

Abstract
HIV-1 Vif inhibits the antiviral activity of APOBEC3G (APO3G) by inducing proteasomal degradation. Here, we studied the effects of Vif on APO3G in vitro. In this system, Vif did not cause APO3G degradation. Instead, Vif induced changes in APO3G that affected immunoprecipitation of the native protein. This effect required wt Vif and was reversed by heat-denaturation of APO3G. Sucrose gradient analysis demonstrated that wt Vif induced the gradual transition of APO3G translated in vitro or expressed in HeLa cells from a low molecular mass conformation to puromycin-sensitive high molecular mass (HMM) complexes. In the absence of Vif or the presence of biologically inactive Vif APO3G failed to form HMM complexes. Our results expose a novel function of Vif that promotes the assembly of APO3G into presumably packaging-incompetent HMM complexes and may explain how Vif can overcome the APO3G-imposed block to HIV replication under conditions of no or inefficient APO3G degradation.
Keywords: Vif, APOBEC3G, high molecular mass complexes, protein degradation
Introduction
HIV-1 and most other lentiviruses encode Vif (Virion infectivity factor), an accessory protein that is required for virus replication in primary CD4+ T cells and monocytes. The host cell factor that inhibits virus replication in the absence of Vif was identified as APOBEC3G (APO3G) (Sheehy et al., 2002). APO3G belongs to a family of cytidine deaminases that in humans include APOBEC1, APOBEC2, seven genes designated APO3A through APO3H, as well as the activation-induced deaminase (AID) gene (for review see (Jarmuz et al., 2002; Wedekind et al., 2003)). In the absence of Vif, APO3G is active against a broad range of retroviruses but can also block hepatitis B virus whose replication cycle involves the reverse transcription of a pregenomic RNA intermediate (Seeger and Mason, 2000). Recently, another member of the APOBEC family, APOBEC3F, was shown to inhibit HIV-1 replication by a mechanism similar to that of APO3G (Wiegand et al., 2004; Zheng et al., 2004). Interestingly, Vif displays species specificity in its ability to interact with APO3G and the results from several independent reports suggest that the determinants of this species specificity map to amino acid 128 of APO3G (Bogerd et al., 2004; Mangeat et al., 2004; Schrofelbauer et al., 2004; Xu et al., 2004). It is now well established that Vif can reduce the cellular expression of APO3G and inhibit its incorporation into virions although the precise mechanism of the latter activity of Vif is still under investigation (Kao et al., 2003; Mariani et al., 2003; Marin et al., 2003; Sheehy et al., 2003; Stopak et al., 2003). The reduction in cellular expression has been attributed to the degradation of APO3G by cytoplasmic proteasomes (Conticello et al., 2003; Liu et al., 2004; Marin et al., 2003; Mehle et al., 2004b; Sheehy et al., 2003; Stopak et al., 2003; Yu et al., 2003). Based on these reports, degradation of APO3G involves binding of Vif to APO3G but also requires the interaction of Vif with components of an Skp1-cullin-F-box (SCF)-like complex that recognizes a conserved sequence motif located near the C-terminus of Vif (Mehle et al., 2004b; Yu et al., 2003). Other studies, however, found that Vif can prevent APO3G encapsidation even in the absence of noticeable degradation suggesting that intracellular degradation of APO3G may not be the sole mechanism by which Vif neutralizes its antiviral activity (Kao et al., 2007; Kao et al., 2003; Kao et al., 2004; Mariani et al., 2003; Mehle et al., 2004a; Opi et al., 2007).
APO3G is a cytoplasmic protein. Immunocytochemistry revealed diffuse cytoplasmic staining as well as accumulation of the protein in punctate cytoplasmic structures described as P bodies or stress bodies (Bogerd et al., 2006; Chen et al., 2006a; Gallois-Montbrun et al., 2007; Kozak et al., 2006; Niewiadomska et al., 2007; Opi et al., 2007; Wichroski et al., 2005; Wichroski et al., 2006). FPLC fractionation of APO3G containing cell lysates further revealed the assembly of APO3G into high molecular mass (HMM) complexes (Chen et al., 2006b; Chiu et al., 2005). Mass spectrometry of immunopurified HMM complexes identified a multi-subunit ribonucleoprotein complex (Chiu et al., 2006; Gallois-Montbrun et al., 2007; Kozak et al., 2006). The formation of high molecular mass complexes is not a unique property of APOBEC3G but was observed for APOBEC3F and APOBEC1 as well (Harris et al., 1993; Wang et al., 2007). Recent data suggest that APO3G antiviral activity is associated with its low molecular mass conformation (Kreisberg et al., 2006; Soros et al., 2007). Thus, the functional significance of HMM APO3G complexes for the control of retroviral replication remains unclear.
In this study, we have analyzed the effects of Vif on APO3G in vitro. We employed rabbit reticulocyte lysates to express Vif and APO3G either alone or in combination and assessed the impact of Vif on APO3G expression and stability. Rabbit reticulocyte lysates contain all the cellular components necessary for protein synthesis and post-translational modifications. In addition, we have previously employed reticulocyte lysates to investigate the ubiquitin-dependent proteasome degradation of CD4 (Chen et al., 1993). We found that Vif did not cause degradation of APO3G in our in vitro system. Instead, we found that binding of wild type (wt) Vif to APO3G interfered with the immunoprecipitation of native APO3G by epitope tag-specific antibodies. This effect was reversed by heat-denaturation of APO3G suggesting an effect of Vif on APO3G conformation. We hypothesized that wt Vif may induce aggregation of APO3G into high molecular mass complexes thereby rendering the protein inaccessible to antibodies for immunoprecipitation. Indeed, sucrose gradient fractionation of APO3G revealed the presence of puromycin-sensitive high molecular mass complexes in samples containing wt Vif. APO3G in the absence of Vif or in the presence of a biologically inactive Vif mutant failed to form similar high molecular mass complexes. Importantly, this effect of Vif on APO3G was not only observed in vitro but was also seen in transfected HeLa cells suggesting that it is a genuine property of Vif. The results from this study expose a novel effect of Vif on APO3G that does not require protein degradation and thus has important implications for our understanding of the role of Vif in the inhibition of APO3G antiviral activity.
Results
In vitro interaction of Vif and APO3G
Co-immunoprecipitation studies performed in tissue culture previously demonstrated physical interactions between Vif and APO3G (Bogerd et al., 2004; Conticello et al., 2003; Mangeat et al., 2004; Mariani et al., 2003; Marin et al., 2003; Stopak et al., 2003; Xu et al., 2004; Yu et al., 2003). To further study functional interactions of Vif and APO3G, we employed an in vitro transcription/translation system where Vif and APO3G genes were separately transcribed using SP6 RNA polymerase and the resulting mRNA was used to direct protein synthesis in rabbit reticulocyte lysates. Our first goal was to verify that Vif and APO3G physically interacted in this in vitro system. To that end, we translated wild type (wt) Vif or a Vif mutant unable to inhibit the antiviral activity of APO3G (VifΔ23–43; (Khan et al., 2001)) as well as APO3G-MycHis in separate translation reactions. A mock reaction without mRNA was prepared in parallel. The two Vif proteins were metabolically labeled with [35S] methionine/cysteine whereas APO3G and the mock sample remained unlabeled. The translation reactions were stopped after 60 min and lysates containing APO3G and either wt Vif or VifΔ23–43 were mixed at 1:1 ratios (20 µl each), supplemented with fresh lysate to a total volume of 150 µl and 50 µl each were incubated at 30°C for 0, 15, or 45 min. Similarly, mock lysates were mixed with lysates containing wt Vif or VifΔ23–43 and processed in the same manner. Samples were either analyzed directly by SDS-PAGE (Fig. 1, Total Protein) or subjected to immunoprecipitation with a polyclonal antibody to the Myc epitope tag present at the C-terminus of APO3G (Fig. 1, IP). As expected, Vif was not precipitated by the Myc-specific antibody in the absence of APO3G (Fig. 1, lanes 1–3 & 7–9). In contrast, wt Vif and VifΔ23–43 co-precipitated with APO3G (Fig. 1, lanes 4–6 & 10–12, respectively). These results confirm that Vif and APO3G are able to interact in vitro and that deletion of residues 23–43 in Vif did not affect its ability to interact with APO3G. Interestingly, pre-incubation of Vif with APO3G containing lysates failed to improve co-immunoprecipitation of wt Vif or VifΔ23–43 suggesting that Vif:APO3G complexes in both cases formed rapidly after mixing the samples.
Fig. 1.
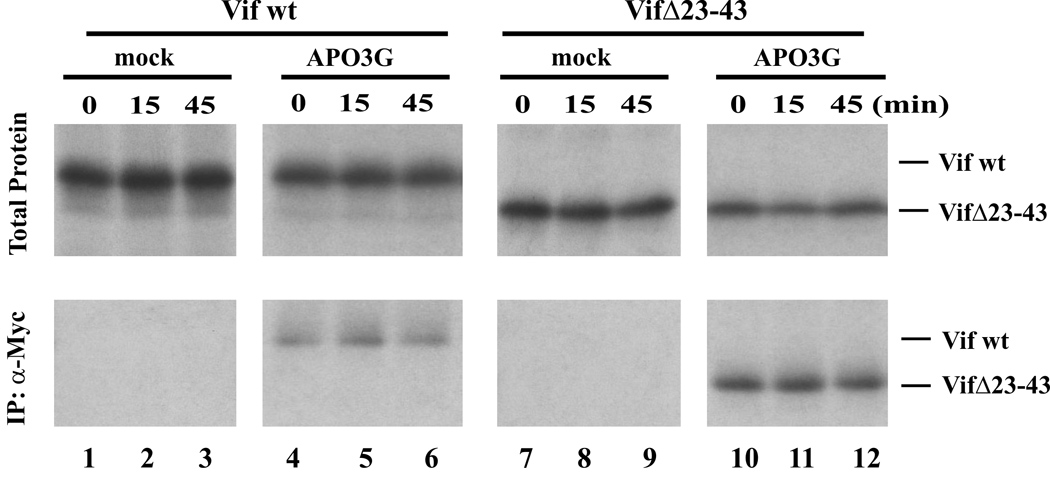
In vitro interaction of Vif and APO3G. In vitro transcription plasmids encoding wt Vif, VifΔ23–43, and human APO3G were linearized at their 3’ ends followed by in vitro transcription and translation in rabbit reticulocyte lysates as described in Materials and Methods. The translation reaction was allowed for 60 min at 30°C and then stopped by addition of RNase A (50 µg/ml). Vif proteins were synthesized in the presence of [35S] methionine/cysteine. APO3G protein and mock samples were not metabolically labeled. Vif-containing lysates were mixed at equal ratios with either mock lysate (mock) or APO3G-containing lysate (APO3G). Samples were divided into equal fractions and incubated for 0, 15, or 45 min at 30°C. An aliquot of each sample (20%) was analyzed directly after TCA precipitation (Total Protein). The remaining samples (80%) were immunoprecipitated with a polyclonal Myc antibody (lower panel). Note that only Vif proteins are visible since APO3G was unlabeled. The positions of wt Vif and VifΔ23–43 in the gel are indicated on the right.
Wt Vif affects immunorecogniton of APO3G
To analyze possible effects of Vif on APO3G stability in vitro, a time-course experiment was performed. To that end, Vif and APO3G were individually translated in reticulocyte lysates. In this case, APO3G protein was labeled with [35S] methionine/cysteine whereas Vif proteins were synthesized in the absence of isotope. The translation reactions were stopped after 60 min, at which time Vif- and APO3G-containing lysates were mixed at 1:1 ratios (20 µl each) and fresh reticulocyte lysate was added to a total volume of 250 µl. The reaction mixture was instantly redistributed into 5 equal aliquots of 50 µl each, which were then incubated for up to 3 hr at 30°C. Individual aliquots were removed at different time points as indicated in figure 2 and stored on dry ice until all samples were collected. Samples (20% of total) from each time point were then either TCA precipitated (Fig. 2A, TCA) or immunoprecipitated (80% of each sample) using a polyclonal antibody against the Myc epitope tag of APO3G (Fig. 2A, IP). APO3G-specific bands were quantified and results from four independent experiments were plotted as percentage of the starting material (Fig. 2B). We found that incubation of wt Vif with APO3G did not have any discernible effects on the stability of APO3G over the three-hour observation period as evidenced by the fact that the amounts of total protein did not decrease with time (Fig. 2B, open circles). Surprisingly, however, there was a gradual decrease in the amount of APO3G immunoprecipitated by the antibody to the Myc epitope tag. Quantitation of the APO3G-specific bands (Fig. 2B, closed circles) showed that the amounts of APO3G recovered by immunoprecipitation rapidly decreased with a "half-life" of about 90 min. The reduction in immunoprecipitated APO3G cannot be explained by Vif-induced degradation of the protein since direct protein analysis (Fig. 2B, open circles) clearly verified the presence of constant amounts of APO3G at all times. This conclusion is further supported by our observation that the presence of 50 µM MG132 during the 3 hr incubation period did not prohibit the gradual loss of immunorecognition of APO3G (Fig. S1). The reduced immunoprecipitation of APO3G can also not be explained by proteolytic removal of the Myc epitope tag since the resulting shift in the mobility of the protein would be apparent in figure 2A. Thus, the most plausible explanation for the progressively reduced immunoprecipitation of APO3G by the Myc-specific antibody is direct or indirect interference by Vif. Of note, the observed effects were epitope tag independent since similar results were obtained with HA-tagged APO3G (data not shown).
Fig. 2.
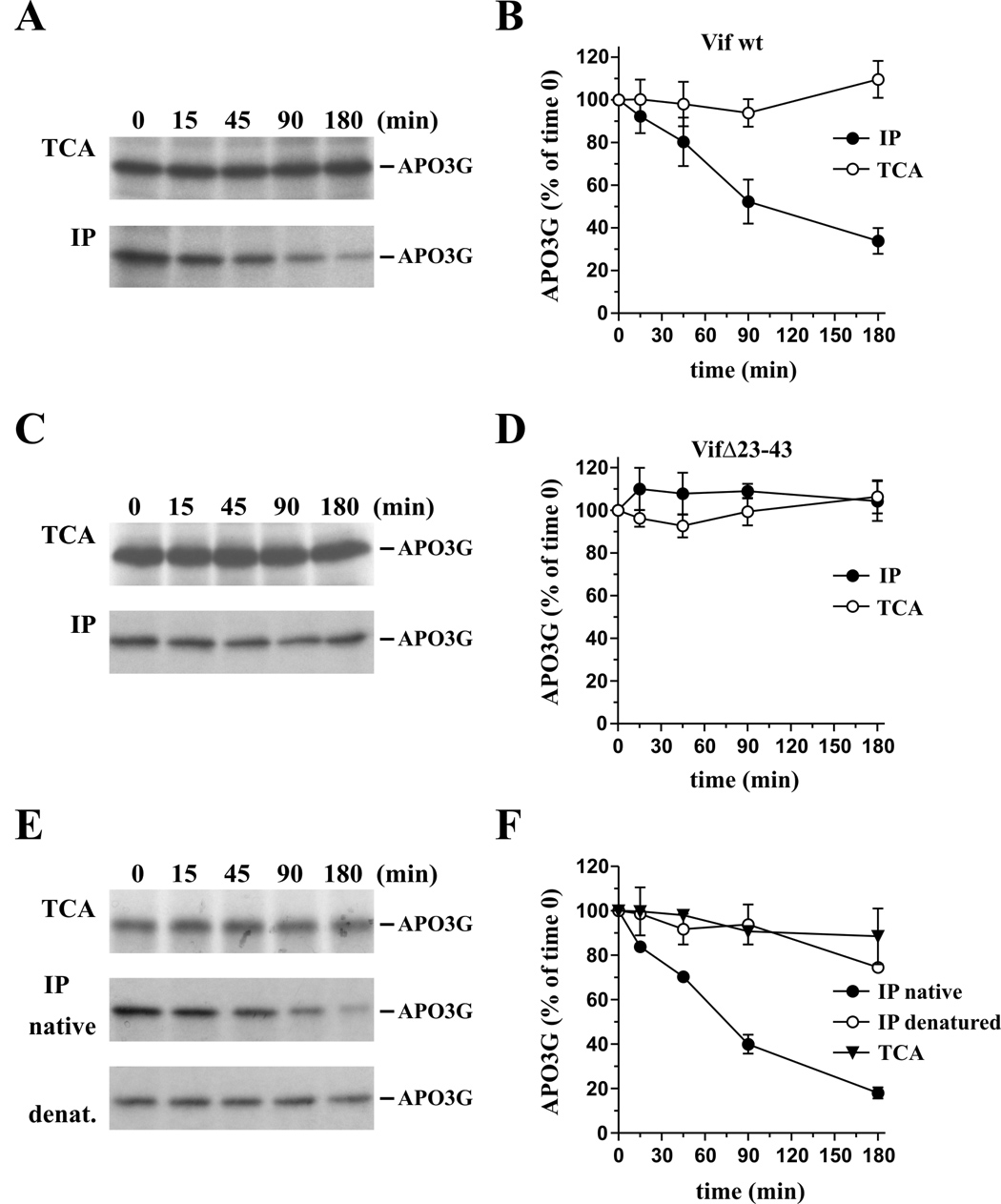
Post-translational effect of Vif on APO3G in vitro. Vif (unlabeled) and APO3G ([35S]-methionine/cysteine-labeled) were translated separately in reticulocyte lysate for 60 min at 30°C. Equal amounts of the lysates were mixed and fresh reticulocyte lysate was added to a total volume of 250 µl. The mixture was immediately divided into 5 samples (50 µl ea.), which were then incubated for 0, 15, 45, 90, or 180 min at 30°C. (A & B) Analysis of wt Vif. (A) For total protein analysis, 20% of the samples from each time point was precipitated with TCA and subjected to SDS-PAGE (top panel). Remaining samples (80%) were immunoprecipitated using a polyclonal Myc antibody prior to SDS-PAGE (lower panel). (B) The APO3G-specific bands from panels A were quantitated using a Fuji BAS 2000 Bio-Image Analyzer and the results were plotted as a function of time. Error bars reflect standard error from four independent experiments. (C & D) Analysis of VifΔ23–43. The experiment is identical to panels A & B except that VifΔ23–43 was employed instead of wt Vif. Error bars reflect standard error from four independent experiments. (E & F) Heat denaturation rescues immunoprecipitation of APO3G exposed to wt Vif. (E) The experiment was done essentially as described for panels A & B. For total protein analysis, 20% of the samples from each time point was precipitated with TCA and subjected to SDS-PAGE (top panel). The remaining samples (80%) were divided into 2 equal parts. One half was immunoprecipitated using a polyclonal Myc antibody as in panel A. The second half was first denatured by adding an equal volume of 2x sample buffer and heating the sample at 95°C for 5 min. Prior to immunoprecipitation, the sample volume was increased to 1 ml using phosphate-buffered saline to reduce detergent and 2-mercaptoethanol levels in the sample. Immunoprecipitation was then carried out as before at 4°C using a polyclonal Myc antibody (lower panel). (F) The APO3G-specific bands from panel E were quantitated as in panel B. Error bars reflect standard error from two independent experiments
A biologically inactive Vif mutant does not interfere with immunorecognition of APO3G
In figure 1 we demonstrated the ability of Vif to interact with APO3G in vitro. This raised the possibility that the reduced immunoprecipitation of APO3G in figure 2A was the result of steric interference by Vif. To address this issue, we performed a kinetic study of APO3G in the presence of the VifΔ23–43 variant, which is biologically inactive (Khan et al., 2001) but still capable of binding to APO3G in vitro (Fig. 1). The experimental procedure and quantitation of the results were the same as described for figure 2A/B. Consistent with the results from figure 2A, direct analysis of APO3G protein did not reveal any effects of VifΔ23–43 on APO3G stability (Fig. 2C, TCA). Interestingly, constant amounts of APO3G were recovered at all times by immunoprecipitation (Fig. 2C, IP) and quantitation of the data confirmed that VifΔ23–43 did not affect immunorecognition of APO3G (Fig. 2D). The different effects of wt Vif and VifΔ23–43 on the immunoprecipitation of APO3G are difficult to explain by steric interference of Vif with Myc-antibody binding to its target epitope since both wt Vif and VifΔ23–43 interacted with APO3G (Fig. 1). Our finding that VifΔ23–43 had no effect on APO3G immunoprecipitation also suggests that the increasing resistance of APO3G to immunoprecipitation in figure 2A was not the result of normal post-translational protein refolding but was caused by the addition of wt Vif to the sample.
As an additional control for the specificity of the effect of HIV-1 Vif on human APO3G, we analyzed the impact of HIV-1 Vif on African Green monkey APO3G (Agm-APO3G), which is resistant to HIV-1 Vif (Bogerd et al., 2004; Mangeat et al., 2004; Mariani et al., 2003; Schrofelbauer et al., 2004). The experiment was performed essentially as described for figure 2A/B employing wild type HIV-1 Vif and Agm-APO3G and the results are shown in figure S2. As expected, Vif had no effect on the stability of Agm-APO3G as judged by direct protein analysis (Fig. S2B, open circles). Importantly, wild type HIV-1 Vif also had no effect on the immunoprecipitation of Agm-APO3G (Fig. S2B, closed circles). Thus, the effect of wild type HIV-1 Vif on the conformation of native human APO3G observed in figure 2 is highly specific and dependent on the direct interaction of wild type HIV-1 Vif with human APO3G.
Heat-denaturation restores immunorecognition of APO3G
To test if the inaccessibility of APO3G to immunoprecipitation in figure 2A was due to a Vif-induced change in APO3G conformation, we subjected APO3G that had previously been exposed to wt Vif to heat-denaturation prior to immunoprecipitation. The experiment was essentially performed as described for figure 2A except that prior to immunoprecipitation by the Myc-specific polyclonal antibody, half of the APO3G sample was heated for 5 min at 95°C as detailed in the legend to figure 2. The results of the experiment are shown in figure 2E and the quantitative analysis of two independent experiments is shown in figure 2F. As expected, direct analysis of TCA precipitated protein confirmed the presence of constant amounts of APO3G throughout the 3 hr observation period (Fig. 2F, closed triangles). Also, immunoprecipitation of non-denatured protein revealed the same gradual loss of APO3G as observed in figure 2A (Fig. 2F, closed circles). Importantly, however, heat-denaturation of the protein samples prior to immunoprecipitation led to the recovery of similar amounts of APO3G by immunoprecipitation at all time points (Fig. 2F, open circles). These results support our model that interaction of wt Vif with APO3G induces a gradual yet reversible conformational change that affects APO3G recognition by epitope tag-specific antibodies.
Analysis of Vif-APO3G complexes
Tissue culture analyses have identified APO3G in two principal conformations: a low molecular mass (LMM) conformation of cytoplasmic APO3G and a high molecular mass (HMM) conformation, in which APO3G is associated with a variety of other host factors in a large RNA:protein complex in P-bodies or stress granules (Chiu et al., 2005; Gallois-Montbrun et al., 2007; Kozak et al., 2006; Wichroski et al., 2006). Newly synthesized APO3G is initially in the LMM conformation but assembles into HMM multi-protein complexes within approximately 30 minutes after synthesis (Soros et al., 2007). While this change of APO3G from LMM to HMM conformation is a natural process that does not require Vif, it is conceivable that the presence of Vif affects the kinetics of APO3G transition from LMM to HMM conformation.
We next studied the possible effects of Vif on APO3G conformation in our in vitro system. In this experiment, APO3G was metabolically labeled with [35S] methionine/cysteine whereas wt Vif and VifΔ23–43 were translated in the absence of isotope as described for figure 2. In vitro synthesized [35S]-labeled APO3G was incubated for 3 hr at 30°C with mock-translated lysate (Fig. 3 A, top panel), lysate containing wt Vif (Fig. 3 A, middle panel), or lysate containing VifΔ23–43 (Fig. 3A, lower panel). Samples were then layered onto a 15–40% sucrose gradient and centrifuged at 86,000 × g for 3 hr as reported (Kozak et al., 2006). Fractions were collected from the top of the gradient and subjected to 12.5% SDS PAGE. Fraction #1 was omitted from the analysis because it contained residual unincorporated isotope. APO3G proteins were visualized by fluorography. In the absence of Vif (Fig. 3A, top panel) or in the presence of VifΔ23–43 (Fig. 3A, bottom panel), APO3G was confined to the top of the gradient (fractions 2–6). In contrast, the presence of wt Vif resulted in a broader distribution of APO3G across the sucrose gradient (Fig. 3A, middle panel) and, in addition, small amounts of APO3G were recovered from the bottom of the gradient (fraction 19). Analysis of Vif in an experiment identical to the one shown in figure 3A except that Vif was metabolically labeled and APO3G remained unlabeled showed that Vif was largely confined to the upper fractions (Fig. 3B, fractions 2–8). We conclude that the presence of wt Vif promotes the assembly of APO3G into HMM complexes in vitro. The formation of in vitro APO3G HMM complexes was detergent-sensitive and complexes could be dissociated by addition of 0.5% SDS (data not shown).
Fig. 3.
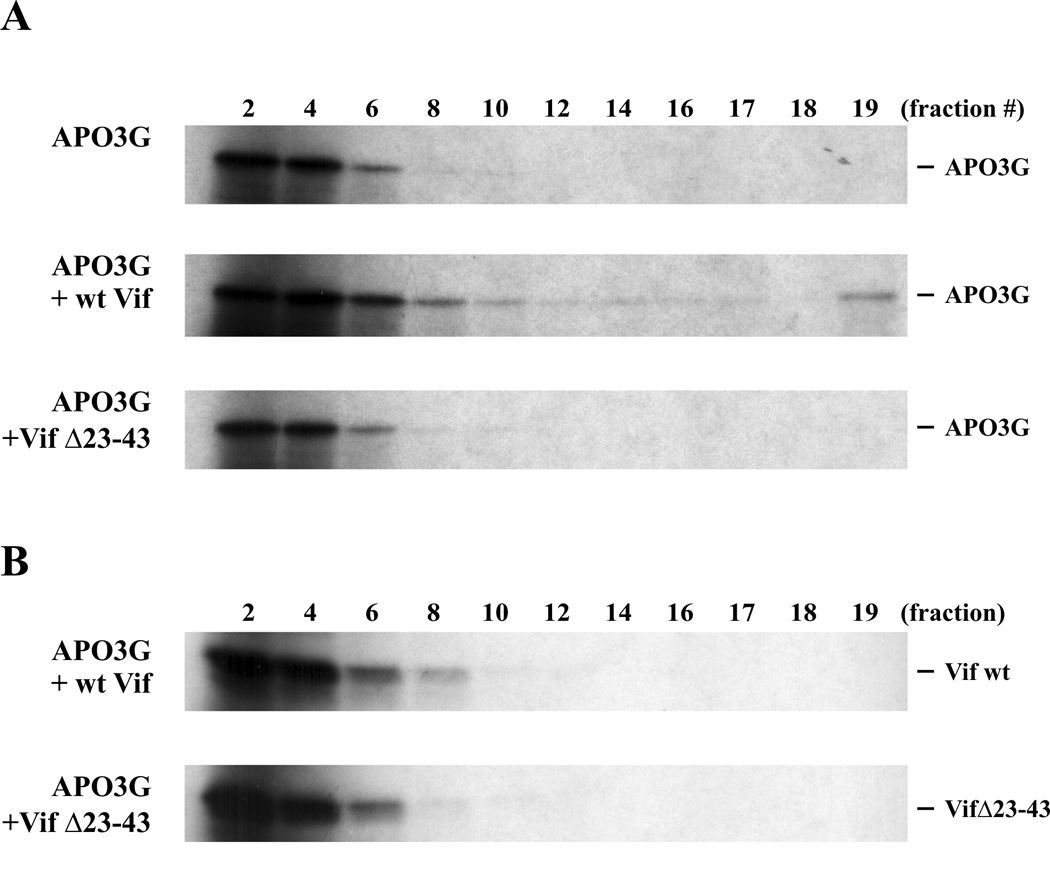
Sucrose gradient analysis of in vitro translated APO3G. (A) In vitro synthesized [35S]-labeled APO3G (20 µl) was incubated for 3 h at 30°C with an equal volume of mock-translated lysate (top panel) or with an equal volume of lysate containing unlabeled wt Vif (middle panel) or VifΔ23–43 (lower panel). After incubation, the samples were layered onto 10 ml gradients of 15–40% sucrose in buffer B (0.01 M Tris-Cl, pH 7.4, 0.25 M KCl, 0.01 M MgCl2 (Kozak et al., 2006)) and centrifuged for 3 h at 86,000 × g. Nineteen fractions (530 µl ea.) were collected from the top of the gradient and combined with 175 µl of 4x sample buffer. Aliquots were separated by SDS-12.5% PAGE and APO3G protein was visualized by fluorography. (B) The experiment was repeated as in panel A except that Vif was now metabolically labeled and APO3G remained unlabeled. The mock-translated control was omitted here since no labeled protein was present in the mock lysate. Sucrose gradient analysis and gel electrophoresis were performed as in panel A.
Vif induces APO3G transition complexes
We next studied the kinetics of Vif-induced formation of HMM APO3G complexes in vitro. The experiment was performed essentially as described for figure 3A except that Vif and APO3G were incubated for various times (30 min, 90 min, 180 min) prior to sucrose gradient centrifugation (Fig. 4). At all times, significant amounts of APO3G were found in the upper fractions of the gradient (Fig. 4, all panels, fractions 2–4). Interestingly, an intermediate APO3G complex was detectable at the 30 minute and 90 minute time points (Fig. 4, fraction 14; top and middle panels) but was absent from the 180 minute sample. At the same time, APO3G in fraction 19 was undetectable in the 30 minute sample (Fig. 4, top panel, fraction 19) but was identified in increasing amounts in the later samples (Fig. 4, middle and lower panel, fraction 19). We conclude that conversion of APO3G from LMM to HMM conformation in vitro in the presence of Vif is a slow and gradual process that involves the formation of a distinct transition complex.
Fig. 4.
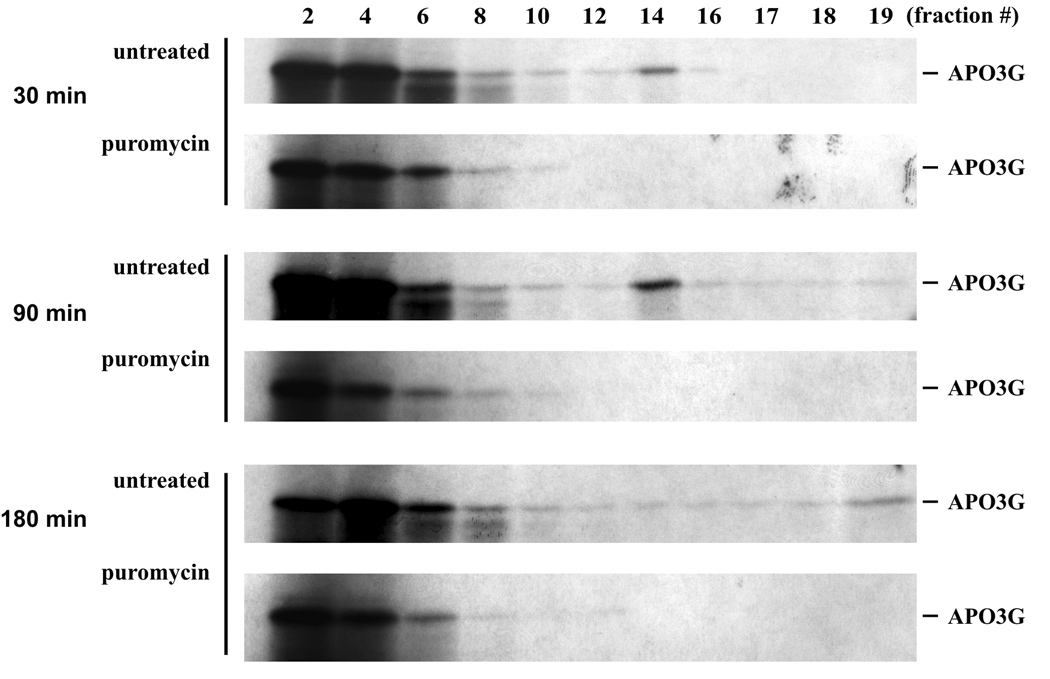
Kinetic analysis of APO3G high molecular mass complexes. In vitro synthesized [35S]-labeled APO3G (20 µl) was incubated for 30, 90, or 180 minutes at 30°C with an equal volume of lysate containing unlabeled wt Vif. Samples were either incubated alone (untreated) or in the presence of puromycin (0.5 µg/ml; puromycin). After incubation, the samples were layered onto 10 ml gradients of 15–40% sucrose and processed as in figure 3A.
Recently, Kozak et al. found that APO3G complexes from transfected 293T cells were sensitive to treatment with puromycin, a drug known to induce the release of nascent proteins from polysomes; their results strongly suggested that APO3G in untreated samples was bound to translationally active polysomes (Kozak et al., 2006). Interestingly, when puromycin was added to our in vitro reactions, the Vif-induced assembly of APO3G into intermediate (fraction 14) and heavy complexes (fraction 19) was completely inhibited as well (Fig. 4, puromycin). Thus, it appears that Vif promotes the association of APO3G with polysomes in vitro.
In Figure 3B we demonstrated that Vif was not part of the heavy APO3G complexes even though the formation of these complexes was Vif-dependent. To see if Vif was part of the APO3G transition complexes identified in figure 4 we metabolically labeled both Vif and APO3G and incubated the combined lysates for 30 or 90 minutes as described for figure 4. Samples were then subjected to sucrose gradient analysis as in figure 4. For comparison we analyzed APO3G in combination with either wt Vif (Fig. 5A) or VifΔ23–43 (Fig. 5B). In this experiment both APO3G and Vif are visible (Fig. 5). As expected, APO3G formed a distinct transition complex in the presence of wt Vif (Fig. 5A, APO3G) following 30 or 90 min incubation. Interestingly, wt Vif was found in fractions of the gradient up to and including fraction 14 (Fig. 5A, wt Vif). Consistent with our previous results, no transition complexes were observed in the presence of VifΔ23–43 and both VifΔ23–43 and APO3G were restricted to fractions 1–10 of the sucrose gradient (Fig. 5B). These results suggest that Vif may be part of APO3G transition complexes but subsequently dissociates as APO3G continues to form increasingly heavy complexes pelleting to the bottom of our gradients.
Fig. 5.
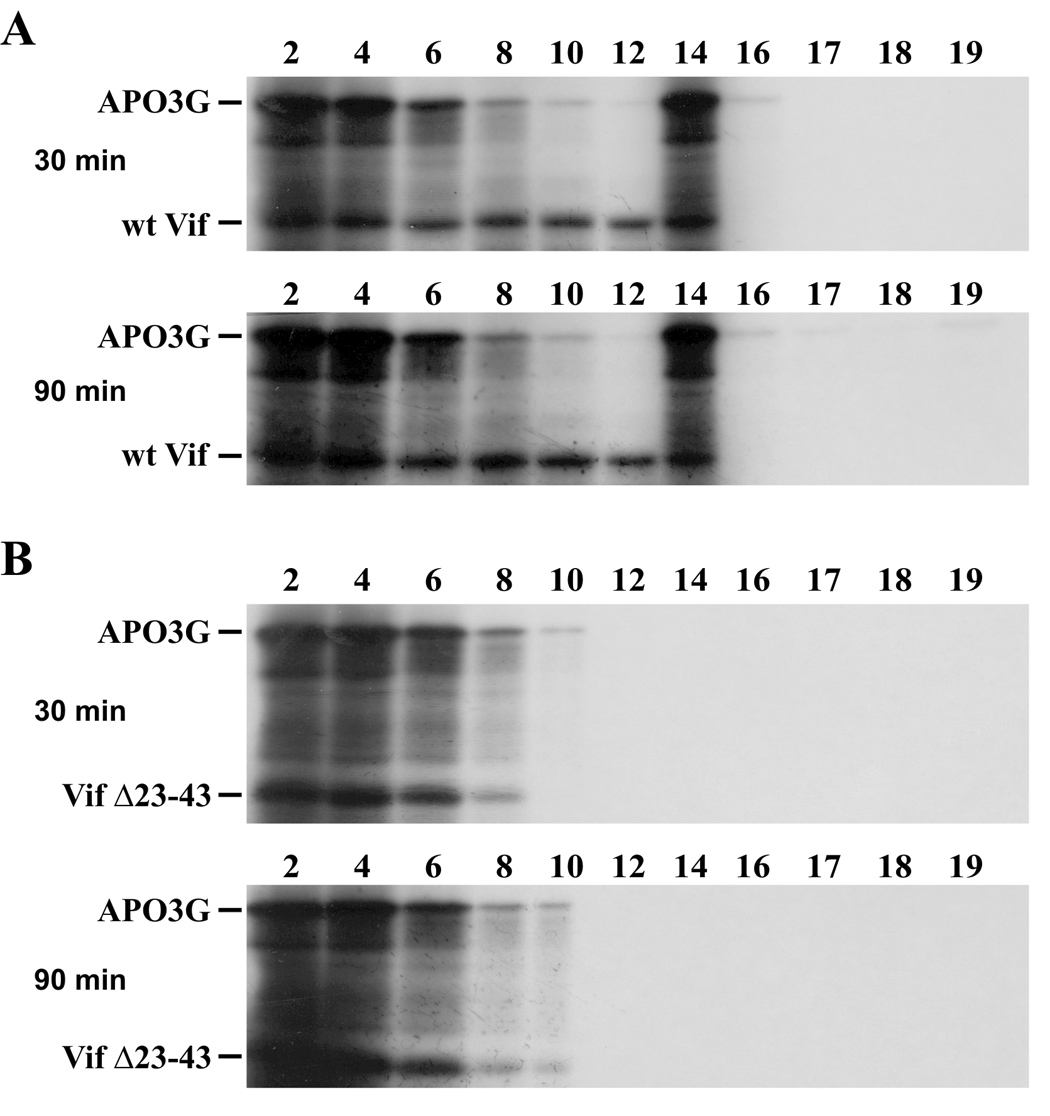
Association of Vif with APO3G complexes. In vitro synthesized [35S]-labeled APO3G (20 µl) was incubated for 30, 90, or 180 minutes at 30°C with an equal volume of lysate containing [35S]-labeled wt Vif (A) or VifΔ23–43 (B). After incubation, the samples were layered onto 10 ml gradients of 15–40% sucrose and processed as in figure 3A.
Effect of Vif on HMM association of APO3G in transfected HeLa cells
We recently identified an APO3G variant, APO3G-C97A, which was resistant to Vif-induced degradation but was still sensitive to Vif with respect to virus incorporation and antiviral activity (Opi et al., 2007). We also found that the ability of Vif to exclude APO3G from virions could be dissociated from its ability to cause proteasomal degradation of APO3G (Kao et al., 2007). From these observations we concluded that Vif had the ability to regulate APO3G encapsidation through degradation-independent mechanism(s). Furthermore, Soros et al. recently provided evidence that virus encapsidated APO3G occurs primarily from the LMM pool of de novo synthesized protein (Soros et al., 2007). Thus, the ability of Vif to promote the transition of APO3G from an LMM to a HMM conformation could be a way to prevent encapsidation of APO3G into HIV-1 virions in the absence of protein degradation.
To study the effect of Vif on HMM association of APO3G in tissue culture, HeLa cells were transfected with pcDNA-APO3G together with either pNL-A1 for the expression of Vif (Fig. 6A) or pNL-A1vif(−) as a vif-negative control (Fig. 6B). To prevent Vif-induced degradation of APO3G, a dominant negative Cul5 mutant, Cul5-Rbx (Yu et al., 2003), was cotransfected in this experiment. Cells were harvested 24 hr later and lysed as described in Materials and Methods. Post-nuclear extracts were overlaid onto 15–40% sucrose gradients and centrifuged for 3 hr at 86,000 × g. Twenty fractions (500 µl each) were collected from the top of the gradients and subjected to immunoblot analysis using an APO3G-specific peptide antibody (Fig. 6A/B, α-APO3G). The same blots were subsequently re-probed with a Vif-specific monoclonal antibody (α-Vif). In general, APO3G from transfected HeLa cells was more broadly distributed in the gradient than in vitro translated APO3G. This suggests that in tissue culture systems, APO3G has a greater propensity to assembly into HMM complexes irrespective of the presence or absence of Vif. Interestingly, the co-expression of Vif resulted in the appearance of a heavy APO3G complex sedimenting to the bottom of the sucrose gradient thus closely mimicking our in vitro results (Fig. 6A, fraction #20). This complex was not seen in the absence of Vif (Fig. 6B). Also, consistent with our in vitro analyses, Vif was absent from the heavy APO3G complexes (Fig. 6A, α-Vif). Our results suggest that the ability of Vif to promote the transition of APO3G from light to heavy complexes is not limited to the in vitro system but can be observed in tissue culture systems as well and is therefore likely to represent a functionally relevant property of Vif.
Fig. 6.
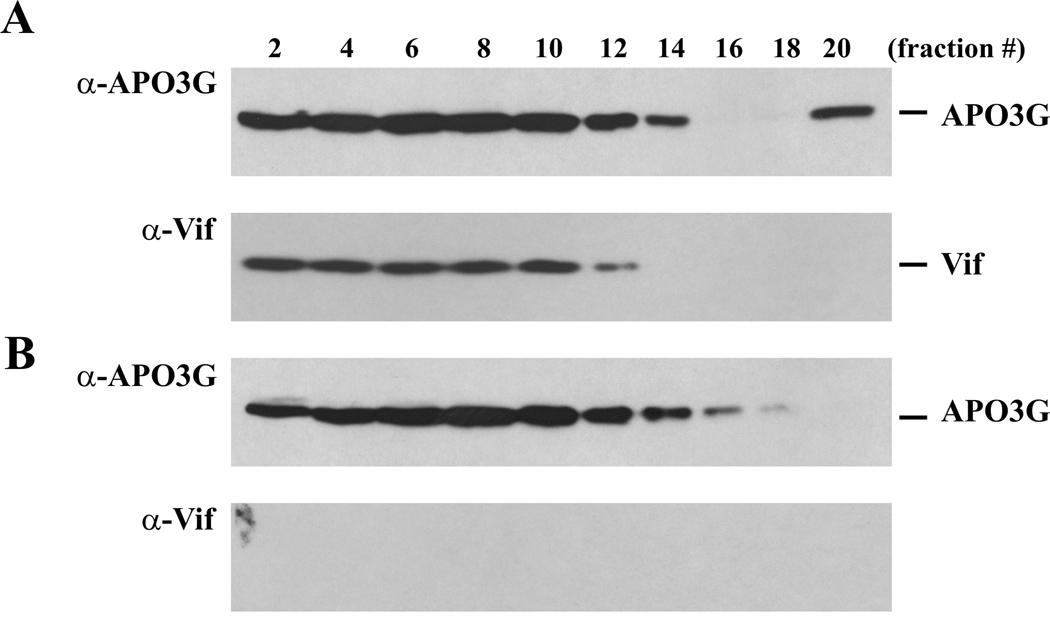
Vif promotes HMM complex formation of APO3G in transfected HeLa cells. HeLa cells were transfected with pcDNA-APO3G (3 µg), together 1 µg of pCul5-Rbx DNA and 2 µg of either pNL-A1 DNA (A) or pNL-A1vif(−) DNA (B). Cells were harvested 24 h after transfection and lysed in 400 µl of NP40 lysis buffer (5 min, 4°C). Nuclei were pelleted (1,500 × g, 5 min), the post-nuclear supernatants were overlaid onto a 15–40% sucrose gradient as described for figure 3, and centrifuged for 3 h at 86,000 × g. Twenty fractions (500 µl each) were collected from the top of the gradient and indicated fractions were analyzed by immunoblotting using an APO3G-specific antibody (α-APO3G). The same blots were then re-probed with a Vif-specific monoclonal antibody (α-Vif).
Discussion
HIV-1 Vif is essential for replication in vivo and in nonpermissive cell types in tissue culture. Several recent studies have reported that Vif can induce the degradation of APO3G when expressed in non-permissive target cells. This effect of Vif is manifested by a reduction of the half-life of APO3G in the presence of Vif when compared to vif-deficient controls (Conticello et al., 2003; Liu et al., 2004; Marin et al., 2003; Mehle et al., 2004b; Sheehy et al., 2003; Stopak et al., 2003; Yu et al., 2003). Furthermore, the decrease in APO3G protein levels was abrogated in the presence of proteasome inhibitors, indicating that Vif targeted APO3G for degradation by proteasomes (Conticello et al., 2003; Liu et al., 2004; Marin et al., 2003; Mehle et al., 2004b; Sheehy et al., 2003; Stopak et al., 2003; Yu et al., 2003). In support of this hypothesis, co-expression of Vif and APO3G was shown to result in poly-ubiquitination of APO3G, which is known to precede the proteasome-dependent degradation of proteins (for review see (Ciechanover et al., 2000)). Indeed, Yu et al demonstrated that Vif interacts with components of an ubiquitin ligase complex through a conserved motif near the C-terminus of Vif providing additional evidence that Vif regulates the degradation of APO3G (Yu et al., 2003). Thus, there is overwhelming evidence that Vif can induce the degradation of APO3G in tissue culture systems.
On the other hand, we and others have found that inhibition of APO3G encapsidation by Vif is not always accompanied by a proportional reduction in cellular APO3G (Kao et al., 2007; Kao et al., 2003; Kao et al., 2004; Mariani et al., 2003) and, more importantly, we have recently identified an APO3G variant that was resistant to Vif-induced degradation but was nonetheless excluded from HIV-1 virions in a Vif-dependent manner (Opi et al., 2007). Thus, there is increasing evidence that Vif can prevent encapsidation of APO3G not only through targeting the protein to a proteasomal degradation pathway but through additional as yet undefined mechanism(s).
Our in vitro study was initiated by the desire to further study the mechanism of Vif-induced degradation of APO3G. We were encouraged in choosing an in vitro system by our previous success in demonstrating proteasome-mediated degradation of CD4 in the presence of Vpu in rabbit reticulocyte lysates (Chen et al., 1993). A significant advantage of the in vitro system was that proteins could be analyzed directly without prior immunoprecipitation since de novo synthesis of metabolically labeled proteins was strictly limited to exogenously supplied mRNA. Our initial immunoprecipitation analyses indeed revealed a gradual decrease of APO3G with time and produced kinetic profiles suggestive of protein degradation (Fig. 2B). However, direct analysis of TCA-precipitated, in vitro-translated proteins revealed that the reduced APO3G signals in the immunoprecipitates were not due to protein degradation but rather reflected an effect of Vif on APO3G that induced a gradual resistance to immunoprecipitation. In fact, APO3G protein was perfectly stable over the 3-hour observation period irrespective of the presence of wt or mutant Vif protein. It is not clear why Vif was unable to induce APO3G degradation in vitro. However, our previous in vitro studies on Vpu-induced degradation of CD4 employed lysates that had been supplemented with canine microsomal membranes (Chen et al., 1993). It is possible that these membrane preparations contributed factors crucial to proteasomal protein degradation. However, this was not further explored in the current study.
Instead, we used the fact that Vif did not cause degradation of APO3G in vitro as an opportunity to study additional degradation-independent effects of Vif on APO3G. Indeed, our in vitro study provided evidence that Vif may promote the sequestration of APO3G into packaging-incompetent HMM complexes. This conclusion was supported by our finding that the presence of Vif induced changes in APO3G that rendered the protein progressively less available to immunoprecipitation. This trend could be reversed by heat denaturation of APO3G prior to immunoprecipitation suggesting that wt Vif induced heat-labile changes in APO3G. Sucrose gradient analyses subsequently showed that the presence of wt Vif was associated with the transition of APO3G into high molecular mass complexes (Fig. 3–Fig. 5). These effects were specific for wt Vif and were not observed with a biologically inactive Vif mutant (Fig. 3A, bottom panel). From the data shown in figure 3A it appears that only a small fraction of the total APO3G assembled into heavy complexes while in figure 2 the majority of APO3G appears to become resistant to immunoprecipitation. This does not rule out a functional correlation between the two effects since it is possible that subjecting the samples to sucrose gradient centrifugation causes partial dissociation of the complexes. This is suggested by our finding that a significant amount of APO3G from transfected HeLa cells is identified in the upper (low molecular mass) fractions in the sucrose gradient shown in figure 6, while FPLC analyses identified the majority of APO3G from transfected HeLa cells in HMM complexes (Opi et al., 2007). Thus, we believe that the gradual transition from a low molecular mass to a high molecular mass conformation of APO3G most likely accounts for its increasing resistance to immunoprecipitation as observed in figure 2.
How Vif might promote the transition of APO3G from LMM to HMM conformations remains to be investigated. Both wt Vif and VifΔ23–43 interacted with APO3G in our in vitro analyses. In fact, VifΔ23–43 appeared to interact slightly more efficiently with APO3G than wt Vif (Fig. 1). These results are consistent with co-immunoprecipitation studies in HeLa cells that revealed solid binding of VifΔ23–43 to APO3G (data not shown). Interestingly, the interaction of Vif and APO3G was not improved by preincubating the proteins at 30°C. This suggested that binding of Vif to APO3G was a rapid process. The rapid binding of Vif to APO3G contrasts the slow and gradual increasing resistance of APO3G to immunoprecipitation as shown in figure 2. These kinetic differences argue against steric interference by Vif with antibody binding. Instead they suggest that the resistance of APO3G to immunoprecipitation is due to the conversion of APO3G from a low molecular mass to a high molecular mass conformation. Why only wt Vif can induce these changes is unclear. Both Vif and APO3G are nucleic acid binding proteins and APO3G HMM complexes were previously found to be sensitive to RNase-treatment (Chiu et al., 2005; Gallois-Montbrun et al., 2007; Kozak et al., 2006; Opi et al., 2007; Wang et al., 2007). Of note, rabbit reticulocyte lysates used in this study had been treated with micrococcal nuclease and our in vitro translation reactions were routinely stopped by addition of RNase A. Nevertheless, it is possible that APO3G or Vif mRNAs used for the in vitro translations were protected from complete RNase digestion and it is possible that the in vitro HMM APO3G complexes represent RNA-protein complexes similar to those observed in vivo. In support of this we found that the APO3G complexes dissociated in the presence of puromycin suggesting that they involved noncovalent interactions of APO3G and polysomes. We therefore propose a model, in which APO3G forms a complex with polysomes in reticulocyte lysates similar to those reported in transfected 293T cells (Kozak et al., 2006; Niewiadomska et al., 2007). We speculate that these Vif induced APO3G-polysome complexes are resistant to immunoprecipitation presumably because the C-terminal epitope tag on APO3G is inaccessible to antibodies in these complexes. Importantly, we were able to demonstrate Vif-induced heavy complexes of APO3G in HeLa cells (Fig. 6) and we observed gradual resistance of APO3G in transfected HeLa cells to immunoprecipitation by conformation-sensitive antibodies (data not shown). We therefore conclude that the effects of Vif on APO3G conformation described in this study reflect a process occurring in vitro as well as in vivo.
Taken together our results provide evidence for a novel function of Vif that is likely to contribute to the exclusion of APO3G from viral particles by sequestering the protein in packaging-incompetent high molecular mass intracellular complexes. Such a function of Vif could be important early in infection when APO3G has not yet been eliminated from virus-producing cells by proteasome-mediated protein degradation.
Materials and Methods
Plasmids
The in vitro transcription vector pSP65 (Promega Biosciences, San Luis Obispo CA) was used for expression of vif and APO3G genes under the control of the SP6 promoter. For the expression of wt Vif, HIV-1 vif was PCR amplified from pNL4-3 and cloned as an EcoRI/BamHI fragment into pSP65 (Chen et al., 1993) resulting in vector pSP-Vif. For the expression of VifΔ23–43, the mutant vif gene was PCR amplified from pNL-Al/VifΔ23–43 (Khan et al., 2001) and cloned between the EcoRI/BamHI sites of pSP65 resulting in pSP-VifΔ23–43. For expression of human APO3G, pSP-hAPO3G was constructed by subcloning APO3G including its C-terminal Myc-His epitope tag from pcDNA-APO3G (Kao et al., 2003) into the pSP65 vector via its EcoRI and XbaI cloning sites. The following plasmids were used for expression in HeLa cells: pcDNA-APO3G for the expression of untagged human wt APO3G; pNL-A1 for the expression of Vif (Strebel et al., 1987), and pNL-A1vif(−) as a vif-negative control (Karczewski and Strebel, 1996). Plasmid pCul5-HA was a gift of Xiao-Fang Yu (Yu et al., 2003). An Rbx binding mutant, pCul5-Rbx-HA, was constructed by deleting residues 566–582 in pCul5-HA as described (Yu et al., 2003). The vector pSP-AgmAPO3G was used for expression of Agm-APO3G. This vector was constructed by subcloning Agm-APO3G including its C-terminal HA tag from pc-AGM-APO3G-HA (gift of Ned Landau) into pSP65.
Antisera
Immunoprecipitation of APO3G was carried out using a polyclonal antibody to the C-terminal Myc epitope tag (Sigma-Aldrich, St. Louis MO). For immunoprecipitation of Agm-APO3G a rat monoclonal antibody to its C-terminal HA tag was used (Roche Diagnostics, Indianapolis IN). A monoclonal antibody to Vif (MAb #319) was used for immunoblot analysis of Vif and was obtained from Michael Malim through the NIH AIDS Research and Reference Reagent Program (Simon et al., 1995). APO3G was identified using a rabbit polyclonal peptide antibody (ApoC17 (Opi et al., 2007)).
In vitro transcription and translation
For the in vitro synthesis of run-off transcripts, all plasmids were initially linearized at the 3’ end of the coding sequences using appropriate restriction enzymes. Plasmids pSP-Vif and pSP-VifΔ23–43 were linearized with BamHI whereas pSP-APO3G was linearized using XbaI. Subsequent in vitro transcription of the linearized DNA was performed using an in vitro transcription kit (SP6 Riboprobe system; Promega Biosciences) according to the manufacturer's instructions. RNA resulting from the transcription of 1 µg of plasmid DNA was suspended in 50 µl of RNase-free water. In vitro transcribed RNA was used to direct the in vitro translation of proteins in rabbit reticulocyte lysates. Typically, 5 µl of RNA was mixed with 40 µl of rabbit reticulocyte lysate (Promega Biosciences) in the presence or absence of 10 µl of [35S] methionine/cysteine (Redivue Pro Mix (10 mCi/ml); Amersham Biosciences Piscataway NJ) and incubated for 60 min at 30°C. Translation reactions were terminated by addition of RNase A (50 µg/ml) to the samples. Proteins were then either subjected to immunoprecipitation or directly analyzed by TCA precipitation and SDS-PAGE.
TCA precipitation and immunoprecipitation
For direct analysis of in vitro translated proteins, proteins were precipitated with trichloroacetic acid (TCA) prior to gel electrophoresis to remove unincorporated isotope. For that purpose, 50 µl of cold 25% TCA was added to 10 µl of translation mix while vortexing. The sample was then incubated on ice for 10 min followed by a brief spin in an Eppendorf microcentrifuge (10 sec, 14,000 × g) to pellet protein aggregates. The supernatant containing unincorporated isotope was discarded and the pellet was washed twice with cold 2% TCA. The pellet was finally suspended in 100 µl of sample buffer (2% SDS, 1% β-mercaptoethanol, 1% glycerol, and 65 mM Tris-hydrochloride (pH 6.8)) and neutralized by addition of 1 N NaOH. The samples were boiled for 5 min and loaded onto 12.5% polyacrylamide-SDS gels.
Immunoprecipitation of in vitro translated proteins was carried out using appropriate antibodies as described in the text. Briefly, polyclonal antibodies to the Myc epitope or to Vif (Vif93) were adsorbed on protein A sepharose beads (Sigma-Aldrich) whereas rat anti-HA antibodies were adsorbed on Protein G sepharose beads (Amersham) for 1 hr at 4°C on a rotating wheel The beads were then washed once with cold Triton-Wash Buffer (50 mM Tris pH 7.4,300 mM NaCl, 0.1% Triton X-100). Antibody-adsorbed beads were then suspended in 1 ml of cold 0.1% BSA/PBS, 40 µl of translation reaction mix was added and beads were incubated for 1 additional hour at 4°C on a rotating wheel. After incubation, the beads were washed 3 times with 750 µl of Triton-Wash Buffer. Bound proteins were solubilized by boiling the beads in 100 µl of sample buffer and were then separated on 12.5% polyacrylamide-SDS gels. Gels were soaked in 1 M sodium salicylic acid for 30 min and dried. Radioactive bands were visualized by fluorography. Quantitation of the radioactivity of the respective bands was performed with a Fuji BAS 2000 Bio-Image Analyzer.
Immunoblotting
For immunoblot analysis of sucrose gradient fractions, 75 µl of each fraction were combined with 25 µl of 4x sample buffer (8% sodium dodecyl sulfate, 250 mM Tris-HCl, pH 6.8, 20% 2-mercaptoethanol, 20% glycerol). Proteins were heated for 10 to 15 min at 95°C with occasional vortexing of the samples prior to SDS-PAGE; proteins were transferred to PVDF membranes and reacted with appropriate antibodies as described in the text. Membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences, Piscataway NJ) and visualized by enhanced chemiluminescence (ECL, Amersham Biosciences).
Sucrose gradient fractionation
For the analysis of in vitro synthesized APO3G, lysates were incubated for different times with lysates containing either wt Vif or the Vif Δ23–43 mutant. After incubation, the samples were layered over 10 ml of 15–40% sucrose gradients made in buffer B (0.01 M Tris-Cl, pH 7.4, 0.25 M KCl,0.01 M MgCl2) and centrifuged at 86,000 × g for 3 h (Kozak et al., 2006). For the analysis of APO3G complexes in HeLa cells, transfected cells were lysed on ice in 400 µl of NP40 lysis buffer (50 mM Tris-HCL pH 7.4, 1% NP40, 0.1% deoxycholate (DOC), 150 mM NaCl, and protease inhibitor cocktail). Nuclei were removed by centrifugation for 5 min at 1,500 × g. Post-nuclear extracts were overlaid onto a 15–40% sucrose gradient and centrifuged for 3 hr at 86,000 × g (Kozak et al., 2006). Nineteen to twenty fractions were collected from the top of the gradient and the samples were combined with 175 µl 4x sample buffer and heated for 5 min at 95°C. Aliquots of each fraction were loaded on 12.5% SDS-PAGE gels. For the analysis of [35S]-labeled in vitro translated proteins, gels were soaked in 1 M sodium salicylic acid for 30 min and dried. Radioactive bands were visualized by fluorography. Quantitation of the radioactivity of the respective bands was performed with a Fuji BAS 2000 Bio-Image Analyzer. Proteins from HeLa extracts were analyzed by immunoblotting.
Supplementary Material
Fig. S1: MG132 does not increase the stability of APO3G in vitro. Wt Vif, VifΔ23–43, and APO3G were separately translated using reticulocyte lysates for 60 min at 30°C. Only APO3G was metabolically labeled. All samples were then pretreated for 20 min at 30°C with MG132 (50 µM). After that, lysates containing APO3G were mixed at equal ratios with lysates containing either wild type Vif (Vif wt) (A & B) or Vif Δ23–43 (C & D). Fresh lysate, supplemented with MG132 (50 µM), was added as described for figure 2A. The samples were then divided into five equal aliquots and incubated for up to 180 min at 30°C. (A & C) After incubation the samples were either analyzed directly following TCA precipitation (top panels) or immunoprecipitated using a polyclonal Myc antibody (bottom panels) as described for figure 2A. (B & D) APO3G-specific bands from panels A & C were quantified and results were plotted as a function of time. Open circles represent TCA-precipitated samples; closed circles represent immunoprecipitated samples.
Fig. S2: Post-translational effect of HIV-1 Vif on Agm-APO3G expression in vitro. HIV-1 Vif and Agm-APO3G were translated separately in reticulocyte lysates for 60 min at 30°C. Equal amounts of the proteins were mixed and fresh reticulocyte lysate was added as described for figure 2A. The samples were incubated for up to 180 min at 30°C. (A) For total protein analysis, 20% of the samples from each time point was precipitated with TCA and subjected to SDS-PAGE. Remaining samples (80%) were immunoprecipitated using a monoclonal antibody to the HA epitope tag of Agm-APO3G prior to SDS-PAGE. (B) Agm-APO3G-specific bands from panel A were quantitated as described for figure 2B.
Acknowledgments
We thank Alicia Buckler-White and Ron Plishka for oligonucleotide synthesis and sequence analysis. We thank Xiao-Fang Yu for pCul5-HA and Ned Landau for pc-AGM-APO3G-HA. A Vif monoclonal antibody (MAb #319) was obtained from Michael Malim through the NIH AIDS Research and Reference Reagent Program. This work was supported by a Grant from the NIH Intramural AIDS Targeted Antiviral Program to K.S. and by the Intramural Research Program of the NIH, NIAID.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bogerd HP, Doehle BP, Wiegand HL, Cullen BR. A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV type 1 virion infectivity factor. Proc Natl Acad Sci U S A. 2004;101:3770–3774. doi: 10.1073/pnas.0307713101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O'Shea KS, Moran JV, Cullen BR. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc Natl Acad Sci U S A. 2006;103:8780–8785. doi: 10.1073/pnas.0603313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, Landau NR, Weitzman MD. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr Biol. 2006a;16:480–485. doi: 10.1016/j.cub.2006.01.031. [DOI] [PubMed] [Google Scholar]
- Chen K, Huang J, Zhang C, Huang S, Nunnari G, Wang FX, Tong X, Gao L, Nikisher K, Zhang H. Alpha interferon potently enhances the anti-human immunodeficiency virus type 1 activity of APOBEC3G in resting primary CD4 T cells. J Virol. 2006b;80:7645–7657. doi: 10.1128/JVI.00206-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MY, Maldarelli F, Karczewski MK, Willey RL, Strebel K. Human immunodeficiency virus type 1 Vpu protein induces degradation of CD4 in vitro: the cytoplasmic domain of CD4 contributes to Vpu sensitivity. J Virol. 1993;67:3877–3884. doi: 10.1128/jvi.67.7.3877-3884.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu YL, Soros VB, Kreisberg JF, Stopak K, Yonemoto W, Greene WC. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature. 2005;435:108–114. doi: 10.1038/nature03493. [DOI] [PubMed] [Google Scholar]
- Chiu YL, Witkowska HE, Hall SC, Santiago M, Soros VB, Esnault C, Heidmann T, Greene WC. High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc Natl Acad Sci U S A. 2006;103:15588–15593. doi: 10.1073/pnas.0604524103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Orian A, Schwartz AL. The ubiquitin-mediated proteolytic pathway: mode of action and clinical implications. J Cell Biochem Suppl. 2000;34:40–51. doi: 10.1002/(sici)1097-4644(2000)77:34+<40::aid-jcb9>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Conticello SG, Harris RS, Neuberger MS. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol. 2003;13:2009–2013. doi: 10.1016/j.cub.2003.10.034. [DOI] [PubMed] [Google Scholar]
- Gallois-Montbrun S, Kramer B, Swanson CM, Byers H, Lynham S, Ward M, Malim MH. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J Virol. 2007;81:2165–2178. doi: 10.1128/JVI.02287-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SG, Sabio I, Mayer E, Steinberg MF, Backus JW, Sparks JD, Sparks CE, Smith HC. Extract-specific heterogeneity in high-order complexes containing apolipoprotein B mRNA editing activity and RNA-binding proteins. J Biol Chem. 1993;268:7382–7392. [PubMed] [Google Scholar]
- Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, Navaratnam N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics. 2002;79:285–296. doi: 10.1006/geno.2002.6718. [DOI] [PubMed] [Google Scholar]
- Kao S, Goila-Gaur R, Miyagi E, Khan MA, Opi S, Takeuchi H, Strebel K. Production of infectious virus and degradation of APOBEC3G are separable functional properties of human immunodeficiency virus type 1 Vif. Virology. 2007 doi: 10.1016/j.virol.2007.08.005. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao S, Khan MA, Miyagi E, Plishka R, Buckler-White A, Strebel K. The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J Virol. 2003;77:11398–11407. doi: 10.1128/JVI.77.21.11398-11407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao S, Miyagi E, Khan MA, Takeuchi H, Opi S, Goila-Gaur R, Strebel K. Production of infectious human immunodeficiency virus type 1 does not require depletion of APOBEC3G from virus-producing cells. Retrovirology. 2004;1:27. doi: 10.1186/1742-4690-1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski MK, Strebel K. Cytoskeleton association and virion incorporation of the human immunodeficiency virus type 1 Vif protein. J Virol. 1996;70:494–507. doi: 10.1128/jvi.70.1.494-507.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Aberham C, Kao S, Akari H, Gorelick R, Bour S, Strebel K. Human immunodeficiency virus type 1 Vif protein is packaged into the nucleoprotein complex through an interaction with viral genomic RNA. J Virol. 2001;75:7252–7265. doi: 10.1128/JVI.75.16.7252-7265.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak SL, Marin M, Rose KM, Bystrom C, Kabat D. The anti-HIV-1 editing enzyme APOBEC3G binds HIV-1 RNA and messenger RNAs that shuttle between polysomes and stress granules. J Biol Chem. 2006;281:29105–29119. doi: 10.1074/jbc.M601901200. [DOI] [PubMed] [Google Scholar]
- Kreisberg JF, Yonemoto W, Greene WC. Endogenous factors enhance HIV infection of tissue naive CD4 T cells by stimulating high molecular mass APOBEC3G complex formation. J Exp Med. 2006;203:865–870. doi: 10.1084/jem.20051856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Yu X, Luo K, Yu Y, Yu XF. Influence of primate lentiviral Vif and proteasome inhibitors on human immunodeficiency virus type 1 virion packaging of APOBEC3G. J Virol. 2004;78:2072–2081. doi: 10.1128/JVI.78.4.2072-2081.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeat B, Turelli P, Liao S, Trono D. A single amino acid determinant governs the species-specific sensitivity of APOBEC3G to Vif action. J Biol Chem. 2004;279:14481–14483. doi: 10.1074/jbc.C400060200. [DOI] [PubMed] [Google Scholar]
- Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003;114:21–31. doi: 10.1016/s0092-8674(03)00515-4. [DOI] [PubMed] [Google Scholar]
- Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med. 2003;9:1398–1403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- Mehle A, Goncalves J, Santa-Marta M, McPike M, Gabuzda D. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 2004a;18:2861–2866. doi: 10.1101/gad.1249904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem. 2004b;279:7792–7798. doi: 10.1074/jbc.M313093200. [DOI] [PubMed] [Google Scholar]
- Niewiadomska AM, Tian C, Tan L, Wang T, Sarkis PT, Yu XF. Differential inhibition of long interspersed element 1 by APOBEC3 does not correlate with high-molecular-mass-complex formation or P-body association. J Virol. 2007;81:9577–9583. doi: 10.1128/JVI.02800-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opi S, Kao S, Goila-Gaur R, Khan MA, Miyagi E, Takeuchi H, Strebel K. Human immunodeficiency virus type 1 Vif inhibits packaging and antiviral activity of a degradation-resistant APOBEC3G variant. J Virol. 2007;81:8236–8246. doi: 10.1128/JVI.02694-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrofelbauer B, Chen D, Landau NR. A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif) Proc Natl Acad Sci U S A. 2004;101:3927–3932. doi: 10.1073/pnas.0307132101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev. 2000;64:51–68. doi: 10.1128/mmbr.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med. 2003;9:1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- Simon JH, Southerling TE, Peterson JC, Meyer BE, Malim MH. Complementation of vif-defective human immunodeficiency virus type 1 by primate, but not nonprimate, lentivirus vif genes. J Virol. 1995;69:4166–4172. doi: 10.1128/jvi.69.7.4166-4172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soros VB, Yonemoto W, Greene WC. Newly synthesized APOBEC3G is incorporated into HIV virions, inhibited by HIV RNA, and subsequently activated by RNase H. PLoS Pathog. 2007;3:e15. doi: 10.1371/journal.ppat.0030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell. 2003;12:591–601. doi: 10.1016/s1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- Strebel K, Daugherty D, Clouse K, Cohen D, Folks T, Martin MA. The HIV 'A' (sor) gene product is essential for virus infectivity. Nature. 1987;328:728–730. doi: 10.1038/328728a0. [DOI] [PubMed] [Google Scholar]
- Wang X, Dolan PT, Dang Y, Zheng YH. Biochemical differentiation of APOBEC3F and APOBEC3G proteins associated with HIV-1 life cycle. J Biol Chem. 2007;282:1585–1594. doi: 10.1074/jbc.M610150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedekind JE, Dance GS, Sowden MP, Smith HC. Messenger RNA editing in mammals: new members of the APOBEC family seeking roles in the family business. Trends Genet. 2003;19:207–216. doi: 10.1016/S0168-9525(03)00054-4. [DOI] [PubMed] [Google Scholar]
- Wichroski MJ, Ichiyama K, Rana TM. Analysis of HIV-1 viral infectivity factor-mediated proteasome-dependent depletion of APOBEC3G: correlating function and subcellular localization. J Biol Chem. 2005;280:8387–8396. doi: 10.1074/jbc.M408048200. [DOI] [PubMed] [Google Scholar]
- Wichroski MJ, Robb GB, Rana TM. Human Retroviral Host Restriction Factors APOBEC3G and APOBEC3F Localize to mRNA Processing Bodies. PLoS Pathog. 2006;2:e41. doi: 10.1371/journal.ppat.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 2004;23:2451–2458. doi: 10.1038/sj.emboj.7600246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Svarovskaia ES, Barr R, Zhang Y, Khan MA, Strebel K, Pathak VK. A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc Natl Acad Sci U S A. 2004;101:5652–5657. doi: 10.1073/pnas.0400830101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- Zheng YH, Irwin D, Kurosu T, Tokunaga K, Sata T, Peterlin BM. Human APOBEC3F Is Another Host Factor That Blocks Human Immunodeficiency Virus Type 1 Replication. J Virol. 2004;78:6073–6076. doi: 10.1128/JVI.78.11.6073-6076.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1: MG132 does not increase the stability of APO3G in vitro. Wt Vif, VifΔ23–43, and APO3G were separately translated using reticulocyte lysates for 60 min at 30°C. Only APO3G was metabolically labeled. All samples were then pretreated for 20 min at 30°C with MG132 (50 µM). After that, lysates containing APO3G were mixed at equal ratios with lysates containing either wild type Vif (Vif wt) (A & B) or Vif Δ23–43 (C & D). Fresh lysate, supplemented with MG132 (50 µM), was added as described for figure 2A. The samples were then divided into five equal aliquots and incubated for up to 180 min at 30°C. (A & C) After incubation the samples were either analyzed directly following TCA precipitation (top panels) or immunoprecipitated using a polyclonal Myc antibody (bottom panels) as described for figure 2A. (B & D) APO3G-specific bands from panels A & C were quantified and results were plotted as a function of time. Open circles represent TCA-precipitated samples; closed circles represent immunoprecipitated samples.
Fig. S2: Post-translational effect of HIV-1 Vif on Agm-APO3G expression in vitro. HIV-1 Vif and Agm-APO3G were translated separately in reticulocyte lysates for 60 min at 30°C. Equal amounts of the proteins were mixed and fresh reticulocyte lysate was added as described for figure 2A. The samples were incubated for up to 180 min at 30°C. (A) For total protein analysis, 20% of the samples from each time point was precipitated with TCA and subjected to SDS-PAGE. Remaining samples (80%) were immunoprecipitated using a monoclonal antibody to the HA epitope tag of Agm-APO3G prior to SDS-PAGE. (B) Agm-APO3G-specific bands from panel A were quantitated as described for figure 2B.