

Abstract
The concentration of intracellular Ca2+ ([Ca2+]i) influences neuronal properties ranging from excitability to neurotransmitter release. Persistent inflammation is associated with changes in the properties of primary afferent neurons ranging from excitability to transmitter release. The purpose of the present study was to determine whether previously described inflammation-induced changes in excitability and transmitter release are associated with changes in the regulation of [Ca2+]i. Acutely dissociated dorsal root ganglion (DRG) neurons harvested from adult rats 3 days following a hindpaw injection of complete Freund’s adjuvant (CFA) or naïve controls, were stimulated with 30 mM K+ (Hi K+). Hi K+ evoked changes in [Ca2+]i were assessed with fura-2 ratiometric microfluorimetry. Subpopulations of DRG neurons were defined by cell body diameter, IB4 binding, capsaicin (CAP) sensitivity and target of innervation (DiI labeling). Inflammation was associated with significant increases in resting [Ca2+]i and increases in the magnitude and decreases in the decay, of the evoked increase in [Ca2+]i. The changes in evoked transients were larger in neurons innervating the site of inflammation. Furthermore, there were differences among subpopulations of DRG neurons with respect to changes in magnitude and/or decay of the evoked transient such that the increase in magnitude was larger in small- and medium-diameter neurons than in large diameter neurons while the decrease in the decay was greater in CAP responsive, IB4 positive, small- and medium-diameter neurons than in CAP unresponsive, IB4 negative and/or large-diameter neurons. These changes in the regulation of [Ca2+]i were not due to inflammation-induced changes in passive or active electrophysiological properties. Importantly, an inflammation-induced increase in evoked Ca2+ transients in putative nociceptive afferents may contribute to the pain and hyperalgesia associated with persistent inflammation via facilitation of transmitter release from these afferents.
Keywords: inflammatory pain, nociceptor sensitization, calcium imaging, current clamp, persistent inflammation, retrograde tracer
Introduction
Tissue inflammation is associated with hyperalgesia that reflects, in part, an increase in afferent excitability and/or transmitter release (Richardson and Vasko, 2002, Gold and Flake, 2005). Through its mediation of a number of cellular processes from transcription to ion channel activity, intracellular Ca2+ plays a critical role in the regulation of both neuronal excitability and transmitter release (Berridge, 2005). That changes in the regulation of the concentration of intracellular Ca2+ ([Ca2+]i) may contribute to changes in afferent excitability and transmitter release is suggested by the observations that traumatic nerve injury is associated with decreases in voltage-gated Ca2+ current density (Baccei and Kocsis, 2000, Hogan et al., 2000, McCallum et al., 2003), a decrease in resting [Ca2+]i in injured dorsal root ganglion (DRG) neurons (Fuchs et al., 2005) and a decrease in both the magnitude and decay of evoked Ca2+ transients in injured putative nociceptive DRG neurons (Fuchs et al., 2007). This pattern of nerve injury-induced changes in evoked Ca2+ transients were specific to putative nociceptive DRG neurons as changes in the opposite direction were observed in putative non-nociceptive DRG neurons (Fuchs et al., 2007). Furthermore, diabetic neuropathy is associated with an increase in resting [Ca2+]i levels and a delay in recovery of evoked Ca2+ transients in DRG (Kostyuk et al., 1999, Kostyuk et al., 2001, Huang et al., 2002). And while a number of marked differences in the underlying mechanisms of neuropathic and inflammatory pain have been identified (Amir et al., 2006), the impact of inflammation on the regulation of [Ca2+]i has yet to be described. Therefore, the purpose of the present study was to assess the impact of persistent inflammation on resting [Ca2+]i and evoked Ca2+ transients in DRG neurons.
Experimental Procedures
Animals
Adult (240–340g) male Sprague-Dawley rats (Harlan, Indianapolis, IN) were used for this study. Rats were housed in University of Maryland Dental School or University of Pittsburgh animal facilities in groups of three on a 12:12 light dark schedule. Food and water are available ad lib. All the experiments were approved by both the University of Maryland Dental School and the University of Pittsburgh Institutional Animal Care and Use Committees and performed in accordance with National Institutes of Health guidelines as well as guidelines established by the International Association of the Study of Pain for the use of laboratory animals in research. Rats were deeply anesthetized for both tissue labeling and tissue harvest (details provided below). The depth of anesthesia was assessed by monitoring for the presence or absence of corneal reflexes and withdrawal responses from noxious pinch of the hindpaw. Respiration rate was also monitored. Supplemental anesthesia was administered until animals were areflexive. Following tissue harvest, rats were killed by decapitation. All efforts were made to minimize the number of animals used and their suffering.
Labeling of DRG neurons innervating the glabrous skin of the hindpaw
DRG neurons innervating the glabrous skin of left hindpaw were labeled with the retrograde tracer DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbo-cyanine perchlorate, Invitrogen, Carlsbad CA). Labeling was performed as previously described (Katz and Gold, 2006). DiI was dissolved in DMSO (170 mg/ml, Sigma-Aldrich, St Louis MO) and diluted 1:10 in 0.9% of sterile saline. Diluted dye was injected subcutaneously at 3–5 sites in glabrous skin (10 μl total volume) with a 30 g injection needle under anesthesia induced by 5% isoflurane (Abbott Laboratories, North Chicago, IL, USA) and maintained with 1.5% isoflurane. DiI-labeled neurons were easily identified under epifluorescence illumination with a Texas-red filter set.
Tissue inflammation
Two to three weeks after labeling, inflammation was induced at the site of DiI injection in the left hindpaw with a subcutaneous injection of complete Freund’s adjuvant (CFA, Sigma-Aldrich). CFA was diluted 1:1 in 0.9% of sterile saline and injected (100 μl) with a 25 g needle under isoflurane anesthesia. DRG were harvested for study 3 days after CFA injection.
Cell dissociation
Rats were deeply anaesthetized with a subcutaneous injection of a mixture (1 ml/kg of 55 mg/ml ketamine, 5.5 mg/ml xylazine, and 1.1 mg/ml acepromazine) (ketamine was from Fort Dodge Animal Health, Fort Dodge, WI, USA; xylazine and acepromazine were from Phoenix Scientific Inc., St Joseph, MO, USA). L4–5 DRGs (ipsilateral to the labeling and/or inflamed site) from inflamed or naïve rats were harvested, enzymatically treated and mechanically dissociated as described previously (Lu et al., 2006). Dissociated DRG neurons were plated onto glass coverslips, previously coated by a solution of 5 μg/ml mouse laminin (Invitrogen) and 0.1 mg/ml poly-l-ornithine (Sigma-Aldrich). All experiments were performed within 8 h of tissue harvest.
Subclassification of DRG neurons
DRG neurons were sub-classified on the basis of several criteria. These included cell body size, binding to the plant (Griffonia simplicifolia) lectin isolectin B4 (IB4, Sigma-Aldrich), the responsiveness to the algogenic compound capsaicin (CAP, Sigma-Aldrich) and DiI labeling. Neurons were divided into small (<30μm), medium (30 to 40μm), and large (>40μm) based on cell body diameter as measured with a calibrated eyepiece reticule. Neurons were labeled with IB4 following a 10 minute incubation with 10 μg/ml FITC conjugated IB4 (Lu et al., 2006). Neurons were considered responsive to CAP (CAP+), if application of 500 nM CAP evoked an increase in [Ca2+]i greater than 20% above baseline. Finally, because CFA was injected into the same site as DiI, DiI labeling was used to identify the population of DRG neurons most directly impacted by inflammation. While not all nociceptive afferents are responsive to capsaicin, capsaicin responsive sensory neurons are considered putative nociceptors based on the observations that this compound only elicits the sensation of pain and it selectively activates nociceptive afferents in the rat (Holzer, 1991). Similarly, while not all sensory neurons with a small cell body diameter are nociceptive, a small cell body diameter may also be used to identify putative nociceptive based on the observation that a large majority of high threshold afferents innervating cutaneous tissue in the rat have a small cell body diameter (Lawson, 2002) and that sensory neurons with a small cell body diameter are likely to express other properties of nociceptive afferents (Gold et al., 1996a). Finally, IB4 binding may also be used to identify a subpopulation of putative nociceptive afferents based on the observation that IB4 binding in cutaneous afferents demonstrate properties of nociceptors in vivo (Fang et al., 2006).
Intracellular Ca2+ measurements
DRG neurons were loaded with 2.5 μM Ca2+ indicator fura-2 AM ester with 0.025% pluronic (both from Invitrogen) for 20 min at room temperature as described previously (Lu et al., 2006). They were subsequently labeled with FITC-conjugated IB4. Following fura-2 loading and IB4 labeling, DRG neurons were placed in a recording chamber and were continuously perfused with normal bath solution (containing in mM: 130 NaCl, 3 KCl, 2.5 CaCl2, 0.6 MgCl2, 10 HEPES, 10 glucose, pH adjusted with Tris base to 7.4, osmolality adjusted with sucrose to 325 mOsm). All chemicals were obtained from Sigma-Aldrich. Fluorescence data was acquired on a PC running Metafluor software (Molecular Devices, Sunnyvale, CA) via a CCD camera (Roper Scientific, Trenton, NJ, Model RTE/CCD 1300). The ratio (R) of fluorescence emission (510 nm) in response to 340/380 nm excitation [controlled by a lambda 10–2 filter changer (Sutter Inst.; CA)] was acquired at 1Hz during drug applications. High K+ (30 mM) and CAP (500 nM) were delivered by a computer-controlled fast perfusion system (switching time < 50 ms; ALA Scientific Instruments, Westbury, NY, Model DAD-12).
[Ca2+]i was derived from fluorescent ratios (R) following performance of in situ calibration experiments (Grynkiewicz et al., 1985) according to the following equation: [Ca2+]i (nM) = Kd(Sf2/Sb2)(R−Rmin)/(Rmax−R), where Kd is the dissociation constant of fura-2 for Ca2+ at room temperature (i.e., 224 nM); Sf2/Sb2 is the fluorescence ratio of the emission intensity excited by 380-nm signal in the absence of Ca2+ to that during the presence of saturating Ca2+; Rmin or Rmax is the minimal or maximal fluorescence ratios respectively. Rmin was measured by perfusing the neurons with nominally Ca2+-free bath solution containing 1.0 mM EGTA and 10 μM ionomycin (both from Sigma-Aldrich) with 10 mM Mg2+ for at least 40 min. Rmax was obtained by perfusing neurons with standard bath solution containing 20 mM Ca2+ and 10 μM ionomycin for a few minutes. Background values of F340 and F380 were collected by perfusing the neurons with 50 μM digitonin for a few minutes. Values of Sf2/Sb2, Rmin, or Rmax were 15.51, 0.48, or 14.83 averaged over 65 neurons obtained from 7 separate in situ calibration experiments.
Electrophysiology
Whole-cell patch-clamp recordings were performed using an EPC-9 amplifier (HEKA Elektronik GmbH, Lambrecht, Germany). Series resistance was compensated (>80%) with amplifier circuitry. Data were acquired at 10 kHz and filtered at 2 kHz. Electrodes (1.8–3.0 MΩ) were filled with pipette solution (contained in mM: 100 K-Methansulfonate (MS), 5 Na-MS, 40 KCl, 1 CaCl2, 2 MgCl2, 11 EGTA, 10 HEPES, 2 ATP-Mg, 1 GTP-Li); pH was adjusted to 7.2 with Tris-Base and osmolality was adjusted to 310 mOsm with sucrose. Neurons were continuously perfused with normal bath solution.
Statistical analysis
Neurons were considered “responsive” to high K+, if high K+ evoked an increase in [Ca2+]i greater than 20% above baseline. All data analysis was limited to high K+ responsive neurons. Data are expressed as mean ± S.E.M unless otherwise stated. The “n” for each group refers to the number of neurons. Two-way ANOVA was employed to assess the presence of statistically significant influences of CFA-induced inflammation and the possibility that there were differences between subpopulations of DRG neurons in responses to inflammation, as well as the interaction between the two. Data sets failing normality and/or equal variance tests were transformed by rank and a two-way ANOVA test was run on ranked data. Differences between groups were assessed with a Holm-Sidak post hoc test if there was a statistically significant interaction between main effects. Other statistical tests were employed as indicated. Statistical significance was assessed at p<0.05.
Results
Data were collected from L4 and L5 DRG neurons obtained from 13 naïve and 15 rats in which hindpaw inflammation had been induced with a subcutaneous injection of CFA 72 hours prior to tissue harvest. To determine whether inflammation of the hindpaw influences the magnitude and/or decay of evoked Ca2+ transients in a wider distribution of neurons than those immediately surrounding the site of CFA injection, data was collected from both DiI+ and DiI− DRG neurons. Subpopulations of DiI+ and DiI− DRG neurons were defined by cell body diameter, IB4 binding and capsaicin (CAP) sensitivity. The number of DRG neurons studied from naïve and inflamed rats in each of these subpopulations is indicated (in parenthesis) in each cell of Table 1.
Table 1.
Resting [Ca2+]i (nM) levels among subpopulations of DRG neurons from naïve and inflamed rats
| Subpop. | Stats | DiI+ | Stats | DiI− | ||
|---|---|---|---|---|---|---|
| Naïve (n) | CFA (n) | Naïve (n) | CFA (n) | |||
| Small | S < 0.01
C > 0.05 I < 0.01 |
138 ± 8 (20) | 144 ± 8 (19) | S > 0.05
C < 0.05 I > 0.05 |
122 ± 4 (72) | 138 ± 6 (72) |
| Medium | 114 ± 6 (25) | 145 ± 6 (42) | 120 ± 5 (38) | 135 ± 7 (33) | ||
| Large | 121 ± 8 (18) | 102 ± 8 (12) | 118 ± 4 (39) | 125 ± 5 (31) | ||
| IB4+ | S < 0.01
C > 0.05 I < 0.01 |
125 ± 5 (33) | 150 ± 5 (48) | S > 0.05
C < 0.01 I > 0.05 |
120 ± 3 (81) | 140 ± 6 (72) |
| IB4− | 123 ± 7 (30) | 114 ± 6 (25) | 120 ± 4 (68) | 128 ± 4 (64) | ||
| CAP+ | S > 0.05
C > 0.05 I > 0.05 |
128 ± 5 (36) | 141 ± 5 (56) | S > 0.05
C < 0.05 I > 0.05 |
121 ± 3 (74) | 139 ± 5 (80) |
| CAP− | 118 ± 7 (27) | 126 ± 10 (17) | 120 ± 3 (75) | 128 ± 5 (56) | ||
Small, Medium and Large are cell body diameter. IB4 is isolectin B4. CAP is capsaicin. Data are mean ± SEM. The number in parentheses is the number of neurons studied in each group. Two-way ANOVA was used to assess the presence of main effects associated with subpopulation (S: cell body diameter, IB4 binding or CAP sensitivity) and inflammation (C: Naïve versus CFA) as well as a significant interaction (I) between the two. DiI+ and DiI− neurons were analyzed separately. Results of this analysis are indicated in columns labeled Stats. Post-hoc analysis (Holm-Sidak test) indicated that resting [Ca2+]i was significantly (p < 0.05) higher medium diameter DiI+ neurons from inflamed rats than in those from naïve rats. Similarly, post-hoc analysis indicated that resting [Ca2+]i was significantly (p < 0.01) higher medium diameter DiI+ neurons from inflamed rats than in those from naïve rats.
Impact of inflammation on subpopulations of DRG neurons
There is evidence that persistent inflammation can increase sensory neuron cell body size (Flake et al., 2005), as well as the proportion of neurons responsive to CAP (Breese et al., 2005) and/or that are labeled by IB4 (Xu et al., 2005). We therefore sought to determine whether similar changes were detectable in populations of neurons defined by DiI labeling. The size distribution of DRG neurons from naïve rats was similar to results from previous studies with 43.4% (92 out of 212), 29.7% (63 out of 212) and 26.9% (57 out of 212) of neurons classified as small-, medium- and large-diameter neurons respectively (Lawson et al., 1993). Inflammation had no statistically significant influence on cell body diameter whether data were analyzed by proportion (with a Chi-Square test) or median (with a Mann-Whitney U test): the size distribution of DRG neurons collected from inflamed rats was 43.5% (91 out of 209), 35.9% (75 out of 209) and 20.6% (43 out of 209) as small-, medium- and large-diameter, respectively (p > 0.05); the median cell body diameter of DRG neurons from naïve rats was 31.3 μm (with 27.5 μm and 41.0 μm as 25th and 75th percentiles, respectively) while that from inflamed rats was 30.0 μm (with 26.9 μm and 37.5 μm as 25th and 75th percentiles, respectively) (p > 0.05). Similarly, when DiI+ (Figure 1A) and DiI− (Figure 1B) neurons were analyzed independently, no statistically significant influence of inflammation on cell body diameter was detected.
Figure 1.

Histograms of cell body diameter for naïve (CTRL) and inflamed (CFA) DiI+ (A) and DiI− (B) DRG neurons. Bin size is 5 μm. In order to facilitate comparing control and inflamed neurons, they have been plotted together and normalized with respect to the total number of neurons studied in each group. The median (+/− 25th and 75th percentiles) cell body diameter was 34.3 (29.1 and 41.0) and 32.0 (29.5 and 34.5) for DiI+ neurons from naïve and inflamed rats, respectively (p > 0.05, Mann-Whitney U test). The median cell body diameter was 30.8 (27.0 and 41.0) and 28.8.0 (25.0 and 37.5) for DiI− neurons from naïve and inflamed rats, respectively (p > 0.05, Mann-Whitney U test).
No statistically significant influence of inflammation was detected on the distribution of IB4 binding. Data from DiI+ and DiI− neurons were analyzed separately. The percentage of IB4+ neurons in the DiI+ group from naïve rats (52% (33 of 63)) was similar to that in neurons from inflamed rats (64% (48 of 73)) (p > 0.05, Chi-square test). Similar numbers were obtained for the DiI− group where IB4+ neurons accounted for 54% (81 of 149) and 53% (72 of 136) of neurons from naïve and inflamed rats, respectively (p > 0.05, Chi-square test).
A similar analysis was performed on the distribution of CAP sensitivity among DRG neurons from naïve and inflamed rats. There was a statistically significant (p < 0.01, Chi-square test) inflammation-induced increase in the percentage of DiI+ neurons that were CAP+ from 57% (36 of 63) in naïve rats to 77% (56 of 73) in inflamed rats. In contrast, there was no statistically significant (p > 0.05) difference between naïve (50% (74 of 149)) and inflamed (59% (80 of 136)) rats with respect to the percentage of CAP+ DiI− neurons.
The Ca2+ transients evoked by CAP were also analyzed. Two second application of 500 nM CAP was used to evoke Ca2+ transients in DRG neurons at the end of each experiment. There were no statistically significant (p > 0.05, student’s t-test) differences between naïve and inflamed rats with respect to the magnitude of CAP evoked transients in either DiI+ or DiI−neurons. The magnitude of CAP-evoked Ca2+ transients from DiI+ neurons was 577 ± 71 nM (n = 36) and 637 ± 59 nM (n = 48) from naïve and inflamed rats, respectively. The magnitude of CAP-evoked transients from DiI− neurons was 476 ± 44 nM (n = 74) and 570 ± 41 nM (n = 73) from naïve and inflamed rats, respectively. In most cases, the CAP-evoked increase in [Ca2+]i failed to fully recover to baseline levels within 2 minutes of CAP application, therefore, the decay of the CAP evoked transient was not analyzed.
In summary, persistent inflammation had no statistically significant influence on cell body diameter, IB4 binding or the magnitude of the CAP evoked Ca2+ transient. However, there was a statistically significant increase in the proportion of DiI+ neurons that were CAP+ in the presence of inflammation.
Inflammation induced increase in resting [Ca2+]i in DRG neurons
Resting [Ca2+]i in subpopulations of DiI+ and DiI− DRG neurons from naïve and inflamed rats are shown in Table 1. Data were analyzed with a two-way ANOVA in order to assess the presence of main effects associated with subpopulation or the presence of inflammation as well as an interaction between the two. There was a significant main effect associated with size and a significant interaction between size and inflammation among DiI+ neurons with post-hoc analysis (Holm-Sidak) indicating that resting [Ca2+]i was higher (p < 0.05) in medium diameter neurons following inflammation. The impact of inflammation on resting Ca2+ among DiI+ neurons as a function of cell body diameter is illustrated in a scatterplot in Figure 2A. In subpopulations of DiI+ neurons defined by IB4 binding there was also a significant interaction between IB4 binding and inflammation that was largely due to the higher resting [Ca2+]i in IB4+ neurons following inflammation (Table 1, p < 0.01). Among DiI− neurons, there was a main effect (p < 0.01) associated with inflammation in subpopulations defined by size, IB4 binding and CAP sensitivity with resting Ca2+ levels increased following inflammation. However, in none of these analyses was there a significant main effect associated with subpopulation, nor was there a significant interaction between subpopulation and inflammation. The impact of inflammation on resting Ca2+ among DiI− neurons as a function of cell body diameter is illustrated in a scatterplot in Figure 2B.
Figure 2.

Scatter plots of resting [Ca2+]i versus cell body diameter for DiI+ (A) and DiI− (B) neurons from naïve (CTRL) and inflamed (CFA) rats. Inflammation was associated with an increase in resting [Ca2+]i in a subpopulation of small and medium diameter DiI− neurons. Note, scale of Y-Axis in A and B differ.
Inflammation increases evoked Ca2+ transients in DRG neurons
Application of high K+ (30 mM) resulted in an increase (>20% above baseline) in [Ca2+]i in over 90% of neurons tested from both naïve (212 of 231) and inflamed (209 of 230) rats; these values were not significantly different (p > 0.05, Chi-square test). However, evoked Ca2+ transients were larger and decayed more slowly in subpopulations of DRG neurons following inflammation (Figure 3A and B). To quantify these changes, the magnitude of evoked Ca2+ transient was analyzed as the difference between peak and baseline [Ca2+]i and the decay of the Ca2+ transient analyzed as time to 50% from peak (T50). Pooled data for both DiI+ and DiI− neurons are plotted in Figure 4 and were analyzed with a two-way ANOVA. Results of this analysis for DiI+ neurons revealed significant main effects associated with both inflammation (p < 0.01) and subpopulation (p < 0.01) whether subpopulations were defined by cell body diameter (Figure 4A), IB4 binding (Figure 4C) or CAP sensitivity (Figure 4E). Furthermore, there was a significant (p < 0.01) interaction between these two main effects for both magnitude (Figure 4A right panels) and decay (Figure 4A left panels) of evoked Ca2+ transients when subpopulations of neurons were defined by cell body diameter. There were also significant (p < 0.01) interactions between these two main effects for decay of evoked Ca2+ transients when subpopulations of neurons were defined by CAP sensitivity (Figure 4E left panel). Post-hoc analysis (Holm-Sidak test) indicated that inflammation was associated with an increase (p < 0.01) in the magnitude and a decrease (p < 0.01) in the decay of evoked Ca2+ transients in small and medium diameter neurons. There is also a significant decrease (p < 0.01) in the decay of the evoked Ca2+ transient in CAP+ neurons.
Figure 3.
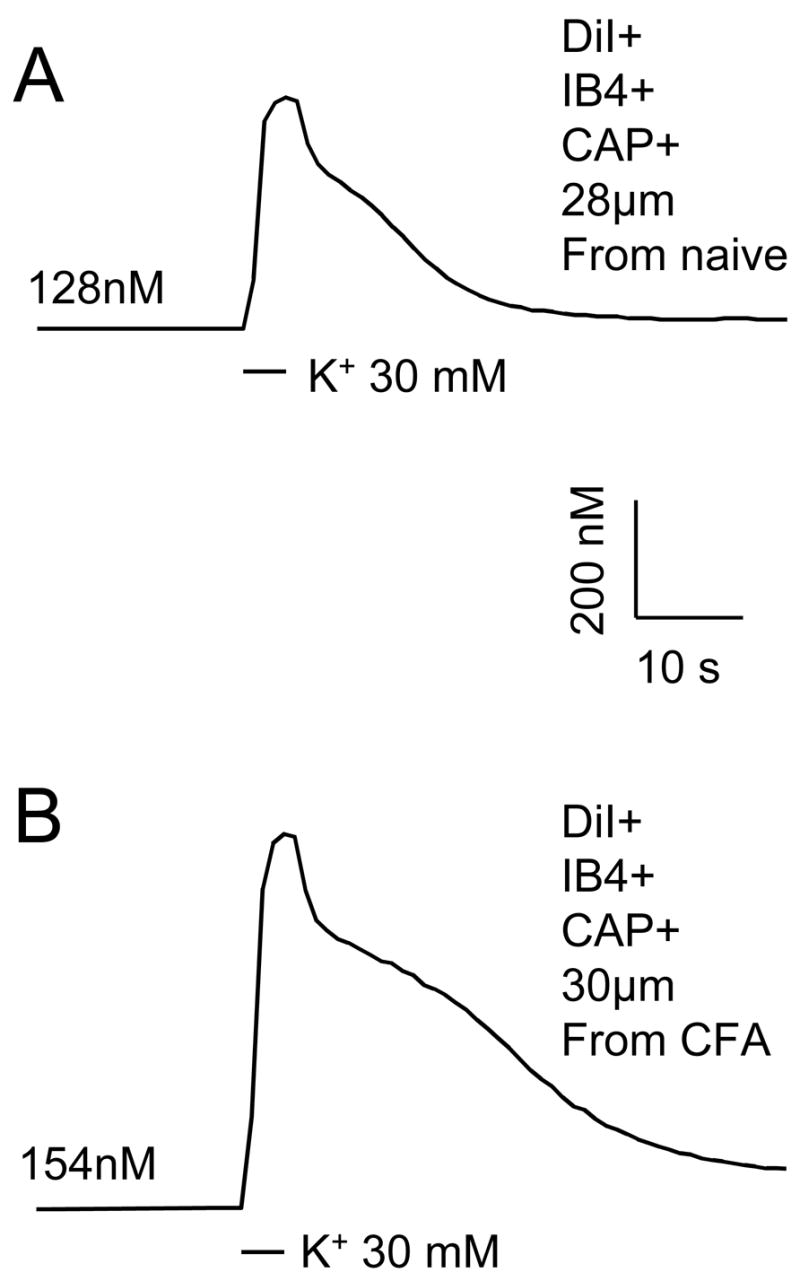
Ca2+ transients were evoked with a 4 second application of 30 mM K+ in putative nociceptive DRG neurons from a naïve (A) rat and a rat in which inflammation had been induced with a subcutaneous injection of CFA 3 days prior to harvesting ganglia (B). High K+ was applied at points indicated by black bars. Scale bars for A and B are the same. The resting [Ca2+]i in each neurons is indicated.
Figure 4.

Differential impact of persistent inflammation on evoked Ca2+ transients among subpopulations of DRG neurons. In each panel, data were pooled from neurons in each subpopulation define by cell body size (A, B where S = small, M = medium and L = large)), IB4 binding (C, D) and capsaicin (CAP) sensitivity (E, F). Neurons were further sub-divided according to the presence (A, C, and E) or absence (B, D and F) of DiI labeling and whether they were from naïve (filled bars) or inflamed (open bars) rats. The magnitude of evoked Ca2+ transient (left) was analyzed as Δ [Ca2+]i (peak value – baseline), and the decay of the evoked Ca2+ transient (right) was analyzed as time to 50% of peak (T50), of evoked Ca2+ transients. Data in each panel were analyzed with a two-way ANOVA in order to assess main effects associated with subpopulation (S), inflammation (C) or an interaction between the two (I). Post-hoc analyses (Holm-Sidak) were performed if there was a significant interaction between main effects, and statistically significant differences between naïve and inflamed groups are indicated. The number of neurons in each group is listed in Table 1. ** is p < 0.01.
In DiI− neurons, there were also significant main effects associated with subpopulation for magnitude and decay of evoked Ca2+ transients when subpopulations were defined by cell body diameter (p < 0.01, Figure 4B), IB4 binding (p < 0.01, Figure 4D) and CAP sensitivity (p < 0.01, Figure 4F). There were also significant main effects associated with inflammation for magnitude and decay when subpopulations were defined by cell body diameter (p < 0.01, Figure 4B), IB4 binding (p < 0.01, Figure 4D) and CAP sensitivity (p < 0.01, Figure 4F). In addition, there was a significant interaction between these two main effects for both magnitude (p < 0.01) and decay (p < 0.01) of evoked transients in subpopulations of neurons defined by cell body diameter and for the magnitude (p < 0.01) of evoked transients in subpopulations of neurons defined by CAP sensitivity. As with DiI+ neurons, the significant interactions (Holm-Sidak post-hoc test) were associated with an increase (p < 0.01) in the magnitude and decrease (p < 0.01) in the decay of evoked transients in small and medium diameter neurons from inflamed rats and an increase in the magnitude (p < 0.01) of evoked Ca2+ transients in CAP+ neurons from inflamed rats.
To determine whether there were differences between DiI+ and DiI− neurons with respect to the influence of inflammation on evoked Ca2+ transients, data in each subpopulation of DRG neurons was analyzed as a percent change from mean value for naïve neurons in each subpopulation. Pooled data were analyzed with a two-way ANOVA to assess the presence of main effects associated with subpopulation (S), DiI labeling (D) or an interaction (I) between the two (Figure 5). Results of this analysis revealed the presence of a statistically significant main effect associated with DiI labeling for the percent change in both the magnitude and the decay of the evoked increase in [Ca2+]i for subpopulations defined by cell body size (Figure 5A), IB4 binding (Figure 5B) or CAP sensitivity (Figure 5C). In each case, the percent change was larger (p < 0.01) in DiI+ neurons than in DiI− neurons. There was a statistically significant main effect associated with subpopulation in the percent increase in the magnitude of the evoked Ca2+ transient in subpopulations defined by cell body size and capsaicin sensitivity. In both cases, the percent increase in the magnitude of the evoked Ca2+ transient was larger in DiI+ neurons. Finally, there was a statistically significant interaction between subpopulation and DiI labeling for the percent change in both the magnitude and decay of the evoked Ca2+ transient in subpopulations defined by cell body size and for the decay of the evoked Ca2+ transient in subpopulations defined by IB4 binding. Post-hoc (Holm-Sidak) analysis indicated that the percent increase in the magnitude of the evoked Ca2+ transient was larger (p < 0.01) in medium diameter DiI+ neurons than in medium diameter DiI− neurons, while the decay of the evoked Ca2+ transient was larger (p < 0.01) in DiI+ small diameter and IB4+ neurons than in the respective subpopulation of DiI− neurons.
Figure 5.
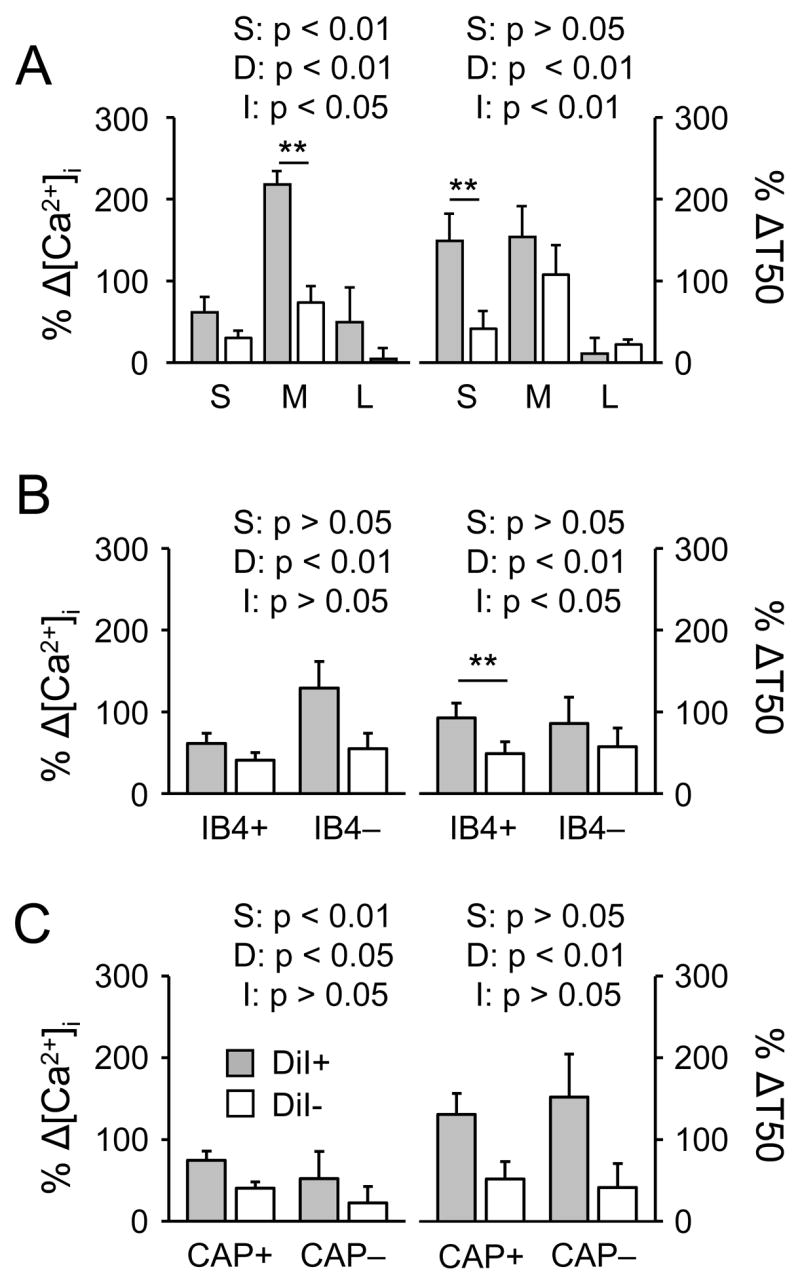
Differential impact of persistent inflammation on evoked Ca2+ transients among subpopulations of DiI+ (filled bars) and DiI− (open bars) DRG neurons. Data for each neuron was analyzed as a percent change from the mean value for magnitude (Δ [Ca2+]i; Left panels) or decay (Δ T50; Right panels) obtained from naïve neurons in each subpopulation, and pooled. Subpopulations were defined by cell body size (A, where S = small, M = medium and L = large)), IB4 binding (B) and capsaicin (CAP) sensitivity (C). Data in each panel were analyzed with a two-way ANOVA in order to assess main effects associated with subpopulation (S), DiI labeling (D) or an interaction between the two (I). Post-hoc analyses (Holm-Sidak) were performed if there was a significant interaction between main effects, and statistically significant differences between naïve and inflamed groups are indicated. The number of neurons in each group is listed in Table 1. ** is p < 0.01.
Excitability
We (Flake et al., 2005, Harriott et al., 2006, Katz and Gold, 2006) and others (Moore et al., 2002, Dang et al., 2005) have previously demonstrated that persistent inflammation results in an increase in the excitability of dissociated DRG neurons. It is therefore possible that the inflammation-induced increase in the high K+ evoked Ca2+ transient reflects an increase in DRG neuron excitability. There is also evidence of an inflammation-induced depolarization of resting membrane potential (Flake et al., 2005), suggesting that the inflammation-induced increase in resting [Ca2+]i may reflect a depolarization-induced activation of voltage-gated Ca2+ channels. In order to explore these possibilities, we recorded from small and medium diameter DiI+ DRG neurons from three naïve and 3 inflamed rats in current clamp mode in order to assess the impact of inflammation on resting membrane potential and the response to a 4 second 30 mM K+ application. Data was collected from 10 small and 10 medium diameter neurons, and 9 small and 15 medium diameter neurons from naïve and inflamed rats, respectively. High K+ application to neurons from inflamed rats resulted in a rapid membrane potential depolarization that was effectively “clamped” to a depolarized potential in the majority of neurons studied (Figure 6). Membrane depolarization was associated with few, if any, action potentials, although 1 small diameter neuron from an inflamed rat continued to fire action potentials throughout the duration of the high K+ application. Following termination of the high K+, resting membrane potential was rapidly re-established. None of the neurons demonstrated spontaneous activity. Resting membrane potential and input resistance as well as parameters describing the response to high K+ (30 mM) are summarized in Table 2. Data from naïve rats are similar to those described previously. (Lu et al., 2006). Statistical comparisons (t test or Mann-Whitney U) of data from naïve and inflamed rats revealed no statistically significant differences between subpopulations. Of note, while we have previously described an inflammation-induced increase in the excitability of cutaneous afferents (Katz and Gold, 2006), excitability was assessed in this previous study with depolarizing stimuli at or around the action potential threshold. Thus, the apparent absence of an inflammation-induced increase in the excitability of neurons studied with a 4 second application of 30 mM K+ is likely due to the nature of the stimulus, and the impact of the stimulus on resting membrane potential.
Figure 6.
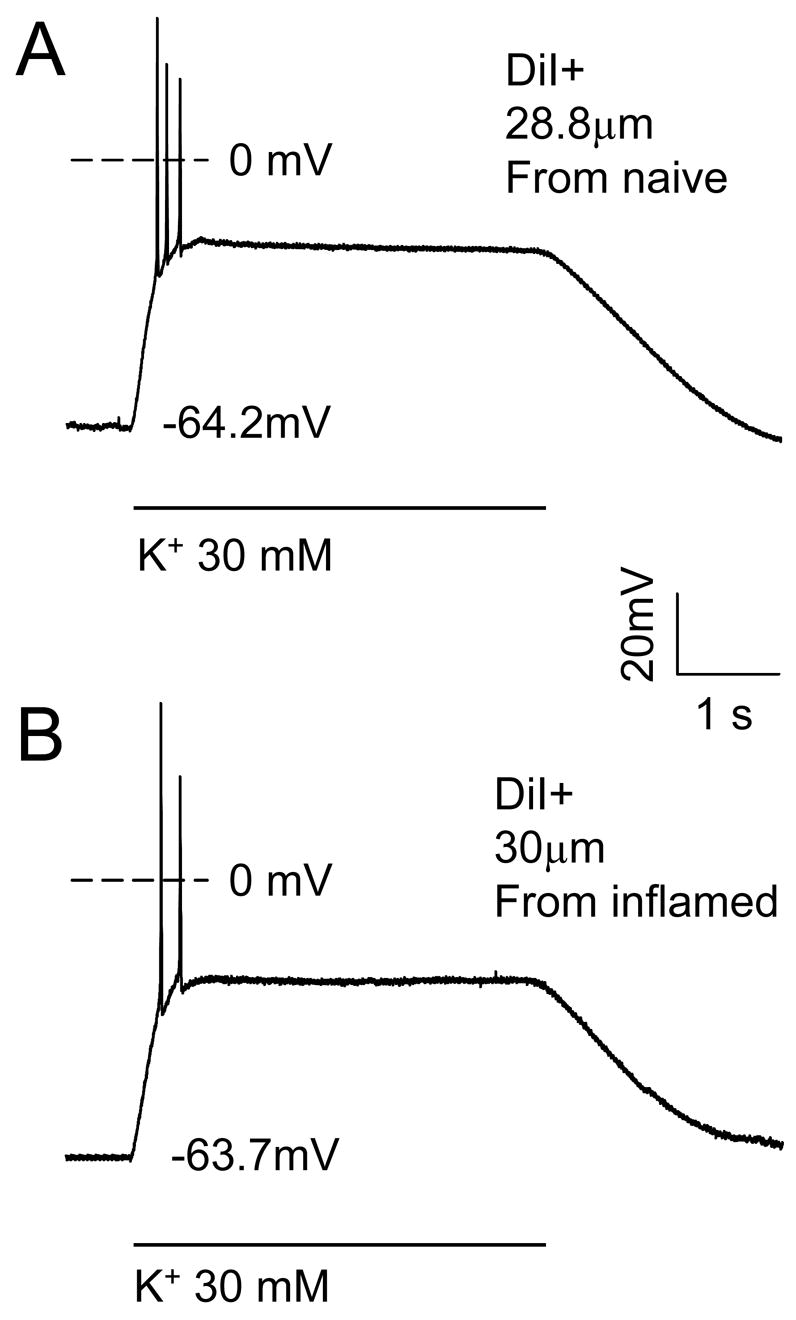
Current clamp recording of the response of typical putative nociceptive DRG neurons from a naïve (top trace (28.8 μm diameter CAP responsive neuron) and inflamed (bottom trace, 30 μm diameter CAP responsive neuron) rats to 30 mM K+ (high K+). Voltage traces show a rapid depolarization that was associated with a brief burst of action potentials. The membrane potential was then stably “clamped” for the remainder of the high K+ application, after which, membrane potential returned to baseline. The resting membrane potential for each neuron is indicated.
Table 2.
Resting and high K+ evoked changes in electrophysiological properties of DRG neurons from naïve and inflamed rats
| Group | Subpop. (n) | Vrest (mV) | Rin (MΩ) | Tstable (ms) | V(K+) (mV) | AP # | Recov (s) |
|---|---|---|---|---|---|---|---|
| Naïve | Small(10) | −61 ± 3.7 | 362 ± 84 | 986 ± 322 | −16.7 ± 1.0 | 4.5 (1,15) | 1.5 ± 0.2 |
| Medium(10) | −60 ± 4.1 | 357 ± 83 | 701 ± 133 | −18.8 ± 4.2 | 1 (0,4) | 1.2 ± 0.1 | |
| Inflamed | Small(9) | −63 ± 3.9 | 599 ± 192 | 689 ± 66 | −16.6 ± 1.8 | 1 (0,7) | 1.3 ± 0.24 |
| Medium(15) | −64 ± 2.8 | 596 ± 90 | 1063 ± 117 | −18.7 ± 1.3 | 1 (0,3) | 1.5 ± 0.11 |
Subpop: subpopulation; n: number of neurons studied; Vrest: resting membrane potential; Rin: Input resistance at rest; Tstable: time to membrane stabilization at new membrane potential in the presence of 30 mM K+, V(K+): Membrane potential in the presence of 30 mM K+; AP#: Median number of action potentials evoked in response to 30 mM K+ application, numbers in parentheses are 25th and 75th percentile (all other data are mean ± SEM); Recov: time of recovery to 50% of resting membrane potential following wash of 30 mM K+; Small: small diameter DiI+ neurons, Medium: Medium diameter DiI+ neurons.
Discussion
The purpose of this study was to determine whether persistent inflammation impacts resting and evoked Ca2+ transients in DRG neurons. In order to identify neurons innervating the site of inflammation, DiI was injected prior to the induction of inflammation. Consistent with our previous observations (Lu et al., 2006), there were marked differences between subpopulations of DRG neurons defined by cell body size, IB4 binding and CAP sensitivity with respect to the magnitude and decay of evoked Ca2+ transients: small diameter, IB4+ and CAP+ neurons had the largest and most slowly decaying evoked Ca2+ transients. Three days after the induction of inflammation, there was a significant increase in the magnitude and decrease in the decay of evoked Ca2+ transients in these subpopulations. Similar increases were also observed in medium diameter and IB4− neurons but were not observed in large diameter and CAP– neurons. These changes were larger in neurons innervating the site of inflammation. Finally, the inflammation-induced changes in Ca2+ handling were not associated with changes in resting membrane potential or an inflammation-induced change in the impact of high K+ on membrane potential.
The present results constitute the first description of persistent inflammation-induced changes in resting and evoked Ca2+ transients. The observation that changes were observed in specific subpopulations of sensory neurons and were larger in neurons innervating the site of inflammation (i.e., DiI+), suggests these changes are not due to a generalized illness response impacting the over-all health of the neurons in vitro. This suggestion is further supported by current clamp results indicating that neurons from inflamed rats had a “normal” resting membrane potential, and input resistance, and did not demonstrate abnormal spontaneous activity. Our current clamp results also argue against the possibility that changes in resting and evoked [Ca2+]i reflect inflammation-induced changes in resting membrane potential or excitability. Furthermore, we have preliminary evidence of an inflammation-induced decrease in the density of voltage-gated Ca2+ currents (Lu and Gold, 2007), suggesting that the increase in the evoked Ca2+ transient is not due to an increase in initial Ca2+ influx. Rather, we suggest that our observations indicate that persistent inflammation results in changes specific Ca2+ regulatory mechanism(s). These would involve a decrease in Ca2+ efflux, uptake and/or binding and/or an increase in Ca2+ release or capacitative Ca2+ entry.
Given the impact of both inflammatory mediators such as nerve growth factor (Woolf, 1996) and neural activity (Fields et al., 2005) on afferent properties, it is likely that these factors underlie the inflammation-induced changes the regulation of [Ca2+]i. Nevertheless, it is important to consider other factors that may have contributed to the observed inflammation-changes in [Ca2+]i regulation. One of these factors is the retrograde tracer used to identify neurons innervating the site of the CFA injection. That is, differences between DiI+ and DiI− neurons may reflect the influence of DiI labeling on the cellular properties of sensory neurons. Several of lines of evidence suggest, however, that the influence of DiI on cellular properties of DRG neurons is likely to be minimal. First, and most importantly, data from the present study indicate that in the absence of inflammation there are no statistically significant differences between DiI+ and DiI− neurons with respect to any of the properties measured (from resting [Ca2+]i to cell body diameter). Second, comparing data sets in the literature between DiI labeled and unlabeled neurons, there appears to be no consistent influence of DiI on the density of voltage-gated Na+ currents (Gold et al., 2003) or K+ currents (Gold et al., 1996b, Harriott et al., 2006). Third, while there do appear to be differences between subpopulations of sensory neurons labeled by DiI with respect to both baseline, and inflammation-induced changes in excitability (i.e., (Gold and Traub, 2004)), these differences appear to reflect properties of the afferents labeled, rather than an influence of the DiI, as similar patterns of excitability changes are observed in unlabeled neurons (Gold et al., 1996a).
A second factor that may have contributed to the observed inflammation-induced change in [Ca2+]i regulation is the impact of inflammation on the properties (cell body diameter, IB4 binding and capsaicin sensitivity) used to define subpopulations of sensory neurons ((Breese et al., 2005, Flake et al., 2005, Xu et al., 2005). Consistent with at least some of these previous results, we observed a statistically significant increase in the proportion of CAP+ neurons. The potential problem here is that if neurons that were unresponsive to capsaicin in the absence of inflammation become responsive to capsaicin, than an inflammation-induced change in the physiology of “capsaicin responsive” neurons may simply reflect the impact of the previously capsaicin unresponsive neurons. For example, if a subpopulation of capsaicin unresponsive neurons has a large response to high K+ in the absence of inflammation and these neurons become capsaicin responsive in the presence of inflammation, they may be the “cause” of an average increase in the response to high K+ in capsaicin responsive neurons in the presence of inflammation. This would represent a change in the neurons that make up the subpopulation of capsaicin responsive neurons and not a change in mechanisms underlying the regulation of [Ca2+]i, as we have suggested. Several lines of evidence support the latter, despite an apparent increase in CAP receptor (TRPV1) expression. These include: 1) resting [Ca2+]i, and evoked Ca2+ transients were smaller in the IB4− and CAP– subpopulations, indicating that the emergence of IB4 binding and CAP sensitivity in these subpopulations, alone, could not have contributed to the observed changes in [Ca2+]i regulation; 2) the largest or most slowly decaying evoked transients in all but one of the IB4− and CAP– neurons from naïve rats were smaller than the mean values for transients in IB4+ and CAP+ neurons from naïve rats, suggesting that there is no subpopulation of IB4− or CAP– neurons, that could have contributed to the observed changes in the evoked Ca2+ transient if this subpopulation became IB4+ or CAP+; and 3), inflammation-induced changes in the regulation of [Ca2+]i were present in DiI− neurons in the absence of detectable changes in cell body diameter, IB4 binding or capsaicin sensitivity.
The basis for the observation that persistent inflammation was associated with a different pattern of changes in resting [Ca2+]i in DiI+ and DiI− neurons is not immediately clear. However, two possible explanations arise from the observation that the average increase in resting [Ca2+]i in DiI− neurons appeared to reflect a relatively large increase in a small subpopulation of DiI−neurons (i.e., see Figure 2B). Given that at least some DiI− neurons were impacted by CFA-induced inflammation, one possibility is that a distinct subpopulation of neurons (i.e., those innervating muscle or hairy skin) not labeled with the DiI injection, has a unique response to inflammation (with an increase in resting Ca2+). A second possibility is that the specific response to inflammation depends on the inflammatory milieu (i.e., relative concentrations of pro- and anti-inflammatory mediators): the inflammatory milieu around a subpopulation of neurons outside the region of the DiI injection may be both different from that inside the region of DiI injection and sufficient to drive an increase in resting Ca2+.
Results from previous studies on the impact of traumatic nerve injury or diabetes on the regulation of [Ca2+]i, in combination with those of the present study on the impact of persistent inflammation suggest that the specific pattern of injury and/or disease-induced changes in the regulation of [Ca2+]i depends on the nature of the injury and/or disease. For example, traumatic nerve injury appears to impact resting [Ca2+]i in specific subpopulations of sensory neurons where there are consistent and relatively large (~60%) decreases in resting [Ca2+]i in injured large diameter CAP insensitive neurons and a smaller (~30%), if detectable decrease in small diameter CAP sensitive neurons (Fuchs et al., 2005). Traumatic nerve injury also results in decrease in both the peak and the duration of evoked Ca2+ transients in injured nociceptive neurons and an increase in peak and decrease in duration of evoked Ca2+ transients in large-diameter (presumably non-nociceptive neurons) (Fuchs et al., 2007). In contrast, data from animal models of diabetic neuropathy suggest resting [Ca2+]i is increased (Kostyuk et al., 1999, Kostyuk et al., 2001, Huang et al., 2002) (but see (Li et al., 2005)) while the rate of recovery from evoked Ca2+ transients is decreased (Kostyuk et al., 1999, Kostyuk et al., 2001, Huang et al., 2002, Li et al., 2005). Thus, traumatic nerve injury results in one pattern of changes in the regulation of [Ca2+]i, inflammation results in another pattern of changes, and diabetes, which has properties in common with both inflammation (i.e., see (Hartge et al., 2007)) and traumatic nerve injury, results in changes common to both.
Inflammation-induced changes in the regulation of [Ca2+]i were manifest in putative nociceptive afferents (i.e., CAP responsive and/or IB4+ neurons with a small cell body diameter). Because a change in [Ca2+]i can influence neuronal properties via changes in ion channel activity (Gold et al., 1996c, Scholz et al., 1998), enzymatic activity (Zhang et al., 2001) and/or gene expression (Eshete and Fields, 2001), the physiological impact of the inflammation-induced changes in resting and evoked increases in [Ca2+]i will depend on where in the neuron they are manifest. For example, an increase in [Ca2+]i close to site of transmitter release should facilitate transmitter release, augmenting neurogenic inflammation in the periphery and the transmission of nociceptive information at the central terminal. On the other hand, an increase in [Ca2+]i close to functional Ca2+ dependent K+ channels should increase K+ channel activity resulting in a decrease in afferent excitability and the transmission of nociceptive information. While a change in the dynamics of Ca2+ signaling in the cell body may result in changes in gene expression that are either pro- or anti-nociceptive (Fields et al., 2005). Thus, the inflammation-induced changes in the regulation of [Ca2+]i may contribute to the hyperalgesia associated with persistent inflammation, or serve as a feedback inhibitory mechanism functioning as a “break” to a host of pro-nociceptive inflammatory processes. Additional experiments will be needed to distinguish between these possibilities.
Acknowledgments
We thank A. Harriott for critical reading of this manuscript. This work was supported by NIH grant NS 44992 (MSG).
List of abbreviations
- AVOVA
Analysis of variance
- CAP
Capsaicin
- CFA
Complete Freund’s Adjuvant
- [Ca2+]i
Concentration of intracellular calcium
- DiI
1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbo-cyanine perchlorate
- DMSO
Dimethyl sulfoxide
- DRG
Dorsal root ganglion
- Hi K+
30 mM potassium
- IB4
Isolectin B4
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amir R, Argoff CE, Bennett GJ, Cummins TR, Durieux ME, Gerner P, Gold MS, Porreca F, Strichartz GR. The role of sodium channels in chronic inflammatory and neuropathic pain. J Pain. 2006;7:S1–29. doi: 10.1016/j.jpain.2006.01.444. [DOI] [PubMed] [Google Scholar]
- Baccei ML, Kocsis JD. Voltage-gated calcium currents in axotomized adult rat cutaneous afferent neurons. J Neurophysiol. 2000;83:2227–2238. doi: 10.1152/jn.2000.83.4.2227. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Unlocking the secrets of cell signaling. Annu Rev Physiol. 2005;67:1–21. doi: 10.1146/annurev.physiol.67.040103.152647. [DOI] [PubMed] [Google Scholar]
- Breese NM, George AC, Pauers LE, Stucky CL. Peripheral inflammation selectively increases TRPV1 function in IB4-positive sensory neurons from adult mouse. Pain. 2005;115:37–49. doi: 10.1016/j.pain.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Dang K, Bielfeldt K, Lamb K, Gebhart GF. Gastric ulcers evoke hyperexcitability and enhance P2X receptor function in rat gastric sensory neurons. J Neurophysiol. 2005;93:3112–3119. doi: 10.1152/jn.01127.2004. [DOI] [PubMed] [Google Scholar]
- Eshete F, Fields RD. Spike frequency decoding and autonomous activation of Ca2+-calmodulin-dependent protein kinase II in dorsal root ganglion neurons. J Neurosci. 2001;21:6694–6705. doi: 10.1523/JNEUROSCI.21-17-06694.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Djouhri L, McMullan S, Berry C, Waxman SG, Okuse K, Lawson SN. Intense isolectin-B4 binding in rat dorsal root ganglion neurons distinguishes C-fiber nociceptors with broad action potentials and high Nav1.9 expression. J Neurosci. 2006;26:7281–7292. doi: 10.1523/JNEUROSCI.1072-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD, Lee PR, Cohen JE. Temporal integration of intracellular Ca2+ signaling networks in regulating gene expression by action potentials. Cell Calcium. 2005;37:433–442. doi: 10.1016/j.ceca.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Flake NM, Bonebreak DB, Gold MS. Estrogen and inflammation increase the excitability of rat temporomandibular joint afferent neurons. J Neurophysiol. 2005;93:1585–1597. doi: 10.1152/jn.00269.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs A, Lirk P, Stucky C, Abram SE, Hogan QH. Painful nerve injury decreases resting cytosolic calcium concentrations in sensory neurons of rats. Anesthesiology. 2005;102:1217–1225. doi: 10.1097/00000542-200506000-00023. [DOI] [PubMed] [Google Scholar]
- Fuchs A, Rigaud M, Hogan QH. Painful Nerve Injury Shortens the Intracellular Ca2+ Signal in Axotomized Sensory Neurons of Rats. Anesthesiology. 2007;107:106–116. doi: 10.1097/01.anes.0000267538.72900.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Dastmalchi S, Levine JD. Co-expression of nociceptor properties in dorsal root ganglion neurons from the adult rat in vitro. Neuroscience. 1996a;71:265–275. doi: 10.1016/0306-4522(95)00433-5. [DOI] [PubMed] [Google Scholar]
- Gold MS, Flake NM. Inflammation-mediated hyperexcitability of sensory neurons. Neurosignals. 2005;14:147–157. doi: 10.1159/000087653. [DOI] [PubMed] [Google Scholar]
- Gold MS, Shuster MJ, Levine JD. Characterization of six voltage-gated K+ currents in adult rat sensory neurons. J Neurophysiol. 1996b;75:2629–2646. doi: 10.1152/jn.1996.75.6.2629. [DOI] [PubMed] [Google Scholar]
- Gold MS, Shuster MJ, Levine JD. Role of a slow Ca2+-dependent slow afterhyperpolarization in prostaglandin E2-induced sensitization of cultured rat sensory neurons. Neurosci Lett. 1996c;205:161–164. doi: 10.1016/0304-3940(96)12401-0. [DOI] [PubMed] [Google Scholar]
- Gold MS, Traub RJ. Cutaneous and colonic rat DRG neurons differ with respect to both baseline and PGE2-induced changes in passive and active electrophysiological properties. J Neurophysiol. 2004;91:2524–2531. doi: 10.1152/jn.00866.2003. [DOI] [PubMed] [Google Scholar]
- Gold MS, Weinreich D, Kim CS, Wang R, Treanor J, Porreca F, Lai J. Redistribution of Na(V)1.8 in uninjured axons enables neuropathic pain. J Neurosci. 2003;23:158–166. doi: 10.1523/JNEUROSCI.23-01-00158.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Harriott AM, Dessem D, Gold MS. Inflammation increases the excitability of masseter muscle afferents. Neuroscience. 2006 doi: 10.1016/j.neuroscience.2006.03.049. [DOI] [PubMed] [Google Scholar]
- Hartge MM, Unger T, Kintscher U. The endothelium and vascular inflammation in diabetes. Diab Vasc Dis Res. 2007;4:84–88. doi: 10.3132/dvdr.2007.025. [DOI] [PubMed] [Google Scholar]
- Hogan QH, McCallum JB, Sarantopoulos C, Aason M, Mynlieff M, Kwok W, Bosnjak ZJ. Painful neuropathy decreases membrane calcium current in mammalian primary afferent neurons. Pain. 2000;86:43–53. doi: 10.1016/s0304-3959(99)00313-9. [DOI] [PubMed] [Google Scholar]
- Holzer P. Capsaicin: cellular targets, mechanisms of action, and selectivity for thin sensory neurons. Pharmacology Reviews. 1991;43:143–201. [PubMed] [Google Scholar]
- Huang TJ, Sayers NM, Fernyhough P, Verkhratsky A. Diabetes-induced alterations in calcium homeostasis in sensory neurones of streptozotocin-diabetic rats are restricted to lumbar ganglia and are prevented by neurotrophin-3. Diabetologia. 2002;45:560–570. doi: 10.1007/s00125-002-0785-x. [DOI] [PubMed] [Google Scholar]
- Katz EJ, Gold MS. Inflammatory hyperalgesia: a role for the C-fiber sensory neuron cell body? J Pain. 2006;7:170–178. doi: 10.1016/j.jpain.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Kostyuk E, Svichar N, Shishkin V, Kostyuk P. Role of mitochondrial dysfunction in calcium signalling alterations in dorsal root ganglion neurons of mice with experimentally-induced diabetes. Neuroscience. 1999;90:535–541. doi: 10.1016/s0306-4522(98)00471-0. [DOI] [PubMed] [Google Scholar]
- Kostyuk E, Voitenko N, Kruglikov I, Shmigol A, Shishkin V, Efimov A, Kostyuk P. Diabetes-induced changes in calcium homeostasis and the effects of calcium channel blockers in rat and mice nociceptive neurons. Diabetologia. 2001;44:1302–1309. doi: 10.1007/s001250100642. [DOI] [PubMed] [Google Scholar]
- Lawson SN. Phenotype and function of somatic primary afferent nociceptive neurones with C-, Adelta- or Aalpha/beta-fibres. Exp Physiol. 2002;87:239–244. doi: 10.1113/eph8702350. [DOI] [PubMed] [Google Scholar]
- Lawson SN, Perry MJ, Prabhakar E, McCarthy PW. Primary sensory neurones: neurofilament, neuropeptides, and conduction velocity. Brain Res Bull. 1993;30:239–243. doi: 10.1016/0361-9230(93)90250-f. [DOI] [PubMed] [Google Scholar]
- Li F, Obrosova IG, Abatan O, Tian D, Larkin D, Stuenkel EL, Stevens MJ. Taurine replacement attenuates hyperalgesia and abnormal calcium signaling in sensory neurons of STZ-D rats. Am J Physiol Endocrinol Metab. 2005;288:E29–E36. doi: 10.1152/ajpendo.00168.2004. [DOI] [PubMed] [Google Scholar]
- Lu S-G, Gold MS. Inflammation induced decrease in voltage-gated calcium currents in subpopulations of rat DRG neurons. Soc Neurosci Abs. 2007 [Google Scholar]
- Lu SG, Zhang X, Gold MS. Intracellular calcium regulation among subpopulations of rat dorsal root ganglion neurons. J Physiol. 2006;577:169–190. doi: 10.1113/jphysiol.2006.116418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum JB, Kwok WM, Mynlieff M, Bosnjak ZJ, Hogan QH. Loss of T-type calcium current in sensory neurons of rats with neuropathic pain. Anesthesiology. 2003;98:209–216. doi: 10.1097/00000542-200301000-00032. [DOI] [PubMed] [Google Scholar]
- Moore BA, Stewart TM, Hill C, Vanner SJ. TNBS ileitis evokes hyperexcitability and changes in ionic membrane properties of nociceptive DRG neurons. Am J Physiol Gastrointest Liver Physiol. 2002;282:G1045–G1051. doi: 10.1152/ajpgi.00406.2001. [DOI] [PubMed] [Google Scholar]
- Richardson JD, Vasko MR. Cellular mechanisms of neurogenic inflammation. J Pharmacol Exp Ther. 2002;302:839–845. doi: 10.1124/jpet.102.032797. [DOI] [PubMed] [Google Scholar]
- Scholz A, Gruss M, Vogel W. Properties and functions of calcium-activated K+ channels in small neurones of rat dorsal root ganglion studied in a thin slice preparation. J Physiol. 1998;513(Pt 1):55–69. doi: 10.1111/j.1469-7793.1998.055by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ. Phenotypic modification of primary sensory neurons: the role of nerve growth factor in the production of persistent pain. Philos Trans R Soc Lond B Biol Sci. 1996;351:441–448. doi: 10.1098/rstb.1996.0040. [DOI] [PubMed] [Google Scholar]
- Xu P, Van Slambrouck C, Berti-Mattera L, Hall AK. Activin induces tactile allodynia and increases calcitonin gene-related peptide after peripheral inflammation. J Neurosci. 2005;25:9227–9235. doi: 10.1523/JNEUROSCI.3051-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Kenyon JL, Nicol GD. Phorbol ester-induced inhibition of potassium currents in rat sensory neurons requires voltage-dependent entry of calcium. J Neurophysiol. 2001;85:362–373. doi: 10.1152/jn.2001.85.1.362. [DOI] [PubMed] [Google Scholar]