

Abstract
Vascular cell adhesion molecule-1 (VCAM-1) is an adhesion molecule expressed by endothelial cells for recruitment of leukocytes during inflammation. It is also abundantly expressed by smooth muscle cells in atherosclerotic lesions and in injured arteries. In this study, we examined the role of VCAM-1 in smooth muscle cell migration. Smooth muscle cells were isolated from the aorta of C57BL/6 mice and transfected with short interfering RNAs (siRNAs) targeting VCAM-1. Inhibition on VCAM-1 expression by siRNAs was assessed by Western blot analysis, RT-PCR and by measuring soluble VCAM-1 concentrations in the incubation medium. One siRNA that showed greater suppression on VCAM-1 expression was used for migration assay. A single scratch wound was made on 70% confluent cells and cells migrated from wounded monolayer were counted 24 and 48 h after injury. Treatment with VCAM-1 siRNA resulted in a significant reduction in the number of migrated cells. This siRNA also exhibited a minor effect on smooth muscle cell proliferation. Thus, our findings indicate that VCAM-1 is necessary for the migration of smooth muscle cells and interfering VCAM-1 expression could be an effective approach to prevention and treatment of atherosclerosis and restenosis.
Keywords: siRNA, VCAM-1, Smooth muscle cells, Migration
1. Introduction
During the development of atherosclerosis and neointimal hyperplasia, vascular smooth muscle cells (SMC) in the media transform from a quiescent contractile phenotype into a synthetic phenotype that proliferate and migrate from the media to the intima. Once SMC arrive in the intima of adjacent arterial walls, they start to replicate and produce extracellular matrix, leading to the formation of intimal lesions. SMC migration is regulated by several adhesion molecules. Activated SMC exhibit an altered set of adhesion receptors and modulated actin cytoskeletal reorganization [1,2]. Among cell adhesion receptors, the integrin super family plays an important role in SMC migration by interacting with the actin cytoskeleton and the extracellular matrix [1].
Vascular cell adhesion molecule-1 (VCAM-1) is a member of the immunoglobulin super family known to be expressed by endothelial cells for recruitment of leukocytes during inflammation [3]. It is also expressed by cytokine-stimulated SMC in vitro and by injured arteries in vivo [4–6]. VCAM-1 interacts with the integrin α4β1 (very late-acting antigen 4 [VLA-4]) that is constitutively expressed on the surface of leukocytes. Blockade of the VCAM-1/VLA-4 pathway reduces monocyte adhesion and infiltration and neointimal formation after arterial injury [6,7]. VCAM-1 domain 4-deficiency markedly reduced arterial VCAM-1 expression, monocyte adherence in the aortic root, and fatty streak formation in apolipoprotein E-deficient mice [8]. Recent studies have shown that VCAM-1 is required for accumulation of SMC in the neointima in the mouse injury model [7] and VLA-4 is expressed on SMC [9], thus suggesting a possibility that VCAM-1 may act directly on SMC in addition to inflammatory cells.
RNA interference (RNAi) is a sequence-specific, post-transcriptional gene silencing mechanism initiated by the introduction of double stranded RNA (dsRNA) homologous in sequence to the gene to be silenced [10]. The introduction of small interfering RNA (siRNA) with 21 to 22 nucleotides in length does not stimulate an immune response in mammalian cultured cells and can effectively target specific mRNA for gene silencing [11]. The ability of siRNA to silence individual gene expression with a high degree of specificity is showing promise as a strategy for target-directed therapies in a range of diseases [12,13]. A major advantage of such target-specific therapies is a lower risk of systemic side effects [14]. The purpose of this study is to determine the effect of endogenous VCAM-1 on SMC migration by blocking its expression with sequence-specific siRNAs. We have observed direct evidence on the involvement of VCAM-1 in SMC migration.
2. Methods
2.1. Mice
Female C57BL/6J (B6) mice were purchased from Jackson Laboratory, Bar Harbor, Me, and maintained on a regular chow diet. All mice were 3–6 months of age at the time of experiments. All procedures were in accordance with current National Institutes of Health guidelines and were approved by the institutional Animal Care and Use Committee.
2.2. Culture of vascular smooth muscle cells
SMC were isolated from the descending thoracic aorta of mice using an explantation technique [15]. Briefly, under sterile conditions, the anterior chest walls, lungs, and esophagus of mice were removed. The descending aorta was gently cleaned of periadventitial fat and connective tissues and was cut into rings ~3 mm long. The aortic segments of each mouse were placed in a six-well plate and incubated in Dulbecco’s Modified Eagle Medium (DMEM)/F12 medium supplemented with 15% FBS and 1% penicillin/streptomycin. The vessel segments were removed once cell outgrowth was observed. The cells at ~80% confluence were passaged with 0.25% trypsin–EDTA and then plated onto a 0.1% gelatin-coated 60-mm culture dish. In the subsequent passages, cells were split in a 1:4 ratio. All experiments were performed within the first eight passages.
2.3. siRNA selection and transfection
VCAM-1 sense and antisense siRNAs and control siRNAs were designed in conjunction with Qiagen (Valencia, CA). The sequences were as follows: siRNA1, +482CCAUUGUUCUCAUGGAGAA+502, siRNA2, +72GAAUGACUUCAGCCCAGAA+92, siRNA3, +928AAUUGAUUCUACACUCAAA+948, siRNA4, +2585GAUCCUUAAUACUGUUUAU+2605, and control, UUCUCCGAACGUGUCAC GU. For siRNA transfection, SMC were cultured in six-well plates with 2 ml of culture medium until 70% confluence was reached. siRNAs and transfection reagent (Qiagen, Valencia, CA) were added to the medium and then incubated for 24 or 48 h at 37 °C.
2.4. Assessment of VCAM-1 expression
Inhibition of VCAM-1 expression by siRNAs was assessed by ELISA, Western blot analysis, and RT-PCR. The concentration of soluble VCAM-1 in the medium that was incubated with SMC for 24 or 48 h after siRNA transfection was measured with an ELISA kit from the R&D Systems (Minneapolis, MN). The expression level of VCAM-1 on SMC 48 h after transfection was analyzed by Western blot analysis. Briefly, cell lysates were separated by electrophoresis on a 5–12% gradient polyacrylamide gel and electrophoretically transferred onto a nitrocellulose membrane. The membrane was incubated overnight with primary antibodies against mouse VCAM-1 (R&D systems) or β-actin (ICN Pharmaceuticals Inc.) and then incubated for 1 h with alkaline–phosphatase-conjugated secondary antibodies (ICN Pharmaceuticals Inc.). Signals were detected by the chemiluminescence method (ECL, Amersham International). For RT-PCR, total RNA was extracted with Trizol reagents (Invitrogen) from SMC 48 h after transfection. The first-strand cDNA was reverse-transcribed from the total RNA with the ThermoScript RT-PCR System (Invitrogen). The cDNA product was amplified by PCR for 28 cycles of 30 s at 94 °C, 60 s at 55 °C and 60 s at 72 °C. The PCR products were separated in 2% agarose gels and visualized under UV light following ethidium bromide staining. GAPDH was amplified simultaneously in a separate set of tubes under the same conditions and used as control. The primers used for VCAM-1 amplification were 5-AAGCAGAGACTTGAAATGCC-3′/5′-CCCTTGAACAGATCAATCTCC-3′and for GAPDH were 5′ACCACAGTCCATGCCATCAC-3′/5′-TCCACCACCCTGTTGCTGTA-3′.
2.5. Cell migration assay
Once 70% confluency was achieved, SMC were transfected with either siRNA4 or control siRNA. After a 24-h incubation, a single uniform scratch was made using a 1000 μl blue plastic pipet tip. The scratch resulted in a cell-free gap of approximately 1.0 mm between two adjoining areas of SMC [16]. The SMC were then incubated in a 95% air/5% CO2 environment. Each scratch was randomly photographed at four separate sites along the length of the scratch, starting proximally and ending distally. The photographs were taken on an inverted microscope immediately after the scratch and then again at 24 and 48 h. Migrating cells across the wound edges were counted manually.
In a separate experiment, once a single uniform scratch was made on 70% confluent SMC, the medium was replaced with 5% FBS medium, the medium containing 50 μg/ml anti-mouse VCAM-1 antibody or isotype IgG (R&D systems). Photographs were taken immediately and then 48 h after the scratch.
2.6. Cell proliferation assay
The effect of siRNA4 or control siRNA on SMC proliferation was determined using a colorimetric (MTT-tetrazolium) assay kit from Chemicon International. In brief, at 70% confluency, SMC were transfected with either siRNA4 or control siRNA for 24 or 48 h before the MTT assay was run. The tetrazolium reagent was reduced by living cells to form insoluble blue formazan product. The cells were then washed, solubilized with a detergent provided by the company, and quantified at 570 nm.
2.7. Data analysis
Data are presented as means ± S.D. ANOVA was used to compare differences among different siRNAs in the effect on VCAM-1 expression. When only two means were compared, the Student’s t-test was applied. Differences were considered statistically significant at P < 0.05.
3. Results
SMC isolated from the descending thoracic aorta of B6 mice were transfected with either siRNAs targeting VCAM-1 or control siRNA. Inhibition of VCAM-1 expression by the siRNAs was evaluated by measuring soluble VCAM-1 in the medium that was incubated with SMC. As shown in Fig. 1, all four siRNAs showed significant inhibition on VCAM-1 expression by SMC within the time frame studied (P < 0.05), although their effects varied dramatically. siRNA1 and siRNA4 exhibited strong inhibition on VCAM-1 expression compared to siRNA3. siRNA1 decreased soluble VCAM-1 levels by 79% at 24 h and 89% at 48 h relative to the control (P < 0.001 vs. control, n = 4 for each group) and siRNA4 reduced VCAM-1 by 82% and 91% at 24 and 48 h respectively (P < 0.001 vs. control, n = 4). In contrast, siRNA3 only reduced VCAM-1 levels by 29% at 24 h and 42% at 48 h (P < 0.001 vs. control, n = 4). siRNA2 had an intermediate effect, decreasing VCAM-1 levels by 59% at 24 h and 72% at 48 h (P < 0.001 vs. control, n = 4 for each group). The effect of siRNA1 and siRNA4 on VCAM-1 expression was further determined by Western blot analysis and RT-PCR (Fig. 2). Compared to the control siRNA, both siRNAs resulted in marked reductions of VCAM-1 expression at protein and transcript levels. In contrast, the expression levels of β-actin and GAPDH in SMC were not affected by the siRNAs.
Fig. 1.
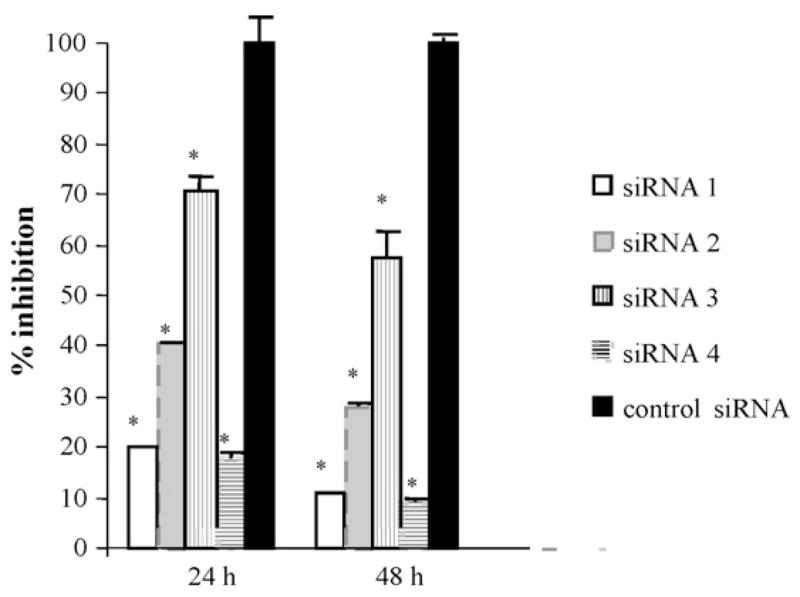
siRNA inhibition of VCAM-1 expression by smooth muscle cells (SMC) harvested from the aorta of C57BL/6 (B6) mice. SMC were transfected with siRNA 1–4 or a control siRNA targeting the VCAM-1 mRNA. Soluble VCAM-1 concentrations in the medium after 24 and 48 h incubation with SMC was measured by ELISA and expressed as % of the control. Values are means ± S.D. of four separate experiments. *P < 0.05 vs. control.
Fig. 2.
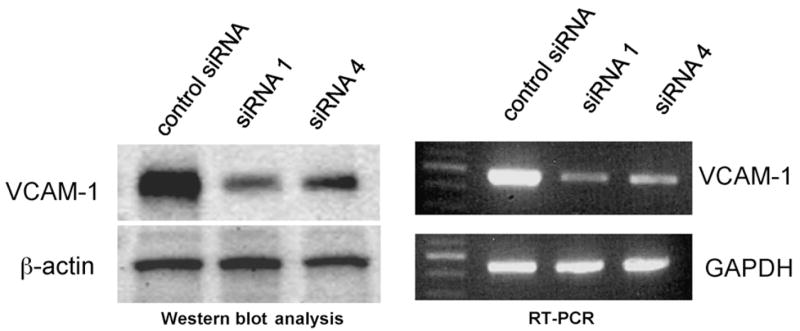
Western blot (left panel) or RT-PCR (right panel) analysis of VCAM-1 expression by SMC. SMC were incubated for 48 h with siRNA 1, 4 or control siRNA targeting VCAM-1 mRNA.
Given the largest capacity to reduce VCAM-1 expression, siRNA4 was used for the migration assay. At 70% confluence, SMC were transfected with either siRNA4 or control siRNA for 24 h before a scratch was made using a pipet tip to remove a band of adherent SMC for each well. This procedure resulted in a cell-free gap of approximately 1.0 mm between two adjoining areas of cells. As shown in Fig. 3, cells migrated across the wound edge into the scratch area at 24 and 48 h. At either time point, SMC transfected with siRNA4 underwent less migration into the scratch area compared to those transfected with control siRNA. To quantitate migrated SMC in the scratch area, each scratch was divided into two 25% border regions and one 50% middle region. Those cells in the middle 50% region of the scratch were counted. SMC transfected with siRNA4 had an average of 5 ± 3 cells at 24 h and 29 ± 16 cells at 48 h compared to the average of 38 ± 27 cells at 24 h (P < 0.00005) and 45 ± 23 cells at 48 h (P = 0.0001) in the control group (Fig. 4).
Fig. 3.
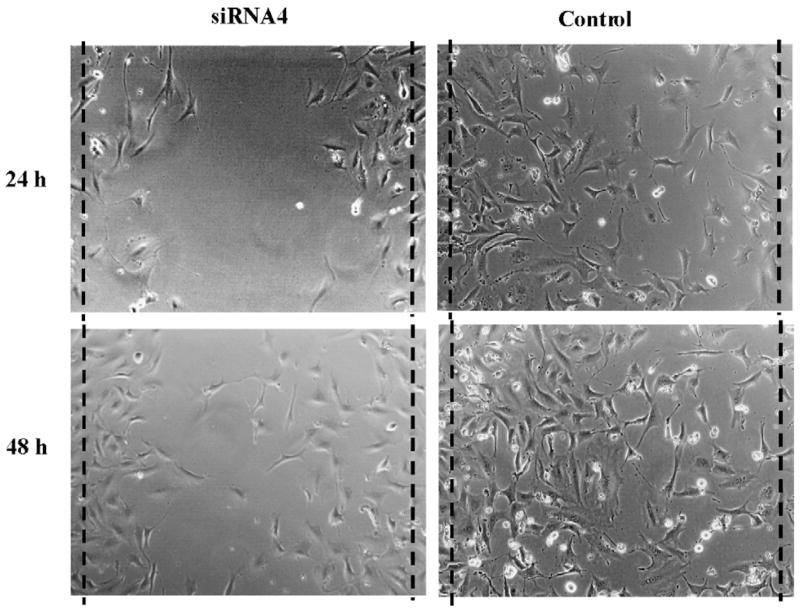
Representative graphs of SMC migration across the wound edges 24 and 48 h after wounding. Seventy percent confluent cells were transfected with siRNA4 or control siRNA targeting VCAM-1 for 24 or 48 h before a cell-free zone of approximately 1 mm in width was created by a pipette tip scratch.
Fig. 4.
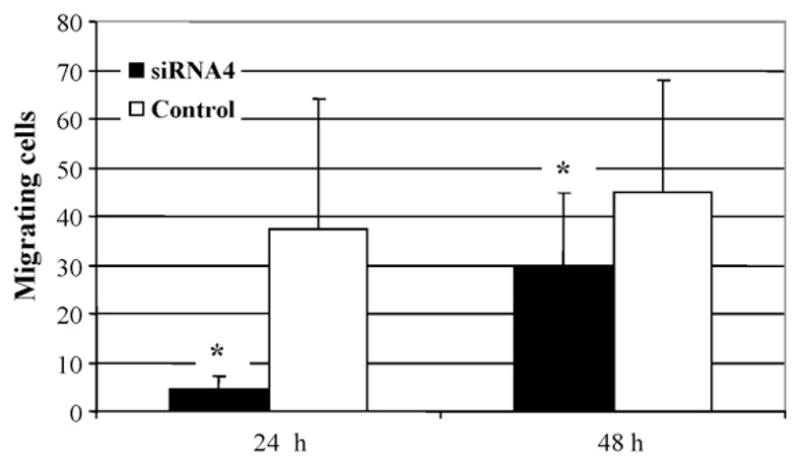
The number of SMC that migrated into the middle 50% area of the wound area 24 and 48 h after wounding. Cells were transfected with VCAM-1 siRNA4 or control siRNA. Values are means ± S.D. of four individual experiments. *P < 0.05 vs. control.
The role of VCAM-1 in the migration of SMC was further investigated by treating the cells with a specific antibody against VCAM-1. As shown in Fig. 5, migrated cells treated with the VCAM-1 antibody for 48 h was 32 ± 11, a number significantly smaller than 48 ± 9 for cells treated with medium only or 47 ± 8 for cells treated with isotype IgG (n =5; P < 0.05).
Fig. 5.
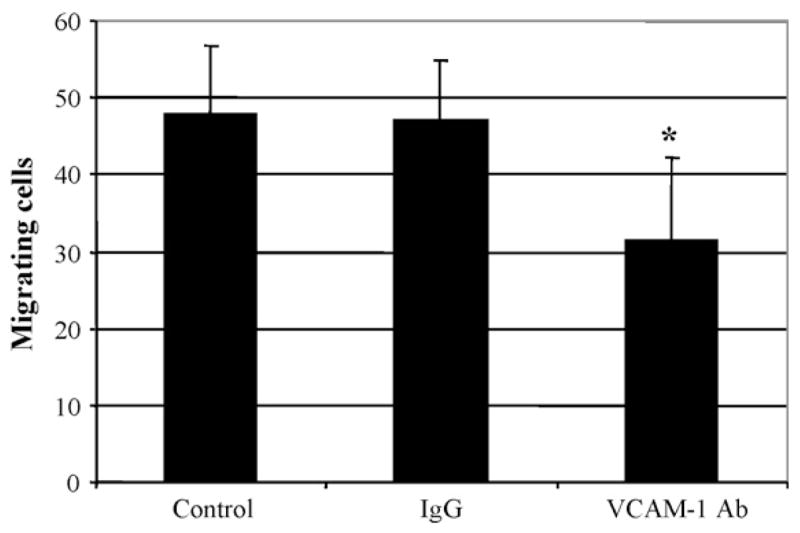
Effect of blocking antibody to VCAM-1 on SMC migration. A cell-free zone was created on 70% confluent SMC with a pipette tip. Cells were then treated with medium only or medium containing 50 μg/ml isotype IgG or an antibody against VCAM-1 for 48 h. The number of SMC migrating into wound area was counted. Values are means ± S.D. of five individual experiments. *P <0.05 vs. treatment with medium only.
The effect of siRNA4 on SMC proliferation was determined using the MTT-tetrazolium assay. Seventy percent confluent SMC were transfected with either siRNA4 or control siRNA for 24 and 48 h before the MTT assay was performed. As shown in Fig. 6, siRNA4 resulted in a smaller but statistically significant reduction in MTT incorporation in SMC, compared to the control siRNA at 24 h (optical density: 0.181 ± 0.008 vs. 0.213 ± 0.025; P = 0.041). At 48 h, MTT incorporation in the cells was also reduced by siRNA4 (optical density: 0.202 ± 0.004 vs. 0.257 ± 0.046), but the reduction was not statistically significant (P = 0.059).
Fig. 6.
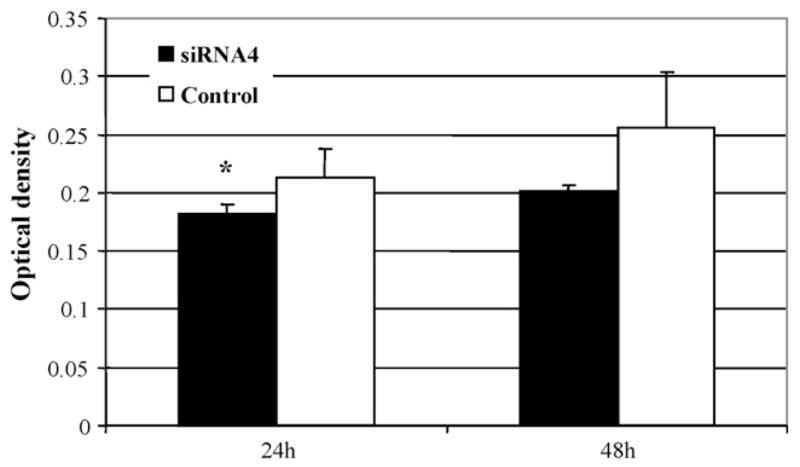
Effect of VCAM-1 siRNA4 on SMC proliferation 24 and 48 h after treatment. Seventy percent confluent cells were transfected with siRNA4 or control siRNA targeting VCAM-1 for up to 48 h before the MTT cell growth assay was run. Values are means ± S.D. of four separate experiments. *P < 0.05 vs. control.
4. Discussion
In the present study, we examined the effect of endogenously produced VCAM-1 on migration and proliferation of vascular SMC in vitro. VCAM-1 is an adhesion molecule that is known to function for the recruitment of leukocytes, such as monocytes and lymphocytes. The present results have demonstrated that VCAM-1 is also involved in the migration of vascular SMC. Furthermore, we demonstrated that blockade of VCAM-1 expression by SMC with siRNA resulted in a slight inhibition on proliferation of the cells.
We used four siRNAs to target different regions of the VCAM-1 mRNA of B6 mice. The inhibition of siRNAs on the expression of VCAM-1 by SMC was confirmed by Western blot analysis and RT-PCR, as well as by measuring soluble VCAM-1 concentrations in the medium incubated with the cells. Recently, we reported that the level of VCAM-1 in the incubation medium reflects its expression level on the cell surface [17]. All four siRNAs tested displayed significant inhibition on VCAM-1 expression, although their effect varied greatly.
VCAM-1 is expressed at high levels in injured arteries and in atherosclerotic lesions [7,18]. Using Western blot analysis, we demonstrated the expression of VCAM-1 in the descending aorta of apoE-deficient mice when fed chow and Western diets [19]. Furthermore, we found that VCAM-1 is expressed by SMC isolated from the aorta of B6 and C3H mice [15]. However, the functional significance of VCAM-1 expression by SMC under both physiological and pathological conditions is unknown. In the current study, we tested hypothesis that SMC produce VCAM-1 and use it to facilitate migration. To investigate this, we studied SMC isolated from the aorta of B6 mice, which are susceptible to diet-induced atherosclerosis.
To migrate toward a defined direction, cells first extend leading lamellopodia, which attach to a substrate, and then contract. After this is the detachment to the substrate at the rear of the cell, allowing tail retraction and forward translocation of the cell body [20,21]. The ability to spread and subsequently migrate depends on a critical value of adhesive strength between cell and substrate: high or low levels of substrate attachment inhibit spreading and migration, whereas maximum migration occurs at intermediate adhesion strengths [22]. We found that SMC transfected by VCAM-1 siRNA displayed significant inhibition in migration compared to the cells transfected with control siRNA. The role of VCAM-1 in the migration of SMC was confirmed by blocking the effect of VCAM-1 with a specific antibody. These findings suggest that SMC produce VCAM-1 that facilitates cell migration.
The inhibition of siRNAs on VCAM-1 expression persisted within 48 h according to the measurement of soluble VCAM-1 levels in the medium incubated with SMC (Fig. 1). However, there were much more migrated cells in wound areas at 48 h relative to the 24 h time point. Since, cell migration is a time-dependent process, it is not unexpected that more cells were present at a later time point. A possibility also exists that SMC treated with VCAM-1 siRNAs had developed a mechanism over time to compensate for the defect due to the reduction in VCAM-1 expression.
Another interesting finding was that VCAM-1 siRNA inhibited SMC proliferation although the effect was small. This finding suggests that endogenously produced VCAM-1 plays a role in SMC proliferation. The mechanism underlying the effect of VCAM-1 on SMC proliferation is unknown. However, it is known that VCAM-1 interacts with the integrin α4β1 (very late-acting antigen 4), which is expressed on the surface of SMC [9], and β-integrins mediate the enhancement of smooth muscle proliferation by collagen and fibronectin [23]. Thus, it is possible that the integrin α4β1–VCAM-1 interaction enhances the proliferative response of SMC to stimuli like collagen. It is noteworthy that VCAM-1 siRNA decreased SMC migration by 82% at 24 h, whereas it only slightly inhibited SMC proliferation. Thus, the effect of VCAM-1 on cell migration may not be attributable to its effect on proliferation. Furthermore, to eliminate the confounding influence of cell proliferation, we performed cell migration assay using Boyden chambers but we failed to observe any significant effect of VCAM-1 siRNA on cell migration (see Supplementary data). To induce cell transmigration across the Boyden membranes, PDGF-β needed to be added to lower chambers. The effect of PDGF-β on cell migration was probably too strong to overwhelm the effect of VCAM-1 siRNA on cell migration. Another explanation is that cell migration in response to wound injury is mediated by a mechanism different from the one induced by PDGF-β.
In summary, the present study identified a new role for VCAM-1 in regulating SMC migration and proliferation. Because excess SMC accumulation and proliferation is a hallmark of atherosclerosis and neointimal hyperplasia, targeting VCAM-1 expression could be effective for prevention and treatment of atherosclerosis and restenosis.
Supplementary Material
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi: 10.1016/j.atherosclerosis.2007.10.015.
Acknowledgments
This work was supported by the National Institutes of Health grant HL71844 and the Society of Interventional Radiology seed grant.
References
- 1.Moiseeva EP. Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc Res. 2001;52:372–86. doi: 10.1016/s0008-6363(01)00399-6. [DOI] [PubMed] [Google Scholar]
- 2.Worth NF, Rolfe BE, Song J, Campbell GR. Vascular smooth muscle cell phenotypic modulation in culture is associated with reorganisation of contractile and cytoskeletal proteins. Cell Motil Cytoskeleton. 2001;49:130–45. doi: 10.1002/cm.1027. [DOI] [PubMed] [Google Scholar]
- 3.Huo Y, Ley K. Adhesion molecules and atherogenesis. Acta Physiol Scand. 2001;173:35–43. doi: 10.1046/j.1365-201X.2001.00882.x. [DOI] [PubMed] [Google Scholar]
- 4.Manka DR, Wiegman P, Din S, et al. Arterial injury increases expression of inflammatory adhesion molecules in the carotid arteries of apolipoprotein-E-deficient mice. J Vasc Res. 1999;36:372–8. doi: 10.1159/000025676. [DOI] [PubMed] [Google Scholar]
- 5.Li H, Cybulsky MI, Gimbrone MA, Jr, Libby P. Inducible expression of vascular cell adhesion molecule-1 by vascular smooth muscle cells in vitro and within rabbit atheroma. Am J Pathol. 1993;143:1551–9. [PMC free article] [PubMed] [Google Scholar]
- 6.Oguchi S, Dimayuga P, Zhu J, et al. Monoclonal antibody against vascular cell adhesion molecule-1 inhibits neointimal formation after periadventitial carotid artery injury in genetically hypercholesterolemic mice. Arterioscler Thromb Vasc Biol. 2000;20:1729–36. doi: 10.1161/01.atv.20.7.1729. [DOI] [PubMed] [Google Scholar]
- 7.Barringhaus KG, Phillips JW, Thatte JS, et al. Alpha4beta1 integrin (VLA-4) blockade attenuates both early and late leukocyte recruitment and neointimal growth following carotid injury in apolipoprotein E (−/−) mice. J Vasc Res. 2004;41:252–60. doi: 10.1159/000078646. [DOI] [PubMed] [Google Scholar]
- 8.Dansky HM, Barlow CB, Lominska C, et al. Adhesion of monocytes to arterial endothelium and initiation of atherosclerosis are critically dependent on vascular cell adhesion molecule-1 gene dosage. Arterioscler Thromb Vasc Biol. 2001;21:1662–7. doi: 10.1161/hq1001.096625. [DOI] [PubMed] [Google Scholar]
- 9.Duplaa C, Couffinhal T, Dufourcq P, et al. The integrin very late antigen-4 is expressed in human smooth muscle cell. Involvement of alpha 4 and vascular cell adhesion molecule-1 during smooth muscle cell differentiation. Circ Res. 1997;80:159–69. doi: 10.1161/01.res.80.2.159. [DOI] [PubMed] [Google Scholar]
- 10.Hammond SM, Boettcher S, Caudy AA, Kobayashi R, Hannon GJ. Argonaute2, a link between genetic and biochemical analyses of RNAi. Science. 2001;293:1146–50. doi: 10.1126/science.1064023. [DOI] [PubMed] [Google Scholar]
- 11.Elbashir SM, Harborth J, Lendeckel W, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–8. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 12.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. EphA2: a determinant of malignant cellular behavior and a potential therapeutic target in pancreatic adenocarcinoma. Oncogene. 2004;23:1448–56. doi: 10.1038/sj.onc.1207247. [DOI] [PubMed] [Google Scholar]
- 13.Song E, Lee SK, Wang J, et al. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat Med. 2003;9:347–51. doi: 10.1038/nm828. [DOI] [PubMed] [Google Scholar]
- 14.Duxbury MS, Matros E, Ito H, et al. Systemic siRNA-mediated gene silencing: a new approach to targeted therapy of cancer. Ann Surg. 2004;240:667–74. doi: 10.1097/01.sla.0000140755.97224.9a. discussion 666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyoshi T, Tian J, Matsumoto AH, Shi W. Differential response of vascular smooth muscle cells to oxidized LDL in mouse strains with different atherosclerosis susceptibility. Atherosclerosis. 2006;189:99–105. doi: 10.1016/j.atherosclerosis.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 16.Zhu N, Lalla R, Eves P, et al. Melanoma cell migration is upregulated by tumour necrosis factor-alpha and suppressed by alpha-melanocyte-stimulating hormone. Br J Cancer. 2004;90:1457–63. doi: 10.1038/sj.bjc.6601698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyoshi T, Matsumoto AH, Shi W. Paradoxical increase in LDL oxidation by endothelial cells from an atherosclerosis-resistant mouse strain. Atherosclerosis. 2007;192:259–65. doi: 10.1016/j.atherosclerosis.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 18.Teupser D, Thiery J, Haas U, et al. Expression of vascular cell adhesion molecule-1 (VCAM-1) in the aortae of hypercholesterolemic rabbits with high (HAR) and low (LAR) atherosclerotic response. Atherosclerosis. 1997;128:157–64. doi: 10.1016/s0021-9150(96)05997-7. [DOI] [PubMed] [Google Scholar]
- 19.Tian J, Pei H, James JC, et al. Circulating adhesion molecules in apoE-deficient mouse strains with different atherosclerosis susceptibility. Biochem Biophys Res Commun. 2005;329:1102–7. doi: 10.1016/j.bbrc.2005.02.090. [DOI] [PubMed] [Google Scholar]
- 20.Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–69. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 21.Ridley AJ, Schwartz MA, Burridge K, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–9. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 22.Palecek SP, Loftus JC, Ginsberg MH, Lauffenburger DA, Horwitz AF. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature. 1997;385:537–40. doi: 10.1038/385537a0. [DOI] [PubMed] [Google Scholar]
- 23.Nguyen TT, Ward JP, Hirst SJ. beta1-Integrins mediate enhancement of airway smooth muscle proliferation by collagen and fibronectin. Am J Respir Crit Care Med. 2005;171:217–23. doi: 10.1164/rccm.200408-1046OC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi: 10.1016/j.atherosclerosis.2007.10.015.