor cells colonize the lung
The poor in vitro migration and invasion capabilities of the p53-deficient cell line raised the question of how these cells were able to form metastases in vivo. We therefore determined whether these cells are able colonize the lung after introduction into the bloodstream. Injection of 105 BL185 cells into nude mice via the tail vein led to the development of lung lesions, identified upon sacrifice of the animals within 28 days after injection (Figure 5C). A similar phenotype was observed with MM189 cells that lack both Trp53 and Ink4a/Arf (Figure 5C). Quantification of the number of lung lesions demonstrated that BL185 cells formed more lesions than MM189 cells (Figure 5D, p<0.001), although the lesions induced by MM189 were on average of larger size. Thus, HCC tumor cells lacking p53 have the capacity to efficiently colonize the lung, a property that is not further enhanced by loss of Ink4a/Arf.
Discussion
The high frequency of intra-hepatic and distant metastases in HCC is a critical issue in the management of patients with this disease. Currently, surgical resection remains the only curative option, yet the presence of metastases eliminates this treatment option. Thus, identifying factors that correlate with the development of metastatic disease and tumor recurrence is of great importance. Indeed, recently published work has identified tumor cell-dependent and tumor microenvironment-dependent gene expression changes associated with the progression to a metastatic state in HCC (46, 47).
Therefore, the development of experimental model systems, both in vivo and in vitro, that allow the dissection of the molecular mechanisms involved in HCC metastasis is of primary importance. We have previously demonstrated the feasibility of modeling metastatic HCC after the somatic and sporadic activation of oncogenes (24). In this paper, we described the refinement of this system to utilize tissue-specific deletion of tumor suppressor genes. This modification allows us to clearly assess contributions that occur in a cell autonomous manner, and we demonstrate here that the effects of Trp53 and Ink4a/Arf deficiency on HCC metastasis occur in a cell autonomous fashion. Further, our data suggest that while loss of p53 enhances the metastatic potential of HCCs, it may not increase the ability of the tumor cells to migrate and invade. A critical caveat for this latter finding is that, to date, we have analyzed only a single cell line derived from a p53 null tumor. Importantly, however, the p53-deficient cell line efficiently colonizes the lung after introduction into the blood stream. This is consistent with the hypothesis that tumor cells develop at an early stage many of the traits required for metastasis (48). It also suggests that in p53-deficient HCCs, the rate-limiting step may be invasion through the basement membrane and intravasation to enter the circulation, because once in the bloodstream, these cells home effectively to the lungs and establish metastatic lesions.
In HCC patients, p53 function is frequently compromised by point mutation and loss of heterozygosity of the wild-type allele (8). Olive et al have recently demonstrated that p53 point mutants commonly found in human tumors display gain of function properties (49). Therefore, it would be intriguing to determine whether the presence of p53 point mutants will enhance the formation of metastasis in our model system and increase the migration and invasion capabilities of cell lines derived from these tumors.
One advantage of our experimental system is the ability to analyze the role of specific domains of oncoproteins and downstream signaling molecules through the rapid generation and introduction of RCAS viruses encoding mutant proteins. We previously exploited this feature to demonstrate the importance of PI3 kinase signaling in the induction of metastasis by PyMT (24). Significantly, we have now observed that inhibition of this pathway with rapamycin reduces the invasion of HCC cell lines, suggesting that one potential mechanism by which PI3 kinase signaling contributes to HCC metastasis may be the induction of tumor cell invasion. Importantly, activating mutations in the catalytic subunit of PI3 kinase are common in HCC (50), and therefore our results suggest that one potential consequence of these activating mutations is the enhancement of cell invasion. Thus, our new results confirm and extend our prior in vivo observations on the role of PI3 kinase signaling in HCC metastasis, and highlight the utility of our experimental model systems for the elucidation of the mechanisms involved in HCC metastasis.
Our data also point to potential roles for the p16 and p19 tumor suppressors in HCC progression. Inactivation of the locus encoding these proteins is a common finding in HCC, yet the roles of these tumor suppressors in the initiation and progression of this disease remain unknown (9-12). Our results indicate that these tumor suppressors may act to enhance both tumor initiation and tumor progression. Indeed, analysis of the TP53 and INK4A/ARF loci in human HCC samples suggested that the loss of these loci might act cooperatively in this disease, although the specific roles of p16INK4A and p14ARF remain to be elucidated (12). Our data suggest that p16 and p19 may act to constrain tumor growth. Concomitant loss of Trp53 and Ink4a/Arf led to the development of palpable PyMT-induced liver tumors at 4 months of age, a phenotype not observed with Trp53 deletion alone. Our results also suggest that p16 and p19 may influence tumor progression by impeding cell invasion, although the mechanisms by which this occurs remain under investigation.
We also observed increased mRNA levels for the transcription factor snail in p53 and Ink4a/Arf double deficient tumors relative to p53 wild-type tumors that have low metastatic potential. Intriguingly, snail mRNA levels are elevated in invasive subclones of the non-invasive BL185 cell line (Y-W.C., J.L.T., and B.C.L., unpublished observations), and previous studies have associated snail with tumor metastasis and poor outcome in HCC (40, 41). It remains to be seen whether snail is required for HCC tumor cell invasion, and whether it is involved in the regulation of Igf2 and cathepsin E, two additional molecules that we have previously associated with metastatic potential in our mouse model (24).
Thus, our results describe a new system for the generation and analysis of HCC invasion and metastasis. The examination of these processes using our model should significantly aid our understanding of malignant progression in this disease.
Acknowledgments
The authors thank Anton Berns for Trp53 and Ink4a/Arf conditional mutant mouse strains; Kirsten Hubbard and Jennifer Morton for critical reading of the manuscript; and members of the Lewis Lab for helpful discussion. The authors apologize to colleagues whose studies were not cited due to space constraints. B.C.L is a Liver Scholar of the American Liver Foundation and the recipient of a Career Development Award in the Biomedical Sciences from the Burroughs Wellcome Fund. Supported by NIH grant CA121171 to B.C.L.
Financial Support: Liver Scholar Award from the American Liver Foundation; Career Development Award in the Biomedical Sciences from the Burroughs Wellcome Fund; NIH grant CA121171 to B.C.L.
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–87. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 3.Lau JWY, Leow CK. Surgical management. In: Leong ASY, Liew CT, Lau JWY, Johnson PJ, editors. Hepatocellular Carcinoma: Diagnosis, investigation and management. London: Arnold; 1999. pp. 147–72. [Google Scholar]
- 4.Robinson WS. Molecular events in the pathogenesis of hepadnavirus-associated hepatocellular carcinoma. Annu Rev Med. 1994;45:297–323. doi: 10.1146/annurev.med.45.1.297. [DOI] [PubMed] [Google Scholar]
- 5.Simonetti RG, Cottone M, Craxi A, et al. Prevalence of antibodies to hepatitis C virus in hepatocellular carcinoma. Lancet. 1989;2:1338. doi: 10.1016/s0140-6736(89)91946-6. [DOI] [PubMed] [Google Scholar]
- 6.Majumder M, Ghosh AK, Steele R, Ray R, Ray RB. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J Virol. 2001;75:1401–7. doi: 10.1128/JVI.75.3.1401-1407.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ueda H, Ullrich SJ, Gangemi JD, et al. Functional inactivation but not structural mutation of p53 causes liver cancer. Nat Genet. 1995;9:41–7. doi: 10.1038/ng0195-41. [DOI] [PubMed] [Google Scholar]
- 8.Buendia MA. Genetics of hepatocellular carcinoma. Semin Cancer Biol. 2000;10:185–200. doi: 10.1006/scbi.2000.0319. [DOI] [PubMed] [Google Scholar]
- 9.Liew CT, Li HM, Lo KW, et al. High frequency of p16INK4A gene alterations in hepatocellular carcinoma. Oncogene. 1999;18:789–95. doi: 10.1038/sj.onc.1202359. [DOI] [PubMed] [Google Scholar]
- 10.Matsuda Y, Ichida T, Matsuzawa J, Sugimura K, Asakura H. p16(INK4) is inactivated by extensive CpG methylation in human hepatocellular carcinoma. Gastroenterology. 1999;116:394–400. doi: 10.1016/s0016-5085(99)70137-x. [DOI] [PubMed] [Google Scholar]
- 11.Jin M, Piao Z, Kim NG, et al. p16 is a major inactivation target in hepatocellular carcinoma. Cancer. 2000;89:60–8. doi: 10.1002/1097-0142(20000701)89:1<60::aid-cncr9>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 12.Tannapfel A, Busse C, Weinans L, et al. INK4a-ARF alterations and p53 mutations in hepatocellular carcinomas. Oncogene. 2001;20:7104–9. doi: 10.1038/sj.onc.1204902. [DOI] [PubMed] [Google Scholar]
- 13.Higashitsuji H, Liu Y, Mayer RJ, Fujita J. The oncoprotein gankyrin negatively regulates both p53 and RB by enhancing proteasomal degradation. Cell Cycle. 2005;4:1335–7. doi: 10.4161/cc.4.10.2107. [DOI] [PubMed] [Google Scholar]
- 14.Beer S, Zetterberg A, Ihrie RA, et al. Developmental context determines latency of MYC-induced tumorigenesis. PLoS Biol. 2004;2:E332. doi: 10.1371/journal.pbio.0020332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deane NG, Lee H, Hamaamen J, et al. Enhanced tumor formation in cyclin D1 x transforming growth factor beta1 double transgenic mice with characterization by magnetic resonance imaging. Cancer Res. 2004;64:1315–22. doi: 10.1158/0008-5472.can-03-1772. [DOI] [PubMed] [Google Scholar]
- 16.Harada N, Oshima H, Katoh M, Tamai Y, Oshima M, Taketo MM. Hepatocarcinogenesis in mice with beta-catenin and Ha-ras gene mutations. Cancer Res. 2004;64:48–54. doi: 10.1158/0008-5472.can-03-2123. [DOI] [PubMed] [Google Scholar]
- 17.Horie Y, Suzuki A, Kataoka E, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113:1774–83. doi: 10.1172/JCI20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jhappan C, Stahle C, Harkins RN, Fausto N, Smith GH, Merlino GT. TGF alpha overexpression in transgenic mice induces liver neoplasia and abnormal development of the mammary gland and pancreas. Cell. 1990;61:1137–46. doi: 10.1016/0092-8674(90)90076-q. [DOI] [PubMed] [Google Scholar]
- 19.Manickan E, Satoi J, Wang TC, Liang TJ. Conditional liver-specific expression of simian virus 40 T antigen leads to regulatable development of hepatic neoplasm in transgenic mice. J Biol Chem. 2001;276:13989–94. doi: 10.1074/jbc.M009770200. [DOI] [PubMed] [Google Scholar]
- 20.Murakami H, Sanderson ND, Nagy P, Marino PA, Merlino G, Thorgeirsson SS. Transgenic mouse model for synergistic effects of nuclear oncogenes and growth factors in tumorigenesis: interaction of c-myc and transforming growth factor alpha in hepatic oncogenesis. Cancer Res. 1993;53:1719–23. [PubMed] [Google Scholar]
- 21.Sandgren EP, Quaife CJ, Pinkert CA, Palmiter RD, Brinster RL. Oncogene-induced liver neoplasia in transgenic mice. Oncogene. 1989;4:715–24. [PubMed] [Google Scholar]
- 22.Terradillos O, Billet O, Renard CA, et al. The hepatitis B virus X gene potentiates c-myc-induced liver oncogenesis in transgenic mice. Oncogene. 1997;14:395–404. doi: 10.1038/sj.onc.1200850. [DOI] [PubMed] [Google Scholar]
- 23.Zender L, Spector MS, Xue W, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125:1253–67. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewis BC, Klimstra DS, Socci ND, Xu S, Koutcher JA, Varmus HE. The absence of p53 promotes metastasis in a novel somatic mouse model for hepatocellular carcinoma. Mol Cell Biol. 2005;25:1228–37. doi: 10.1128/MCB.25.4.1228-1237.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kemp CJ, Donehower LA, Bradley A, Balmain A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell. 1993;74:813–22. doi: 10.1016/0092-8674(93)90461-x. [DOI] [PubMed] [Google Scholar]
- 26.Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 27.Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 28.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29:418–25. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 29.Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature. 2001;413:83–6. doi: 10.1038/35092584. [DOI] [PubMed] [Google Scholar]
- 30.Postic C, Shiota M, Niswender KD, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–15. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 31.Holland EC, Li Y, Celestino J, et al. Astrocytes give rise to oligodendrogliomas and astrocytomas after gene transfer of polyoma virus middle T antigen in vivo. Am J Pathol. 2000;157:1031–7. doi: 10.1016/S0002-9440(10)64615-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewis BC, Klimstra DS, Varmus HE. The c-myc and PyMT oncogenes induce different tumor types in a somatic mouse model for pancreatic cancer. Genes Dev. 2003;17:3127–38. doi: 10.1101/gad.1140403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Himly M, Foster DN, Bottoli I, Iacovoni JS, Vogt PK. The DF-1 chicken fibroblast cell line: transformation induced by diverse oncogenes and cell death resulting from infection by avian leukosis viruses. Virology. 1998;248:295–304. doi: 10.1006/viro.1998.9290. [DOI] [PubMed] [Google Scholar]
- 34.Schaefer-Klein J, Givol I, Barsov EV, et al. The EV-O-derived cell line DF-1 supports the efficient replication of avian leukosis-sarcoma viruses and vectors. Virology. 1998;248:305–11. doi: 10.1006/viro.1998.9291. [DOI] [PubMed] [Google Scholar]
- 35.Jamora C, Lee P, Kocieniewski P, et al. A signaling pathway involving TGF-beta2 and snail in hair follicle morphogenesis. PLoS Biol. 2005;3:e11. doi: 10.1371/journal.pbio.0030011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis BC, Shim H, Li Q, et al. Identification of putative c-Myc-responsive genes: characterization of rcl, a novel growth-related gene. Mol Cell Biol. 1997;17:4967–78. doi: 10.1128/mcb.17.9.4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tickoo SK, Zee SY, Obiekwe S, et al. Combined hepatocellular-cholangiocarcinoma: a histopathologic, immunohistochemical, and in situ hybridization study. Am J Surg Pathol. 2002;26:989–97. doi: 10.1097/00000478-200208000-00003. [DOI] [PubMed] [Google Scholar]
- 38.Scharf JG, Braulke T. The role of the IGF axis in hepatocarcinogenesis. Horm Metab Res. 2003;35:685–93. doi: 10.1055/s-2004-814151. [DOI] [PubMed] [Google Scholar]
- 39.Zhang D, Samani AA, Brodt P. The role of the IGF-I receptor in the regulation of matrix metalloproteinases, tumor invasion and metastasis. Horm Metab Res. 2003;35:802–8. doi: 10.1055/s-2004-814143. [DOI] [PubMed] [Google Scholar]
- 40.Miyoshi A, Kitajima Y, Kido S, et al. Snail accelerates cancer invasion by upregulating MMP expression and is associated with poor prognosis of hepatocellular carcinoma. Br J Cancer. 2005;92:252–8. doi: 10.1038/sj.bjc.6602266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sugimachi K, Tanaka S, Kameyama T, et al. Transcriptional repressor snail and progression of human hepatocellular carcinoma. Clin Cancer Res. 2003;9:2657–64. [PubMed] [Google Scholar]
- 42.Muller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 43.Ohira S, Sasaki M, Harada K, et al. Possible regulation of migration of intrahepatic cholangiocarcinoma cells by interaction of CXCR4 expressed in carcinoma cells with tumor necrosis factor-alpha and stromal-derived factor-1 released in stroma. Am J Pathol. 2006;168:1155–68. doi: 10.2353/ajpath.2006.050204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 45.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 46.Budhu A, Forgues M, Ye QH, et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10:99–111. doi: 10.1016/j.ccr.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 47.Ye QH, Qin LX, Forgues M, et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med. 2003;9:416–23. doi: 10.1038/nm843. [DOI] [PubMed] [Google Scholar]
- 48.Bernards R, Weinberg RA. A progression puzzle. Nature. 2002;418:823. doi: 10.1038/418823a. [DOI] [PubMed] [Google Scholar]
- 49.Olive KP, Tuveson DA, Ruhe ZC, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–60. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 50.Lee JW, Soung YH, Kim SY, et al. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene. 2005;24:1477–80. doi: 10.1038/sj.onc.1208304. [DOI] [PubMed] [Google Scholar]