

Abstract
Activation of hepatic stellate cells (HSC), the key effectors in hepatic fibrogenesis, is characterized by enhanced cell proliferation and overproduction of extracellular matrix. Oxidative stress promotes HSC activation. Glutathione (GSH) is the most important intracellular antioxidant, whose synthesis is mainly regulated by glutamate-cysteine ligase (GCL). We reported previously that (−)-epigallocatechin-3-gallate (EGCG), the major and most active component in green tea extracts, inhibited HSC activation. The aim of this study is to elucidate the underlying mechanisms. We hypothesize that this inhibitory effect of EGCG might mainly result from its antioxidant capability by increasing de novo synthesis of GSH. In this report, we observe that EGCG enhances the levels of cytoplasmic and mitochondrial GSH and increases GCL activity by inducing gene expression of the catalytic subunit GCLc, leading to de novo synthesis of GSH. Real-time polymerase chain reaction and Western blotting analyses show that de novo synthesis of GSH is required for EGCG to regulate the expression of genes relevant to apoptosis and to cell proliferation. Additional experiments demonstrate that exogenous transforming growth factor (TGF)-β1 suppresses GCLc gene expression and reduces the level of GSH in cultured HSC. Transient transfection assays and Western blotting analyses further display that EGCG interrupts TGF-β signaling by reducing gene expression of TGF-β receptors and Smad4, leading to increased expression of GCLc. These results support our hypothesis and collectively demonstrate that EGCG increases the level of cellular GSH in HSC by stimulating gene expression of GCLc, leading to the inhibition of cell proliferation of activated HSC in vitro.
Hepatic stellate cells (HSC) are the major players during liver fibrogenesis. Upon liver injury, normally quiescent HSCs become activated, undergo profound morphological changes, and transdifferentiate into myofibroblast-like cells. This process is termed “HSC activation,” in which loss of vitamin A droplets, de novo expression of α-smooth muscle actin, enhanced cell proliferation, and excessive production of extracellular matrix (ECM) are the most characteristic features (Friedman, 2004; Kisseleva and Brenner, 2006). HSC activation is triggered by the release of mitogenic platelet-derived growth factor and epidermal growth factor (EGF) from activated HSC and fibrogenic transforming growth factor-β1 (TGF-β1), mainly from Kupffer cells (Win et al., 1993). This process is coupled with sequential up-expression of platelet-derived growth factor-β receptor (PDGF-βR) (Wong et al., 1994), type I and II receptors for TGF-β (Friedman et al., 1994), and EGF receptor (EGFR) (Kömüves et al., 2000). It is very important to note that culturing quiescent HSCs on plastic plates causes spontaneous activation, mimicking the process seen in vivo, which provides a good model for elucidating underlying mechanisms of HSC activation and for studying possible therapeutic intervention of the process (Friedman, 2004; Kisseleva and Brenner, 2006).
Oxidative stress reflects the imbalance of pro-oxidants and antioxidants. Oxidative stress-related molecules include reactive oxygen species and lipid peroxidation end products. Increasing evidence has demonstrated that oxidative stress promotes HSC activation and collagen production and plays an important role in the pathogenesis of liver fibrosis (Lee et al., 1995; Tsukamoto et al., 1995; Greenwel et al., 2000). Mammalian cells respond to oxidative stress through antioxidant defense, which includes antioxidant enzymes and nonenzyme molecules. Glutathione (GSH) is the predominant low-molecular-weight thiol and the most important nonenzyme antioxidant. GSH in cells is located in both cytoplasm and mitochondria. The mitochondrial pool of GSH in cells is critical for regulation of thiol and redox status (Kroemer et al., 1998). GSH is sequentially synthesized from glutamate, cysteine, and glycine, which is mainly controlled by the rate-limiting enzyme glutamate-cysteine ligase (GCL). GCL is composed of two subunits: the heavy catalytic subunit GCLc (73 kDa), and the light regulatory subunit GCLm (31 kDa). As an antioxidant, GSH effectively protects cells against damage caused by oxidative stress, including scavenging free radicals, removing hydrogen peroxide (H2O2) and lipid peroxides, and preventing oxidation of molecules in cells.
Application of antioxidants is rational in treatment and prevention of diseases closely associated with oxidative stress (Sueoka et al., 2001). Green tea has been consumed for thousands of years and has displayed numerous beneficial effects to human health (Sueoka et al., 2001). (−)-Epigallocatechin-3-gallate (EGCG), the major component in green tea, possesses potent antioxidant capability (Rice-Evans, 1999). Recent studies have demonstrated the effects of green tea extracts on the protection of the liver against early alcoholic injury in rats (Arteel et al., 2003). We reported previously that EGCG inhibited HSC activation by inhibiting cell proliferation and suppressing gene expression of ECM components (Chen et al., 2002; Chen and Zhang, 2003). In addition, we reported that EGCG inhibited ECM gene expression in activated HSC by interrupting TGF-β signaling through attenuating oxidative stress (Yumei et al., 2006). The aim of this study was to elucidate the mechanisms of EGCG in the inhibition of growth of activated HSC. We hypothesize that the inhibitory effect of EGCG on HSC growth might mainly result from its antioxidant capability by increasing de novo synthesis of GSH. Results presented in the current study support our hypothesis and provide novel insights into the mechanisms of EGCG in the inhibition of HSC activation.
Methods and Materials
Isolation and Culture of Hepatic Stellate Cells
HSCs were isolated using density gradient centrifugation with OptiPrep (Oslo, Norway) from livers of male Sprague-Dawley rats (200–250 g) as we described previously (Chen and Davis, 1999). HSCs were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS). HSCs with approximately four to eight passages were used for experiments. EGCG (purity, >95%), N-Acetyl-cysteine (NAC) and buthionine sulfoximine (BSO) were purchased from Sigma (St. Louis, MO). Glutathione monoethyl ester (GSH-MEE) was purchased from Calbiochem (San Diego, CA).
Western Blotting Analyses
Whole-cell extracts were prepared from cultured HSCs. Protein concentrations were determined using the BCA Protein Assay Kit according to the protocol provided by the manufacturer (Pierce, Rockford, IL). Thirty micrograms of total proteins was subjected to SDS-polyacrylamide gel electrophoresis (10%). Target proteins were detected by primary antibodies and secondary antibodies conjugated with horseradish peroxidase purchased from Santa Cruz Biotechnology (Santa Cruz, CA). β-Actin or β-tubulin was probed as an internal control. Protein bands were visualized by using chemiluminescence reagent (Amersham, Chalfont St. Giles, Buckinghamshire, UK).
RNA Isolation and Real-Time PCR
Total RNA was extracted using TRI-reagent according to the protocol provided by the manufacturer (Sigma). Real-time PCR was performed as we described previously (Chen, 2002). mRNA fold changes of target genes relative to the endogenous glyceraldehyde-3-phosphate dehydrogenase (GAPDH) control were calculated as suggested by Schmittgen et al. (2000). The primers used in these studies are given in Table 1.
TABLE 1.
Primer sequences
| EGFR | |
| Forward | 5′–TGC ACC ATC GAC GTC TAC AT–3′ |
| Reverse | 5′–AAC TTT GGG CGG CTA TCA G–3′ |
| PDGF-αR | |
| Forward | 5′–CTG CCA CAG CAT GAT GAG GAT TGA T–3′ |
| Reverse | 5′–GCC AGG ATG GCT GAG ATC ACC AC–3′ |
| Bax | |
| Forward | 5′–GGG TGG TTG CCC TTT TCT ACT–3′ |
| Reverse | 5′–CCC GGA GGA AGT CCA GTG TC–3′ |
| Bcl-2 | |
| Forward | 5′–ATG GGG TGA ACT GGG GGA TTG–3′ |
| Reverse | 5′–TTC CGA ATT TGT TTG GGG CAG GTC–3′ |
| GCLc | |
| Forward | 5′–GTC TTC AGG TGA CAT TCC AAG C–3′ |
| Reverse | 5′–TGT TCT TCA GGG GCT CCA GTC–3′ |
| GCLm | |
| Forward | 5′–CTG CTA AAC TGT TCA TTG TAG G–3′ |
| Reverse | 5′–CTA TGG GTT TTA CCT GTG–3′ |
| GAPDH | |
| Forward | 5′–GGC AAA TTC AAC GGC ACA GT–3′ |
| Reverse | 5′–AGA TGG TGA TGG GCT TCC C–3′ |
Determination of Cell Proliferation
Cell proliferation was determined by using the CellTiter 96 Aqueous Nonradioactive Cell Proliferation Assay Kit, following the protocol provided by the manufacturer (Promega, Madison, WI).
Plasmid Constructs and Transient Transfection
The GCLc promoter luciferase reporter plasmid pGCLc-luc was generated by inserting a promoter region (−1758/+2 base pairs) of rat GCLc into the pGL3-enhancer luciferase reporter plasmid (Yang et al., 2001). The cDNA expression plasmid pdn-Tβ-RII was a gift from Dr. Robert J. Lechleider (National Cancer Institute, Bethesda, MD), containing a full-length cDNA encoding the dominant-negative form of Tβ-RII (de Caestecker et al., 1998). The cDNA expression plasmid pSmad4-cDNA encodes full-length constitutively active form of Smad4, which was kindly provided by Dr. Lechleider as well (de Caestecker et al., 1998). Transient transfection assays were performed using Lipofectamine (Life Technologies, Carlsbad, CA) by following the protocol provided by the manufacturer. Transfection efficiency was controlled by cotransfection of the β-galactosidase reporter plasmid pSV-β (0.5~0.8 μg/well) (Promega). Luciferase activities were expressed as relative units after normalization with β-galactosidase activity. Results were combined from multiple independent experiments (n ≥ 6).
Isolation of Cytoplasmic and Mitochondrial Fractions for GSH Determination
Cytosol and mitochondria from cultured rat HSCs were prepared by using a Mitochondria Isolation Kit for Cultured Cells purchased from Pierce (Pierce Biotechnology, Inc., Rockford, IL). Mitochondrial pellets were resuspended in 1× phosphate-buffered saline containing 0.1% Triton X-100, disrupted by sonication, and centrifuged at 15,000g for 30 min. The supernatant was assayed for mitochondrial GSH.
GSH Assays
Levels of GSH and oxidized GSH were determined by using the enzyme immune assay kit GSH-400 (Cayman Chemical, Ann Arbor, MI) following the protocol provided by the manufacturer. The concentration of total GSH was calculated according to the equation in the protocol.
Analyses of GCL Activity
GCL activity was spectrophotometrically determined as described previously with slight modifications (Fraser et al., 2003). In brief, a sample of cell extracts (20 μl) was mixed with the reaction solution (0.21 ml) containing 100 mM Tris-HCl, pH 8.0, 150 mM KCl, 20 mM MgCl2, 2 mM Na2EDTA, 5 mM Na2ATP, 2 mM phosphoenolpyruvate, 10 mM l-glutamate, 10 mM l-α-aminobutyrate, 0.27 mM NADH, 2 μg of type II rabbit muscle pyruvate, and 2 μg of lactate dehydrogenase. The reaction was initiated by the addition of ATP to a final concentration at 5 mM. The decrease in the absorbance of NADH at 340 nm was monitored for 30 min with an interval of 2 min by using a SpectraMax 190 plate reader (Molecular Devices, Sunnyvale, CA) and expressed as millimoles of NADH oxidized per minute. Protein concentration was quantitated by BCA assay (Pierce). The final GCL activity was calculated and expressed as millimoles of NADH oxidized per minute per milligram of protein.
Statistical Analyses
Differences between means were evaluated using an unpaired two-sided Student’s t test (p < 0.05 was considered significant). Where appropriate, comparisons of multiple treatment conditions with control were analyzed by analysis of variance with the Dunnett’s test for post hoc analysis.
Results
EGCG Elevated the Level of Cellular GSH and Increased the Activity of GCL in HSCs
EGCG is a potent antioxidant (Rice-Evans, 1999). To evaluate the effect of EGCG on the level of GSH, cultured HSCs were treated with EGCG at 50 μM for various times. Cytosol and mitochondria were prepared from these cells for the determination of GSH. As shown in Fig. 1A, EGCG enhanced the levels of GSH in both cytoplasm and mitochondria in cultured HSCs. It was observed that EGCG had no significant effect on the elevation of the cellular GSH content in the first several hours, indicating a delayed response to the natural antioxidant. In addition, the EGCG enhancement of the GSH contents in cytoplasm and in mitochondria was biphasic at the beginning. We hypothesized that EGCG might increase the level of cellular GSH in HSCs by enhancing the activity of GCL, the rate-limiting enzyme in de novo synthesis of GSH. To test the hypothesis, the activity of GCL was analyzed in passaged HSCs treated with EGCG at indicated concentrations for 24 h. As shown in Fig. 1B, EGCG increased the activity of GCL in a dose-dependent manner in these cells. Taken together, these results suggested that EGCG might elevate the level of cellular GSH in HSCs by increasing the activity of GCL.
Fig. 1.
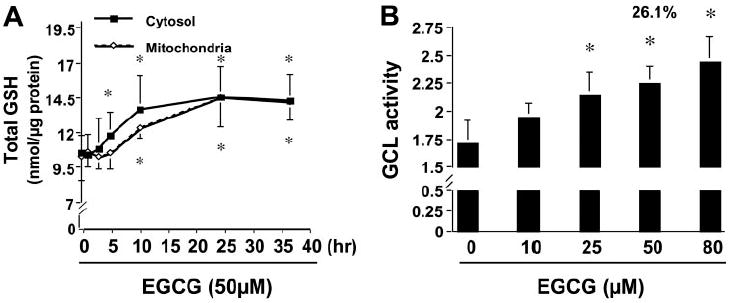
EGCG increases the levels of GSH and GCL activity in activated HSCs in vitro. Passaged HSCs were treated with EGCG at 50 μM for various times (A) or at various concentrations for 24 h (B). Values are means ± S.D. (n ≥ 3). *, p < 0.05 versus cells treated with no EGCG (the first column or point on the left side). A, determination of the levels of GSH in cytoplasm and in mitochondria. Total GSH was expressed as nanomoles per microgram of protein. B, measurement of GCL activity.
The Activity of GCL Was Required for EGCG to Elevate GSH Contents in HSC
Prior studies have demonstrated that BSO, a specific inhibitor of GCL, strongly inhibits GCL activity, leading to the depletion of cellular GSH (Griffith, 1982). To confirm the role of GCL in the EGCG elevation of the content of cellular GSH in activated HSCs, pilot experiments were performed to determine the effective dosage and duration of BSO treatment in passaged HSCs. As shown in Fig. 2, A and B, treatment of cells with BSO reduced the level of cellular GSH in activated HSCs in a dose-dependent (0~0.5 mM) and time-dependent (>8 h) manner. Compared with that in the control cells, the level of cellular GSH was significantly reduced by approximately 40% in cells treated with BSO at 0.25 mM for 24 h. BSO at concentrations higher than 0.5 mM showed no additional impact on the reduction of the level of GSH in cultured HSCs. Exposure of cells to BSO at concentrations lower than 0.5 mM for less than 30 h showed no significant effect on the cell viability (data not shown), which is consistent with other prior observations (Nieto et al., 1999). The treatment of HSCs with BSO at 0.25 mM for 24 h was, therefore, selected for our further experiments. Passaged HSCs were treated with EGCG (50 μM), NAC (5 mM), or GSH-MEE (2 mM) for 24 h with or without the pre-exposure to BSO (0.25 mM) for 1 h. NAC is a precursor in the formation of the antioxidant GSH in cells, whereas GSH-MEE is a cell-permeable derivative of GSH that undergoes hydrolysis by intracellular esterases to release GSH. The levels of GSH in these cells were analyzed. As expected, EGCG, as well as NAC and GSH-MEE, significantly increased the level of total intracellular GSH in the cells (Fig. 2C). The enhancement of cellular GSH concentration by EGCG and NAC but not GSH-MEE was completely blocked by pretreatment with the specific GCL inhibitor BSO, suggesting that GCL activity was required for EGCG and NAC to enhance the level of cellular GSH in cultured HSC. Prior study has demonstrated that the effect of GSH-MEE on increasing the level of cellular GSH was resistant to BSO as a result of transport of GSH-MEE followed by intracellular hydrolysis and release of GSH (Tsan et al., 1989). GCL is not directly involved in the elevation of GSH content from GSH-MEE. These results confirmed the role of GCL in the EGCG elevation of the content of cellular GSH in activated HSCs. It is noteworthy that compared with the GSH precursors, NAC at 5 mM and GSH-MEE at 2 mM, EGCG at 50 μM caused a similar if not greater effect on increasing the level of cellular GSH (Fig. 2C), suggesting that EGCG might use a different and more efficient mechanism to boost GSH synthesis.
Fig. 2.
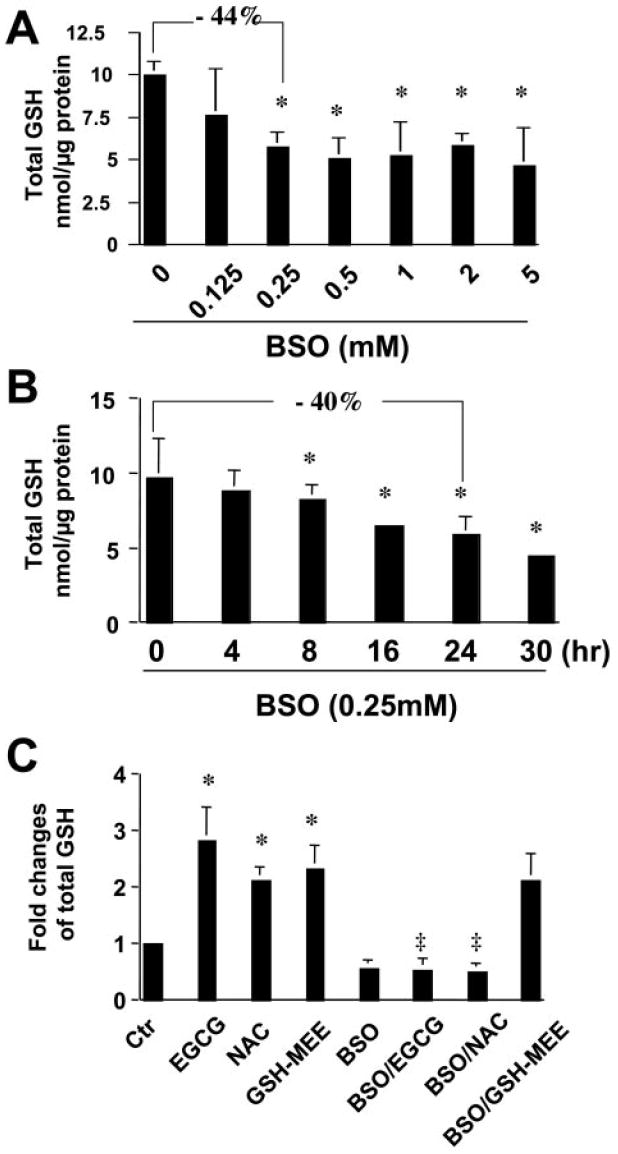
The inhibition of GCL activity by BSO eliminates the role of EGCG in the elevation of total cellular GSH in activated HSCs in vitro. The level of total cellular GSH was measured in passaged HSC. The level of cellular GSH was expressed as nanomoles per microgram of protein. Values are mean ± S.D. (n = 3). *, p < 0.05 versus the control cells (the first bar on the left). A, cells were treated with BSO at various concentrations for 24 h. B, cells were treated with BSO at 0.25 mM for various times. C, cells were treated with EGCG (50 μM), NAC (5 mM), or GSH-MEE (2 mM) for 24 h with or without the pre-exposure to BSO (0.25 mM) for 1 h. ‡, p < 0.05 versus cells treated with EGCG or NAC only (the second or the third bar on the left).
De Novo Synthesis of GSH Played Critical Roles in the EGCG Inhibition of Cell Proliferation and in the Regulation of Expression of Genes Relevant to Cell Proliferation
To test our assumption that the increase in the level of cellular GSH induced by EGCG might lead to the inhibition of cell proliferation of activated HSCs in vitro, HSCs were treated for 24 h with EGCG (50 μM), NAC (5 mM), or GSH-MEE (2 mM), with or without the pre-exposure of the cells to BSO (0.25 mM) for 1 h. As shown in Fig. 3A, EGCG significantly reduced, as expected, the number of viable HSCs by approximately 55% compared with the untreated control. The antioxidants NAC and GSH-MEE mimicked the inhibitory effect and also caused an apparent reduction in the number of viable HSCs. Blocking de novo GSH synthesis by BSO abrogated the inhibitory effects of EGCG and NAC on cell proliferation. However, the pretreatment with BSO could not diminish the role of GSH-MEE in the reduction in the number of viable cells. The production of GSH from GSH-MEE is resistant to BSO as a result of intracellular hydrolysis by esterases without the involvement of GCL (Tsan et al., 1989). These results collectively suggested the requirement of de novo synthesis and enhanced the level of cellular GSH in the EGCG inhibition of HSC growth.
Fig. 3.
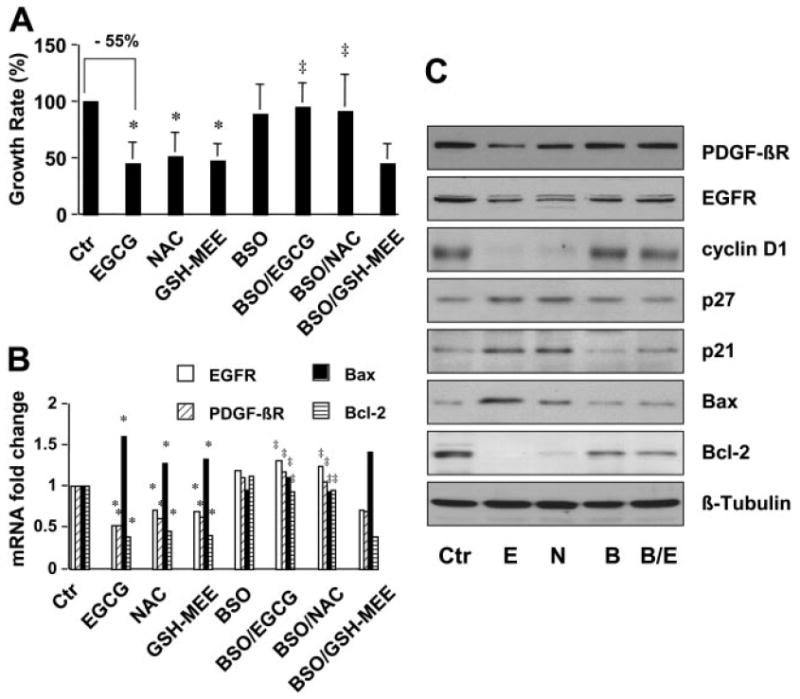
De novo synthesis of GSH is required for EGCG to inhibit HSC growth and to regulate the expression of genes relevant to cell proliferation. HSCs were treated for 24 h with EGCG (50 μM), NAC (5 mM), or GSH-MEE (2 mM) with or without the pre-exposure to BSO (0.25 mM) for 1 h. Values are means ± S.D. (n ≥ 3). *, p < 0.05 versus cells with no treatment (the first column on the left); ‡, p < 0.05 versus cells treated with EGCG or NAC only (the second or third column on the left). A, determination of viable cells. Values are expressed as fold changes compared with the control cells. B, real-time PCR analyses of the steady-state mRNA levels of genes. GAPDH was used as an invariant control for calculating fold changes of target mRNA (n = 3). C, Western blotting analyses of the abundance of proteins. β-Tubulin was used as an invariant control for equal loading. Representative was shown from three independent experiments.
To elucidate the underlying mechanisms of EGCG in the inhibition of cell proliferation of HSCs, we hypothesized that the elevation of the level of cellular GSH by EGCG might alter the expression of genes relevant to cell proliferation and to apoptosis. To test this hypothesis, passaged HSCs were treated with EGCG (50 μM), NAC (5 mM), or GSH-MEE (2 mM) for 24 h with or without the pre-exposure to BSO (0.25 mM) for 1 h. Gene expression was analyzed by real-time PCR and Western blotting analyses, respectively. As shown in Fig. 3B, the steady-state mRNA levels of PDGF-βR and EGFR, both of which mediate the most important mitogenic signaling in promoting HSC proliferation, were significantly reduced by EGCG, NAC, and GSH-MEE. Blocking de novo synthesis of GSH by the pretreatment with BSO eliminated the inhibitory effect of EGCG and NAC. However, BSO had no effect on GSH-MEE. Further experiments in Fig. 3B demonstrated that EGCG, NAC, and GSH-MEE increased the mRNA level of proapoptotic Bax and reduced the abundance of antiapoptotic Bcl-2 in passaged HSCs, suggesting their role in the induction of apoptosis. Blocking GSH synthesis by BSO abolished the role of EGCG and NAC but not GSH-MEE in the regulation of the expression of the genes relevant to apoptosis. These observations were verified by Western blotting analyses (Fig. 3C). In addition, EGCG, mimicking the role of NAC, reduced the level of cyclin D1, a critical regulator of G1 and S-phase transition of cell cycle, and enhanced the abundance of p21(WAF1/Cip1) and p27(Kip1), two critical inhibitory proteins in regulating cell cycle progression, in activated HSC in vitro, suggesting the importance of the antioxidants in cell cycle progression. Pretreatment with BSO abrogated their regulatory roles. Taken together, the observations of the sensitivity of EGCG and NAC to BSO and the GSH-MEE resistance to BSO in Fig. 3 provided strong evidence that the process of the EGCG inhibition of HSC growth, including inducing cell cycle arrest and apoptosis, was mainly mediated by de novo synthesis of cellular GSH.
EGCG Increased Gene Expression of GCLc but not GCLm in Activated HSC
It is worth noting that compared with the effects of NAC at 5 mM and GSH-MEE at 2 mM, EGCG at a much lower concentration (50 μM) showed similar, if not greater, impacts on the elevation of cellular GSH contents (Fig. 2C) on the inhibition of cell proliferation of HSC (Fig. 3A) and on the regulation of gene expression (Fig. 3, B and C), suggesting that EGCG might use a different but more efficient mechanism. We showed that EGCG increased the activity of GCL, the rate-liming enzyme in de novo synthesis of GSH in activated HSC in vitro (Fig. 1B). To explore underlying mechanisms, it was postulated that EGCG might induce gene expression of GCL, leading to the increase of the enzyme activity and to the elevation of the cellular GSH content in activated HSCs. To test the postulation, passaged HSCs were treated with EGCG at various concentrations for 24 h. The effects of EGCG on gene expression of the two subunits of GCL (i.e., GCLc and GCLm) were analyzed by real-time PCR and Western blotting analyses, respectively. Compared with those in untreated control cells, EGCG dose-dependently increased the steady-state level of GCLc transcript (Fig. 4A) and the abundance of GCLc protein (Fig. 4B). In great contrast, EGCG had no apparent impact on gene expression of GCLm in passaged HSCs (Fig. 4, A and B, respectively). These findings collectively suggest that EGCG might increase the level of cellular GSH in passaged HSCs by inducing gene expression of GCLc, leading to the increased activity of GCL.
Fig. 4.
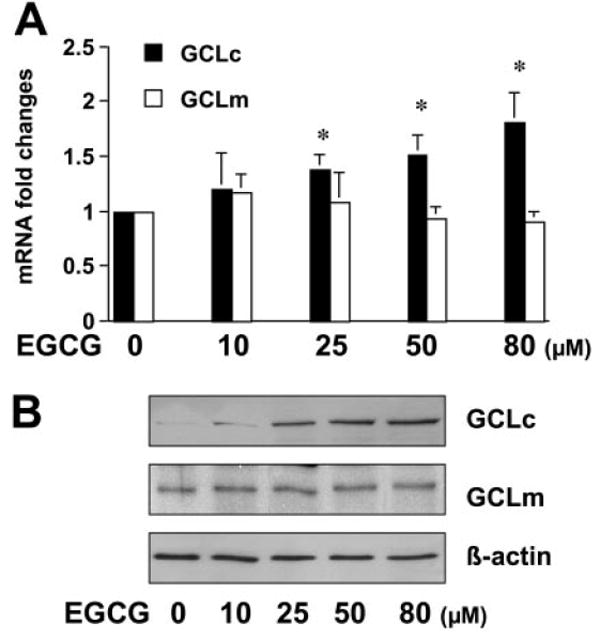
EGCG increases gene expression of GCLc but not GCLm in activated HSCs in vitro. Passaged HSCs were treated with EGCG at various concentrations for 24 h. Total RNA or whole-cell extracts were prepared for real-time PCR (A) or for Western blotting analyses (B), respectively. GAPDH or β-actin was used as an invariant internal control, respectively. Values are means ± S.D. (n = 3). *, p < 0.05 versus the control cells treated with no EGCG. Representative was shown from three independent experiments.
Exogenous TGF-β1 Reduced Cellular GSH Level by Suppressing GCLc Gene Expression in Passaged HSC
Further studies were performed to explore the mechanisms of EGCG in the induction of GCLc gene expression in activated HSC. Prior reports demonstrated that TGF-β deplete intracellular GSH content in some cell types, including fibroblast (NIH3T3) (Liu et al., 2004) and alveolar epithelial cells (Jardine et al., 2002). It was suggested that TGF-β signaling might suppress gene expression of GCL (Arsalane et al., 1997; Jardine et al., 2002). We demonstrated previously that EGCG interrupted TGF-β signaling by suppressing gene expression of TGF-β receptors, leading to the inhibition of gene expression of ECM components in activated HSCs (Yumei et al., 2006). We therefore hypothesized that the EGCG interruption of TGF-β signaling might result in the elimination of its suppressive effect and in the induction of gene expression of GCLc in activated HSCs. To test the hypothesis, we evaluated the effect of TGF-β1 on the levels of GSH in cytoplasm and in mitochondria in passaged HSCs. Cells were treated with TGF-β1 at 10 ng/ml for various hours. Cytosol and mitochondria were prepared from these cells. The levels of GSH in cytoplasm and in mitochondria were determined, respectively. As displayed in Fig. 5, exogenous TGF-β1 significantly reduced the levels of GSH in both cytoplasm and mitochondria in passaged HSCs. However, a biphasic response of the GSH content to TGF-β1 treatment was observed in cytoplasm and mitochondria. The level of GSH in cytoplasm was rapidly reduced in the first 8 h after the exposure to TGF-β1. However, the level of GSH in mitochondria was gradually reduce and was still significantly delayed after treatment for 30 h. Taken together, these results supported our hypothesis and demonstrated the inhibitory role of TGF-β signaling in the level of cellular GSH in passaged HSCs.
Fig. 5.
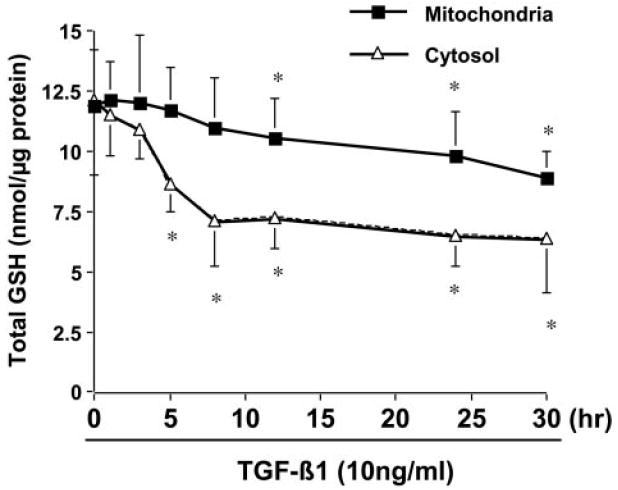
Exogenous TGF-β1 reduces the level of cellular GSH in cultured HSCs. Passaged HSCs were treated with TGF-β1 at 10 ng/ml for various times. The levels of GSH in cytoplasm and in mitochondria were measured as described under Methods and Materials. Total GSH was expressed as nanomoles per micrograms of protein. Values are means ± S.D. (n ≥ 3). *, p < 0.05 versus the control cells with no treatment (the first point on the left).
To further test our hypothesis and explore underlying mechanisms, passaged HSCs were divided into two groups. One group of cells was treated with TGF-β1 at various concentrations for 24 h. Another group of cells was treated with TGF-β1 at 10 ng/ml for various periods of time. Medium was replaced every 24 h with fresh exogenous TGF-β1. The expression of GCLc and GCLm was analyzed by real-time PCR and Western blotting analyses. As shown in Fig. 6, A and B, TGF-β1 dose-dependently reduced gene expression of GCLc but not GCLm at both levels of transcription and translation. In addition, the decrease in gene expression of GCLc lasted no less than 72 h, as displayed in Fig. 6, C and D. Taken together, these results demonstrated that the activation of TGF-β signaling by exogenous TGF-β1 suppressed gene expression of GCLc but not GCLm in activated HSCs in vitro.
Fig. 6.
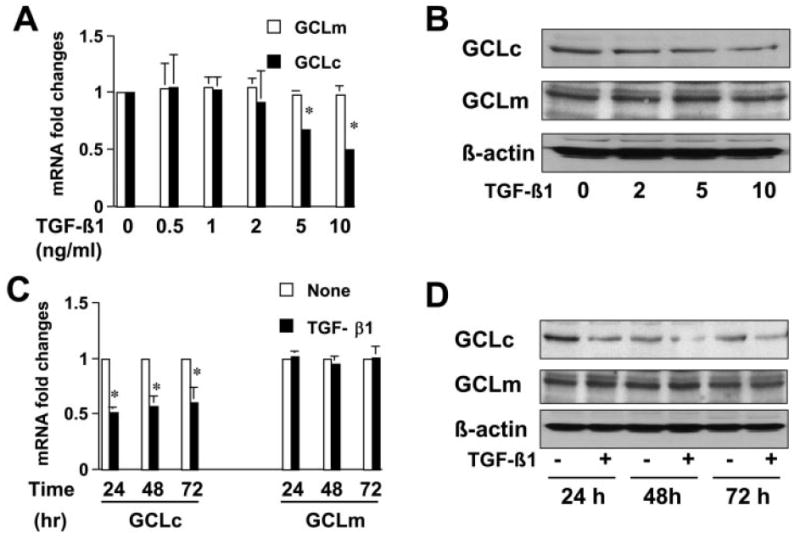
Exogenous TGF-β1 suppresses the expression of GCLc not GCLm in a dose- and time-dependent manner in activated HSCs in vitro. Passaged HSCs were either treated with or without TGF-β1 at indicated concentrations for 24 h (A and B) or treated with TGF-β1 at 10 ng/ml for 24, 48, or 72 h, respectively (C and D). Total RNA or whole-cell extracts were prepared for real-time PCR assays (A and C) or for Western blotting analyses (B and D). Values are means ± S.D. (n ≥ 3). GAPDH was used as an invariant internal control for calculating mRNA fold changes (n = 3). *, p < 0.05 versus the control cells with no treatment. β-Actin was used as an internal control for equal loading in Western blotting analyses. Representative was shown from three independent experiments.
EGCG Induced GCLc Gene Expression by Interrupting TGF-β Signaling
We observed previously strong basal TGF-β signaling in passaged HSCs without the addition of exogenous TGF-β (Yumei et al., 2006). It is presumably activated by paracrine and autocrine action of TGF-β present in medium containing FBS (10%) and secreted by passaged HSCs, respectively. Forced expression of the dominant-negative form of Tβ-RII (dn-Tβ-RII) in transfection assays interrupted TGF-β signaling in passaged HSCs (Yumei et al., 2006). To verify the effect of TGF-β signaling on the inhibition of GCLc gene expression, passaged HSCs were cotransfected with the plasmids pdn-Tβ-RII and pGCLc-luc. The GCLc promoter luciferase reporter plasmid pGCLc-luc contains a fragment of the rat GCLc promoter (−1758/+2 base pairs), subcloned in the luciferase reporter plasmid pGL3 (Yang et al., 2001). The cDNA expression plasmid pdn-Tβ-RII contains a full-length cDNA encoding the dominant-negative form of Tβ-RII (de Caestecker et al., 1998). A total of 4.5 μg of plasmid DNA per well was used for cotransfection of HSC in six-well culture plates. It included 2 μg of pGCLc-luc, 0.5 μg of pSV-β-gal, and 2 μg of pdn-Tβ-RII at indicated doses plus the empty vector pcDNA. The latter was used to ensure an equal amount of total DNA in transfection assays. As shown by luciferase assays in Fig. 7A, forced expression of exogenous dn-Tβ-RII dose-dependently increased luciferase activity in cells transfected with pGCLc-luc. This result suggested that the interruption of TGF-β signaling induced the promoter activity of GCLc gene in activated HSCs in vitro.
Fig. 7.
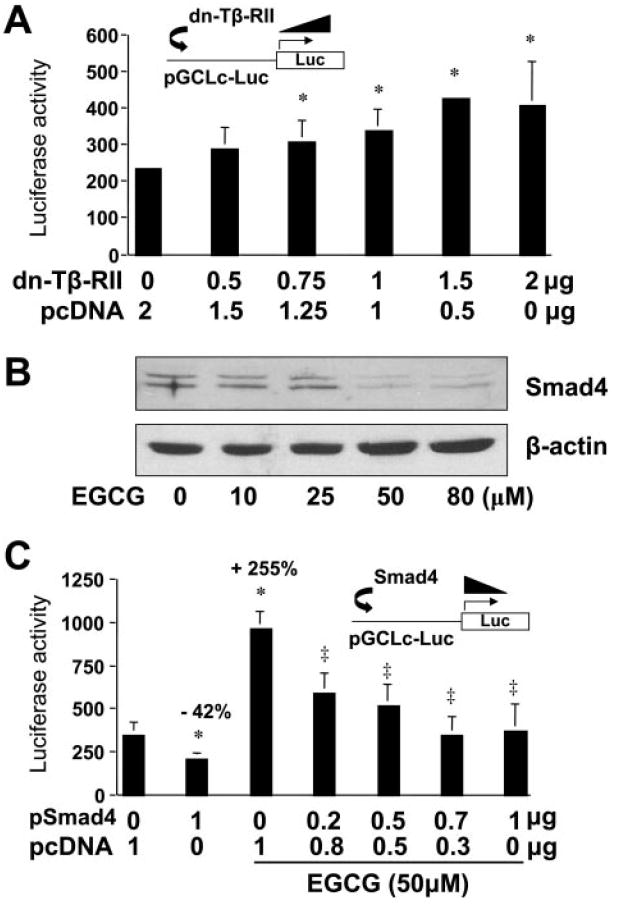
Interrupting TGF-β signaling by EGCG results in the induction of GCLc gene expression in activated HSCs in vitro. Passaged HSCs were cotransfected with the GCLc promoter luciferase reporter plasmid pGCLc-Luc and a cDNA expression plasmid, either pdn-TβRII, encoding the dominant-negative form of Tβ-RII (A), or pSmad4, encoding the constitutively active form of Smad4 (C). The empty vector pcDNA was used to ensure an equal amount of total DNA in transfection assays. Luciferase activities were normalized to β-galactosidase activity. Values are means ± S.D. (n = 3). *, p < 0.05 versus cells transfected with no pdn-Tβ-RII or pSmad4 (the first column on the left in A or C, respectively). ‡, p < 0.05 versus cells treated with EGCG without pSmad4 (the third column on the left in C). B, Western blotting analyses of the abundance of Smad4 in passaged HSCs treated with EGCG at various concentrations for 24 h. β-Actin was used as an internal control for equal loading. Representative was shown from three independent experiments.
Additional experiments were performed to evaluate the impact of EGCG on the protein abundance of Smad4, a key mediator in TGF-β signaling. As shown in Fig. 7B, EGCG apparently reduced the abundance of Smad4 in a dose-dependent manner, suggesting that EGCG interrupted TGF-β signaling not only by suppressing gene expression of TGF-β receptors (Yumei et al., 2006) but also by reducing the abundance of the key mediator Smad4 (Fig. 7B). To confirm the observation, HSCs were cotransfected with the plasmid pGCLc-luc and the plasmid pSmad4, containing a full-length cDNA encoding the constitutively active form of Smad4. After overnight recovery, cells were treated with or without EGCG (50 μM) for an additional 24 h. As shown in Fig. 7C, luciferase activity in cells transfected with pSmad4 (the second column on the left) was significantly reduced by approximately 42% compared with that in the control cells (the first column on the left). In great contrast, EGCG (50 μM) dramatically increased luciferase activity by more than 2.5-fold (the third column on the left). Forced expression of the constitutively active form of Smad4 dose-dependently eliminated the role of EGCG in the elevation of luciferase activity in cells (Fig. 7C), indicating the inhibitory effect of Smad4 on the promoter activity of GCLc gene in activated HSCs. These results collectively demonstrated that EGCG induced GCLc gene expression by suppressing TGF-β signaling and thereby abolished its inhibitory action on the expression of GCLc gene in activated HSCs in vitro.
Discussion
We reported previously that EGCG inhibited HSC activation in vitro by inhibiting cell proliferation and suppressing the expression of ECM genes (Chen et al., 2002). Additional experiments revealed that EGCG reduced ECM gene expression by interrupting TGF-β signaling through attenuating oxidative stress (Yumei et al., 2006). The current study examined the mechanisms of EGCG in the inhibition of cell proliferation of activated HSCs. Results in this report demonstrated that EGCG elevated the level of cellular GSH in activated HSCs by increasing the activity of GCL, leading to the regulation of expression of genes relevant to cell proliferation. Additional experiments indicated that EGCG induced gene expression of GCLc and eliminated the effect of TGF-β signaling on suppressing expression of the gene by interrupting its signaling in activated HSCs in vitro. It was observed recently that the maximum concentration of EGCG in human plasma was approximately 65 ng/ml within 1 to 2 h after oral ingestion of 350 ml of commercial green tea beverage containing 581 mg of total catechins with 0.31 mg/ml EGCG (Masukawa et al., 2006). It was also shown that the absorption rates of catechins in green tea, including EGCG, into human blood were different and very low. Most of them were retained in the blood for only a few hours. We used EGCG at 50 μM (22.92 μg/ml) in most of our in vitro experiments, which is higher than that observed in human blood. However, it bears emphasis that because the in vivo system is multifactorial, directly extrapolating in vitro conditions and results (e.g., effective concentrations) to the in vivo system might be misleading.
Oxidative stress represents an important and novel class of “the third messenger,” leading to the activation of several signal pathways associated with inflammation. Oxidative stress has been implicated in the stimulation of hepatic fibrogenesis. In addition to overproduction of pro-oxidants during liver injury, weakening in antioxidant defense synergistically facilitates oxidative stress. Depletion of GSH, the most abundant antioxidant molecule, in chronic liver injury potentiates oxidative stress, which promotes HSC growth and collagen production. Antioxidants have been proposed in the prevention and treatment of hepatic fibrosis. In the present report, we demonstrated that EGCG and the GSH precursors of NAC and GSH-MEE significantly increased the level of cellular GSH, inhibited cell proliferation of HSC, and regulated the expression of genes relevant to cell proliferation and apoptosis in activated HSCs in vitro. NAC elevates the level of cellular GSH by de novo synthesis of the thiol catalyzed by GCL, which could be specifically inhibited by BSO. However, the increase in the level of cellular GSH from GSH-MEE is resistant to BSO as a result of intracellular hydrolysis by esterases without the involvement of GCL (Tsan et al., 1989). Depletion of GSH alone by BSO was not sufficient to stimulate cell proliferation. However, the pretreatment with BSO, inhibiting GCL activity, apparently abolished the roles of EGCG and NAC but not GSH-MEE in the elevation of cellular GSH content, in the reduction of the number of viable HSCs, and in the regulation of expression of genes relevant to cell proliferation and apoptosis. These results collectively suggested that these effects of EGCG might require the GCL activity to stimulate de novo synthesis of GSH. However, the capability of EGCG in increasing the level of cellular GSH was, in fact, limited. The concentration of EGCG greater than 80 μM showed no such dose-dependent effects (data not shown). It could be explained by the theory of the feedback control (Seelig et al., 1984). GSH is known to be a feedback inhibitor of GCL. EGCG increases the activity of GCL, leading to the de novo synthesis of GSH and the elevation of the level of cellular GSH. The elevated GSH content might suppress, in turn, the activity of GCL in a mechanism of the feedback inhibition, resulting in elimination of the role of EGCG in elevating the level of GSH. The feedback inhibition restricts the ability of EGCG within a limited range of concentrations.
We observed that EGCG enhanced the levels of both cytoplasmic and mitochondrial GSH (Fig. 1A). It was also noticed that the levels of GSH showed no significant response to EGCG within the first several hours. It was a typical delayed response, suggesting the requirement of a rate-limiting enzyme in the process of the de novo synthesis of GSH. Additional experiments supported this suggestion and demonstrated that EGCG increased the GCL activity by inducing gene expression of GCL, the key rate-limiting enzyme in GSH synthesis, in activated HSCs (Figs. 2-4). In addition, the alterations in the level of mitochondrial GSH by either EGCG (Fig. 1A) or TGF-β1 (Fig. 5) were not in the same step with those in cytoplasm. The increase of the GSH content in mitochondria by EGCG was delayed at the beginning and caught up with that in cytoplasm later (Fig. 1A). However, the reduction of the GSH content in mitochondria by exogenous TGF-β1 was still delayed after the treatment for 30 h. Prior work observed the similar biphasic depletion of GSH in cytoplasm and mitochondria after the administration of BSO (Griffith and Meister, 1985). Studies have shown that although free GSH is present at similar millimolar concentrations in both mitochondrial matrix and cytoplasm, no GCL is detectable in mitochondria (Griffith and Meister, 1985). There is little, if any, de novo synthesis of GSH within mitochondria. Mitochondrial GSH in cells mainly arises from cytoplasm (Griffith and Meister, 1985). The exchange of free GSH between mitochondria and cytoplasm is distinct (Griffith and Meister, 1985). GSH is readily transported from cytoplasm into mitochondria. However, it very slowly and difficultly escapes from mitochondria to the cytoplasm (Griffith and Meister, 1985). The observed biphasic responses of GSH contents in cytoplasm and in mitochondria to EGCG or to TGF-β1 might result from the different exchanging rates of free GSH between mitochondria and cytoplasm.
The level of cellular GSH is mainly determined by GSH synthesis (GSH supply) and GSH-consuming (GSH demand). It bears emphasis that our results do not exclude any other mechanisms involved in the antioxidant capacity of EGCG and in the EGCG elevation of the level of cellular GSH in HSCs, including reducing exporting and consuming GSH. This current report focused on the effect of EGCG on GSH synthesis. Additional experiments are ongoing to evaluate the role of EGCG in regulating gene expression and activity of enzymes involved in consuming GSH, including GSH S-transferase and GSH peroxidase. In addition, we could not exclude the roles of any mechanisms and enzymes in the removal of lipid peroxidation products, which requires additional studies.
The concentration of EGCG (50 μM) used in this study is much lower than that of NAC (5 mM) and GSH-MEE (2 mM). However, EGCG produced similar, if not greater, inhibitory effects on HSC growth. These results suggest that EGCG might increase the level of cellular GSH via a different but more efficient mechanism. Either NAC or GSH-MEE primarily functions as one molecule of the GSH precursor in de novo GSH synthesis (Ruffmann and Wendel, 1991). EGCG increased the level of cellular GSH by inducing gene expression of GCLc and enhancing the activity of GCL. This explained why EGCG, compared with NAC and GSH-MEE, was more efficient in the elevation of GSH content in cultured HSC.
GCL is a heterodimer consisting of an active catalytic (GCLc) and a modulatory (GCLm) subunit. GCLc contains all substrate binding sites, whereas the modulatory subunit GCLm modulates the affinity of the GCLc for substrates and inhibitors. EGCG significantly induced gene expression of GCLc and had no apparent impact on GCLm in passaged HSCs. Studies have shown the simultaneous up-regulation of both GCL subunits (Zhang et al., 2007). However, the regulation of Gclc and Gclm is not always coordinated. In many cases, the induction of one gene is favored over the other (Cai et al., 1995, 1997; Moellering et al., 1998). In fact, in some tissues, including the heart and liver, the ratio of GCLc/GCLm is more than 1.0 (Krzywanski et al., 2004). Regulation of GCLc gene expression is affected by many factors, such as oxidative stress, inflammatory cytokines, antioxidants, and insulin (Lu, 1999, 2000).
Our experiments in this study indicated that exogenous TGF-β1 suppressed gene expression of GCLc and reduced the activity of GCL. Our observations are consistent with other prior reports. TGF-β1 depletes cellular GSH content, resulting from the inhibition of gene expression of GCLc (Arsalane et al., 1997; De Bleser et al., 1999; Jardine et al., 2002; Franklin et al., 2003; Liu et al., 2004). TGF-β signaling induces the activity of Smad3-ATF3, leading to the suppression of genes encoding phase II detoxifying proteins, including GCLc (Bakin et al., 2005). Ectopic expression of ATF3 is sufficient to reduce the GCL activity (Bakin et al., 2005). We demonstrated previously the presence of basal TGF-β signaling in cultured HSCs without the addition of exogenous TGF-β. The basal TGF-β signaling is presumably activated by TGF-β derived from FBS (10%) and secreted by passaged HSCs (Yumei et al., 2006). p3xTP-Lux is a TGF-β-inducible luciferase reporter plasmid, containing the plasminogen activator inhibitor gene promoter (Massagué, 1998). This plasmid is often used for studying TGF-β transactivation activity in cells and TGF-β signaling. We demonstrated that the luciferase activity in HSCs transfected with the p3xTP-Lux was significantly high (Yumei et al., 2006). EGCG caused a dose-dependent reduction in luciferase activity in these cells. EGCG suppressed gene expression of TGF-β receptors in activated HSCs in vitro, leading to the interruption of TGF-β signaling (Yumei et al., 2006). We therefore assumed that the EGCG interruption of TGF-β signaling might eliminate its suppressive effect on gene expression of GCLc, leading to the induction of GCLc gene expression in activated HSCs. This assumption was supported by our further observations. The interrupting TGF-β signaling by forced expression of the dominant-negative form of Tβ-RII increased the promoter activity of GCLc, whereas expression of the constitutively active form of Smad4 abrogated the impact of EGCG on the increase in the promoter activity of GCLc gene. It bears emphasis that we could not exclude any alternative mechanisms in the ECGC induction of GCLc gene expression in activated HSCs. In addition, our results could not determine the relationship between the EGCG reduction of the inhibitory effect of TGF-β signaling and the EGCG enhancement of the promoter activity in the induction of GCLc gene expression in activated HSCs in vitro. The two might play additive or synergistic roles in the up-regulation of gene expression of GCLc in passaged HSCs. Additional deletion of gene promoter analyses would be helpful to map out key elements and clarify the underlying mechanism. On the other hand, the enhanced level of cellular GSH by EGCG might not be exclusively derived from the induction of GCLc gene expression. EGCG also shows its impacts on other enzyme systems involved in the defense against oxidative stress (Li et al., 2007).
In summary, results in the current study support our general hypothesis and demonstrate that the inhibitory effect of EGCG on HSC growth might mainly result from its antioxidant capability by increasing de novo synthesis of GSH. These findings provide novel insights into the mechanisms of EGCG in the inhibition of HSC activation and further consolidate the potential application of EGCG as an antifibrotic agent against liver fibrosis.
Acknowledgments
This work was supported by the grant R01-DK047995 from the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases (to A.C.) and R01-DK45334 (to S.C.L.).
ABBREVIATIONS
- HSC
hepatic stellate cell
- FBS
fetal bovine serum
- BSO
buthionine sulfoximine
- ECM
extracellular matrix
- EGCG
(−)-epigallocatechin-3-gallate
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- GCL
glutamate-cysteine ligase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- NAC
N-acetyl-cysteine
- PDGF-βR
platelet-derived growth factor-β receptor
- TGF-β
transforming growth factor-β
- Tβ-R
transforming growth factor-β receptor
- GSH
glutathione
- PCR
polymerase chain reaction
- dn
dominant-negative
- GSH-MEE
glutathione monoethyl ester
- GCLc
catalytic subunit of glutamate-cysteine ligase
- GCLm
modulatory subunit of glutamate-cysteine ligase
References
- Arsalane K, Dubois CM, Muanza T, Begin R, Boudreau F, Asselin C, Cantin AM. Transforming growth factor-beta1 is a potent inhibitor of glutathione synthesis in the lung epithelial cell line A549: transcriptional effect on the GSH rate-limiting enzyme gamma-glutamylcysteine synthetase. Am J Respir Cell Mol Biol. 1997;17:599–607. doi: 10.1165/ajrcmb.17.5.2833. [DOI] [PubMed] [Google Scholar]
- Arteel G, Marsano L, Mendez C, Bentley F, McClain CJ. Advances in alcoholic liver disease. Best Pract Res Clin Gastroenterol. 2003;17:625–647. doi: 10.1016/s1521-6918(03)00053-2. [DOI] [PubMed] [Google Scholar]
- Bakin AV, Stourman NV, Sekhar KR, Rinehart C, Yan X, Meredith MJ, Arteaga CL, Freeman ML. Smad3-ATF3 signaling mediates TGF-beta suppression of genes encoding Phase II detoxifying proteins. Free Radic Biol Med. 2005;38:375–387. doi: 10.1016/j.freeradbiomed.2004.10.033. [DOI] [PubMed] [Google Scholar]
- Cai J, Huang ZZ, Lu SC. Differential regulation of gamma-glutamylcysteine synthetase heavy and light subunit gene expression. Biochem J. 1997;326:167–172. doi: 10.1042/bj3260167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Sun WM, Lu SC. Hormonal and cell density regulation of hepatic gamma-glutamylcysteine synthetase gene expression. Mol Pharmacol. 1995;48:212–218. [PubMed] [Google Scholar]
- Chen A. Acetaldehyde stimulates the activation of latent transforming growth factor-beta1 and induces expression of the type II receptor of the cytokine in rat cultured hepatic stellate cells. Biochem J. 2002;368:683–693. doi: 10.1042/BJ20020949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A, Davis BH. UV irradiation activates JNK and increases αII collagen gene expression in rat hepatic stellate cells. J Biol Chem. 1999;274:158–164. doi: 10.1074/jbc.274.1.158. [DOI] [PubMed] [Google Scholar]
- Chen A, Zhang L. The antioxidant (−)-epigallocatechin-3-gallate inhibits rat hepatic stellate cell proliferation in vitro by blocking the tyrosine phosphorylation and reducing the gene expression of platelet-derived growth factor-β receptor. J Biol Chem. 2003;278:23381–23389. doi: 10.1074/jbc.M212042200. [DOI] [PubMed] [Google Scholar]
- Chen A, Zhang L, Xu J, Tang J. The antioxidant (−)-epigallocatechin-3-gallate inhibits activated hepatic stellate cell growth and suppresses acetaldehyde-induced gene expression. Biochem J. 2002;368:695–704. doi: 10.1042/BJ20020894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bleser PJ, Xu G, Rombouts K, Rogiers V, Geerts A. Glutathione levels discriminate between oxidative stress and transforming growth factor-β signaling in activated rat hepatic stellate cells. J Biol Chem. 1999;274:33881–33887. doi: 10.1074/jbc.274.48.33881. [DOI] [PubMed] [Google Scholar]
- de Caestecker MP, Parks WT, Frank CJ, Castagnino P, Bottaro DP, Roberts AB, Lechleider RJ. Smad2 transduces common signals from receptor serine-threonine and tyrosine kinases. Genes Dev. 1998;12:1587–1592. doi: 10.1101/gad.12.11.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin CC, Rosenfeld-Franklin ME, White C, Kavanagh TJ, Fausto N. TGFbeta1-induced suppression of glutathione antioxidant defenses in hepatocytes: caspase-dependent post-translational and caspase-independent transcriptional regulatory mechanisms. FASEB J. 2003;17:1535–1537. doi: 10.1096/fj.02-0867fje. [DOI] [PubMed] [Google Scholar]
- Fraser JA, Kansagra P, Kotecki C, Saunders RD, McLellan LI. The modifier subunit of Drosophila glutamate-cysteine ligase regulates catalytic activity by covalent and noncovalent interactions and influences glutathione homeostasis in vivo. J Biol Chem. 2003;278:46369–46377. doi: 10.1074/jbc.M308035200. [DOI] [PubMed] [Google Scholar]
- Friedman SL. Stellate cells: a moving target in hepatic fibrogenesis. Hepatology. 2004;40:1041–1043. doi: 10.1002/hep.20476. [DOI] [PubMed] [Google Scholar]
- Friedman SL, Yamasaki G, Wong L. Modulation of transforming growth factor beta receptors of rat lipocytes during the hepatic wound healing response. Enhanced binding and reduced gene expression accompany cellular activation in culture and in vivo. J Biol Chem. 1994;269:10551–10558. [PubMed] [Google Scholar]
- Greenwel P, Dominguez-Rosales JA, Mavi G, Rivas-Estilla AM, Rojkind M. Hydrogen peroxide: a link between acetaldehyde-elicited alpha1(I) collagen gene up-regulation and oxidative stress in mouse hepatic stellate cells. Hepatology. 2000;31:109–116. doi: 10.1002/hep.510310118. [DOI] [PubMed] [Google Scholar]
- Griffith OW. Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J Biol Chem. 1982;257:13704–13712. [PubMed] [Google Scholar]
- Griffith OW, Meister A. Origin and turnover of mitochondrial glutathione. Proc Natl Acad Sci U S A. 1985;82:4668–4672. doi: 10.1073/pnas.82.14.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardine H, MacNee W, Donaldson K, Rahman I. Molecular mechanism of transforming growth factor (TGF)-β1-induced glutathione depletion in alveolar epithelial cells. Involvement of AP-1/ARE and Fra-1. J Biol Chem. 2002;277:21158–21166. doi: 10.1074/jbc.M112145200. [DOI] [PubMed] [Google Scholar]
- Kisseleva T, Brenner DA. Hepatic stellate cells and the reversal of fibrosis. J Gastroenterol Hepatol. 2006;21(Suppl 3):S84–S87. doi: 10.1111/j.1440-1746.2006.04584.x. [DOI] [PubMed] [Google Scholar]
- Kömüves LG, Feren A, Jones AL, Fodor E. Expression of epidermal growth factor and its receptor in cirrhotic liver disease. J Histochem Cytochem. 2000;48:821–830. doi: 10.1177/002215540004800610. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol. 1998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- Krzywanski DM, Dickinson DA, Iles KE, Wigley AF, Franklin CC, Liu RM, Kavanagh TJ, Forman HJ. Variable regulation of glutamate cysteine ligase subunit proteins affects glutathione biosynthesis in response to oxidative stress. Arch Biochem Biophys. 2004;423:116–125. doi: 10.1016/j.abb.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Lee KS, Buck M, Houglum K, Chojkier M. Activation of hepatic stellate cells by TGF alpha and collagen type I is mediated by oxidative stress through c-myb expression. J Clin Invest. 1995;96:2461–2468. doi: 10.1172/JCI118304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YM, Chan HY, Huang Y, Chen ZY. Green tea catechins up-regulate superoxide dismutase and catalase in fruit flies. Mol Nutr Food Res. 2007;51:546–554. doi: 10.1002/mnfr.200600238. [DOI] [PubMed] [Google Scholar]
- Liu RM, Liu Y, Forman HJ, Olman M, Tarpey MM. Glutathione regulates transforming growth factor-beta-stimulated collagen production in fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2004;286:L121–L128. doi: 10.1152/ajplung.00231.2003. [DOI] [PubMed] [Google Scholar]
- Lu SC. Regulation of hepatic glutathione synthesis: current concepts and controversies. FASEB J. 1999;13:1169–1183. [PubMed] [Google Scholar]
- Lu SC. Regulation of glutathione synthesis. Curr Top Cell Regul. 2000;36:95–116. doi: 10.1016/s0070-2137(01)80004-2. [DOI] [PubMed] [Google Scholar]
- Massagué J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Masukawa Y, Matsui Y, Shimizu N, Kondou N, Endou H, Kuzukawa M, Hase T. Determination of green tea catechins in human plasma using liquid chromatography-electrospray ionization mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;834:26–34. doi: 10.1016/j.jchromb.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Moellering D, McAndrew J, Patel RP, Cornwell T, Lincoln T, Cao X, Messina JL, Forman HJ, Jo H, Darley-Usmar VM. Nitric oxide-dependent induction of glutathione synthesis through increased expression of gamma-glutamylcysteine synthetase. Arch Biochem Biophys. 1998;358:74–82. doi: 10.1006/abbi.1998.0854. [DOI] [PubMed] [Google Scholar]
- Nieto N, Friedman SL, Greenwel P, Cederbaum AI. CYP2E1-mediated oxidative stress induces collagen type I expression in rat hepatic stellate cells. Hepatology. 1999;30:987–996. doi: 10.1002/hep.510300433. [DOI] [PubMed] [Google Scholar]
- Rice-Evans C. Implications of the mechanisms of action of tea polyphenols as antioxidants in vitro for chemoprevention in humans. Proc Soc Exp Biol Med. 1999;220:262–266. doi: 10.1046/j.1525-1373.1999.d01-45.x. [DOI] [PubMed] [Google Scholar]
- Ruffmann R, Wendel A. GSH rescue by N-acetylcysteine. Klin Wochenschr. 1991;69:857–862. doi: 10.1007/BF01649460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Zakrajsek BA, Mills AG, Gorn V, Singer MJ, Reed MW. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal Biochem. 2000;285:194–204. doi: 10.1006/abio.2000.4753. [DOI] [PubMed] [Google Scholar]
- Seelig GF, Simondsen RP, Meister A. Reversible dissociation of γ-glutamylcysteine synthetase into two subunits. J Biol Chem. 1984;259:9345–9347. [PubMed] [Google Scholar]
- Sueoka N, Suganuma M, Sueoka E, Okabe S, Matsuyama S, Imai K, Nakachi K, Fujiki H. A new function of green tea: prevention of lifestyle-related diseases. Ann N Y Acad Sci. 2001;928:274–280. doi: 10.1111/j.1749-6632.2001.tb05656.x. [DOI] [PubMed] [Google Scholar]
- Tsan MF, White JE, Rosano CL. Modulation of endothelial GSH concentrations: effect of exogenous GSH and GSH monoethyl ester. J Appl Physiol. 1989;66:1029–1034. doi: 10.1152/jappl.1989.66.3.1029. [DOI] [PubMed] [Google Scholar]
- Tsukamoto H, Rippe R, Niemela O, Lin M. Roles of oxidative stress in activation of Kupffer and Ito cells in liver fibrogenesis. J Gastroenterol Hepatol. 1995;10(Suppl 1):S50–S53. doi: 10.1111/j.1440-1746.1995.tb01798.x. [DOI] [PubMed] [Google Scholar]
- Win KM, Charlotte F, Mallat A, Cherqui D, Martin N, Mavier P, Preaux AM, Dhumeaux D, Rosenbaum J. Mitogenic effect of transforming growth factor-beta 1 on human Ito cells in culture: evidence for mediation by endogenous platelet-derived growth factor. Hepatology. 1993;18:137–145. [PubMed] [Google Scholar]
- Wong L, Yamasaki G, Johnson RJ, Friedman SL. Induction of beta-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. J Clin Invest. 1994;94:1563–1569. doi: 10.1172/JCI117497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang J, Huang ZZ, Ou X, Lu SC. Cloning and characterization of the 5′-flanking region of the rat glutamate-cysteine ligase catalytic subunit. Biochem J. 2001;357:447–455. doi: 10.1042/0264-6021:3570447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yumei F, Zhou Y, Zheng S, Chen A. The antifibrogenic effect of (−)-epigallocatechin gallate results from the induction of de novo synthesis of glutathione in passaged rat hepatic stellate cells. Lab Invest. 2006;86:697–709. doi: 10.1038/labinvest.3700425. [DOI] [PubMed] [Google Scholar]
- Zhang H, Court N, Forman HJ. Submicromolar concentrations of 4-hydroxynonenal induce glutamate cysteine ligase expression in HBE1 cells. Redox Rep. 2007;12:101–106. doi: 10.1179/135100007X162266. [DOI] [PMC free article] [PubMed] [Google Scholar]