

Abstract
The deposition of fibrillar structures (amyloids) is characteristic of pathological conditions including Alzheimer's and Parkinson's diseases. The detection of protein deposits and the evaluation of their kinetics of aggregation are generally based on fluorescent probes such as thioflavin T and Congo red. In a search for improved fluorescence tools for studying amyloid formation, we explored the ability of N-arylaminonaphthalene sulfonate (NAS) derivatives to act as noncovalent probes of α-synuclein (AS) fibrillation, a process linked to Parkinson's disease and other neurodegenerative disorders. The compounds bound to fibrillar AS with micromolar Kds, and exhibited fluorescence enhancement, hyperchromism, and high anisotropy. We conclude that the probes experience a hydrophobic environment and/or restricted motion in a polar region. Time- and spectrally resolved emission intensity and anisotropy provided further information regarding structural features of the protein and the dynamics of solvent relaxation. The steady-state and time-resolved parameters changed during the course of aggregation. Compared with thioflavin T, NAS derivatives constitute more sensitive and versatile probes for AS aggregation, and in the case of bis-NAS detect oligomeric as well as fibrillar species. They can function in convenient, continuous assays, thereby providing useful tools for studying the mechanisms of amyloid formation and for high-throughput screening of factors inhibiting and/or reversing protein aggregation in neurodegenerative diseases.
INTRODUCTION
The aggregation of proteins and peptides is a fundamental feature of several disorders collectively designated as conformational or misfolding in origin. These conditions are often neurodegenerative, such as in Alzheimer's and Parkinson's diseases and spongiform encephalopathies, as well as nonneuropathic, as in type II diabetes and different forms of cancer. A ubiquitous cytopathological finding is the presence of intracellular or extracellular highly organized fibrillar aggregates (1). Although the polypeptide chains involved in these diseases lack sequence homology or strong similarities in amino acid composition and size, the resultant amyloid fibrils are remarkably uniform with respect to both external morphology and internal structure. The mature fibrils are rodlike structures with a diameter of 7–13 nm and formed from interwound protofilaments. In the core structure of stacked β-sheets, the strands are perpendicular to the longitudinal axis. This canonical cross-β-structure is also adopted by nondisease-associated proteins upon induction of aggregation in vitro (1). The kinetics of amyloid formation is consistent with a nucleation-propagation mechanism originating from as yet poorly defined nucleation centers, and progressing via oligomerization to an elongation phase leading to the formation of the characteristic fibrils (2).
The detection of amyloid deposits in affected brain tissue, as well as the evaluation of fibrillation kinetics in vitro, is generally based on the use of fluorescent probes such as thioflavin T (ThioT) and Congo red (3). These compounds interact with the cross-β-amyloid fibrils, exhibiting significant changes in fluorescent and absorption signals. The exact mechanism of binding is not fully understood (4–7). Other compounds, some structurally related to Congo red and ThioT, have been evaluated and employed as histochemical stains for amyloid (8–12). However, the in vitro assays based on these probes demonstrate significant drawbacks. For example, the ThioT signal can be suppressed in densely packed aggregates that exclude the dye, whereas false positives can arise from interaction with amorphous aggregates or bacteria (3). Uncharged high-affinity ThioT derivatives, although optimal for amyloid detection in positron emission tomography, do not possess useful fluorescent properties (11,12). The ThioT assay is poorly reproducible, more qualitative than quantitative, and does not always report the presence of amyloid fibrils (5). Furthermore, it requires periodic sampling and is thus not amenable to continuous and/or highly parallelized determinations. Congo red is less specific, binding to proteins with different types of secondary structure (13). Finally, the structural features of prefibrillar structures arising during the early oligomerization steps of aggregation are generally not revealed by these indicators, although it has been shown recently that certain analogs of Congo red and ThioT are able to detect soluble oligomers of the amyloid β-peptide (Aβ) in vitro and in vivo (12). This last feature is of central importance because of the cytotoxicity attributed to such oligomeric intermediates (1).
The deficiencies outlined above have led to major efforts directed at the design of new dyes and spectroscopic techniques for detecting amyloid structures as well as for elucidating the dynamics of protein association in general. Fluorescence spectroscopy is particularly useful in such studies due to its inherent sensitivity and versatility. Numerous approaches based on steady-state and time-resolved fluorescence determinations of extrinsic probes (14–16), residue-specific fluorescence (17), resonance energy transfer (18), correlation spectroscopy (19), the dual-color SIFT assay (20), and anisotropy measurements (21–25) have been employed to characterize the conformational ensemble of amyloid proteins.
Some of the studies mentioned above have involved protein engineering for introduction of site-specific residues or peptide segments for labeling with specific reagents. In parallel studies, we employed pyrene (see (60)), and other organic dyes (C. W. Bertoncini, E. A. Jares-Erijman, and T. M. Jovin, unpublished) for recording structural features of monomeric, oligomeric, and fibrillar species of α-synuclein in vitro and in cells. However, extrinsic probes are more generally applicable in that they do not require costly protein design and validation and are particularly suitable for screening assays. Furthermore, they potentially permit a ready comparison of wild-type proteins and disease-related familial or other mutants, including samples in situ or directly isolated from biological specimens.
N-arylaminonaphthalene sulfonate (NAS) derivatives are widely used as fluorescent probes providing information about the molecular microenvironment and conformation of proteins, mainly due to their interaction with partially folded intermediates and hydrophobic patches. They exhibit a very low quantum yield in water, but become highly fluorescent in nonpolar media or when placed in a milieu that limits the mobility of the surrounding solvent dipoles. These properties make them sensitive indicators of processes that modify the exposure of the probes to water, including protein self-assembly (26,27). Several naphthalene sulfonate derivatives have been employed in studies of protein oligomerization (26,28,29), the mechanism(s) and kinetics of amyloid formation (14,24,30–37), and the modulation of oligomerization pathways (38,39).
In this work, we explored the ability of some NAS derivatives to act as noncovalent markers for aggregated forms of the presynaptic protein α-synuclein (AS). AS has been linked with the pathogenesis of a number of neurodegenerative disorders, including Parkinson's disease, dementia with Lewy bodies, multiple system atrophy, and amyotrophic lateral disease. AS is the major component of neuronal and glial cytoplasmatic inclusions found in patients suffering from these diseases (40). Three domains can be distinguished in the AS sequence: 1), the amphipathic N-terminus (residues 1–60), with five imperfect repeats of a KTKEGV consensus motif; 2), the central hydrophobic amyloidogenic region (residues 61–95), also known as the non-Aβ component (NAC); and 3), the highly acidic and praline-rich C-terminus (residues 96–140). Structurally, AS can be classified as a natively disordered protein (41), although it has recently been shown to possess a long-range interaction between the C-terminal domain and the NAC region leading to an aggregation autoinhibited state (42). A significant rearrangement of protein structure, involving the population of partially folded intermediates and oligomeric species (18,32), occurs during AS fibrillation. We anticipated that NAS probes would be responsive to these conformational changes due to the polarity sensitivity of their fluorescence properties. We performed a systematic study of selected members of this family, namely 2-anilinonaphthalene-6-sulfonate (2,6-ANS), 2-(p-toluidinio)-naphthalene-6-sulfonate (2,6-TNS), 4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonate (bis-ANS), and bis(8-p-toluidino-1-naphthalenesulfonate) (bis-TNS) exposed to monomeric and purified fibrillar forms of AS. The interactions of these dyes (and ThioT) were characterized by steady-state and time-resolved fluorescence spectroscopy. Binding affinities for the fibrils were also determined and additional measurements during the process of aggregation were carried out. The results are discussed in terms of available structural and spectroscopic data.
MATERIALS AND METHODS
Chemicals
2,6-ANS and bis-ANS were obtained from Molecular Probes (Eugene, OR), and 2,6-TNS was from Acros Organics (Geel, Belgium). ThioT was purchased from Sigma Chemical (St. Louis, MO). bis-TNS was the kind gift of Dr. Gregorio Weber (deceased).
NAS stock solutions were prepared in ethanol (TNS derivatives were first dissolved in a small volume of DMF). The concentrations were determined by UV absorption from a dilution prepared in methanol, except in the case of bis-TNS, which was diluted into water, using the following extinction coefficients: ɛ319 = 27,000 M−1 cm−1 (2,6-ANS), ɛ318 = 26,000 M−1 cm−1 (2,6-TNS), ɛ395 = 23,000 M−1 cm−1 (bis-ANS) and ɛ390 = 16,540 M−1 cm−1 (bis-TNS). The later value was from (43) and the others from (44). ThioT solutions were in water.
Buffer solutions were of analytical grade and filtered through a 0.22-μm size membrane.
Preparation of monomeric and fibrillar AS
The bacterial expression and purification of recombinant human AS were as described previously (33). Protein purity was assessed by sodium dodecyl sulfate polyacrylamide gel electrophoresis.
Monomeric AS stock solutions were prepared in 25 mM Tris-HCl, pH 7.5, 100 mM NaCl, and centrifuged 30 min at 105 × g before use to remove aggregated material. Protein concentrations were determined by absorbance using an ɛ275 of 5600 M−1 cm−1 (41).
Fibril suspensions were obtained by incubating 300 μM AS at 70°C with constant shaking at 800 rpm in an Eppendorf Thermomixer comfort (Hamburg, Germany). At this elevated temperature, the pH of the Tris-HCl buffer drops from 7.5 (at 25°C) to 6.2. Fibrils were centrifuged at 14,000 rpm during 30 min in an Eppendorf MiniSpin Plus bench-top centrifuge. After removal of the supernatant, the pellet was resuspended in 25 mM Tris-HCl, pH 7.5, 100 mM NaCl. This cycle was repeated three times. The protein concentration in monomeric units was determined by absorbance from an aliquot incubated in 6 M Gdm-HCl at 25°C for 24 h, a procedure leading to complete disaggregation of the fibrils.
Protein aggregation assay
The aggregation of AS (100 μM) was carried out in 25 mM Tris-HCl, pH 7.2 (at 37°C), 100 mM NaCl, 0.01% Na-azide (Sigma) according to standard procedures (33). The centrifuged (13,000 × g, 30 min) and filtered protein sample (350 μL) was incubated in glass vials (Zinsser Analytik, Frankfurt, Germany) at 37°C under continuous stirring (350 rpm) in a Kendro incubator using Teflon magnetic microbars. Samples were withdrawn at different time points and mixed with the fluorescent dyes to final concentrations of ∼97 μM protein and 10 μM probe. Fibril formation was monitored by both steady-state and time-resolved fluorescence spectroscopy. Samples were run in duplicate and analyzed independently.
The analysis of the aggregation traces measured by steady-state fluorescence spectroscopy was performed as described in Fernández et al. (45), assuming a nucleation polymerization model according to an equation for the fractional normalized amyloid conversion, 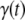 :
:
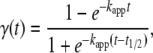 |
(1) |
where 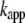 is an apparent rate constant for the incorporation of monomers at growth points located in aggregates and
is an apparent rate constant for the incorporation of monomers at growth points located in aggregates and 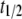 corresponds to the time point of 50% apparent conversion, i.e.,
corresponds to the time point of 50% apparent conversion, i.e., 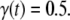 In practice,
In practice, 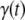 refers to the fractional ThioT signal without independent determinations of monomer conversion.
refers to the fractional ThioT signal without independent determinations of monomer conversion.
Fluorescence spectroscopy
Steady-state measurements
Corrected emission spectra were acquired with a Cary Eclipse spectrofluorimeter (Varian, Australia) equipped with a thermally controlled cuvette holder. Spectral slits were set at a 10-nm bandwidth for both excitation and emission. Emission spectra during aggregation were recorded in a Perkin Elmer LS50B spectrofluorimeter with a bandwidth of 2.5 nm and a 3-mm dual light path. Spectra were acquired at 25°C. 2,6-ANS and 2,6-TNS were selectively excited at 320 nm, bis-ANS and bis-TNS at 395 nm, and ThioT at 446 nm.
Steady-state anisotropy values, 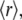 were determined from the parallel (VV) and perpendicular (VH) polarization signals (V, vertical; H, horizontal, the symbol order indicating excitation and emission) according to the standard expression
were determined from the parallel (VV) and perpendicular (VH) polarization signals (V, vertical; H, horizontal, the symbol order indicating excitation and emission) according to the standard expression
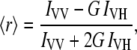 |
(2) |
where G is a correction constant (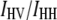 ) taking into account the differential polarization sensitivity of the detection system.
) taking into account the differential polarization sensitivity of the detection system.
In titration experiments, increasing volumes of 500 μM dye were added to a solution containing 2–8 μM AS or to buffer alone (blank) under continuous stirring. A 5-mm path cuvette was used. The spectra were recorded after equilibration for 5 min. Titration curves were analyzed assuming a single-site model according to
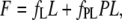 |
(3) |
where 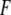 is the fluorescence intensity corrected for inner filter effects, and
is the fluorescence intensity corrected for inner filter effects, and 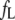 and
and 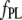 are the apparent specific fluorescence signals for the free ligand L and the complex PL, respectively. By solving the mass balance and conservation equations assuming one accessible binding site per monomer in the aggregated state, we obtain
are the apparent specific fluorescence signals for the free ligand L and the complex PL, respectively. By solving the mass balance and conservation equations assuming one accessible binding site per monomer in the aggregated state, we obtain
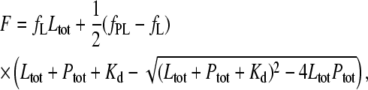 |
(4) |
where Ltot and Ptot are the total dye and protein concentrations, after corrections for dilution, and Kd is the equilibrium dissociation constant. (Note that unitary stoichiometric factors were assumed for both the protein and probe, corresponding to common practice. With fibrillar targets, it is difficult to implement more sophisticated kinetic and thermodynamic techniques for evaluating the number, nature, and distribution of binding sites. Stoichiometric factors other than unity would enter into Eq. 4 as multiplicative coefficients of Ptot, with the effect of displacing the fractional binding of the ligand to higher values.) The titration curves were fit by nonlinear regression with Eq. 4, after correction for inner filter effects by multiplication with a factor of 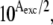 thereby compensating for the strong absorption of the dyes at the fluorescence excitation wavelengths. Absorption spectra at a number of dye concentrations were recorded in a 5-mm light path. Calibration curves at the appropriate wavelengths were constructed. No deviations from the Lambert-Beer equation were observed in the range of concentrations used. The UV-visible spectra were not substantially modified by the presence of fibrillar AS at concentrations <10 μM. The slopes obtained after linear regression were used to calculate the absorbance at the corresponding concentration of dye in the titration experiments.
thereby compensating for the strong absorption of the dyes at the fluorescence excitation wavelengths. Absorption spectra at a number of dye concentrations were recorded in a 5-mm light path. Calibration curves at the appropriate wavelengths were constructed. No deviations from the Lambert-Beer equation were observed in the range of concentrations used. The UV-visible spectra were not substantially modified by the presence of fibrillar AS at concentrations <10 μM. The slopes obtained after linear regression were used to calculate the absorbance at the corresponding concentration of dye in the titration experiments.
Time-resolved fluorescence measurements
Fluorescence intensity and time-resolved anisotropy decays were acquired with an IBH 5000U fluorescence lifetime spectrometer (Glasgow, UK) equipped with a TBX-04-A picosecond photon detection module. Measurements were performed in a time-correlated single-photon counting (TCSPC) mode using the Data Station, v 2.1, with DAS6 software. The excitation source was a 337-nm (2,6-ANS and 2,6-TNS), 373-nm (bis-ANS and bis-TNS),or 457-nm (ThioT) wavelength pulsed NanoLEDs with a repetition rate of 1 MHz. Bandpass filters used in the emission channel to minimize light scattering were Corion ss420 and ss430 for 2,6-ANS and 2,6-TNS, respectively, and Balzers B20 for bis-ANS, bis-TNS, and ThioT in the presence of purified fibrils. In the latter case, an additional Corion ss420 bandpass filter was placed in the excitation channel. For 2,6-ANS and 2,6-TNS, during protein aggregation, the emission filter was a Balzers K40 bandpass. The impulse response function (IRF) was obtained at the corresponding excitation wavelength using a Ludox scattering suspension.
The emission decays were obtained under magic-angle conditions (polarizers: excitation 0°, emission 54.7°) to eliminate polarization effects. The data were analyzed using the IBH DAS6 software. Reconvolution analysis was performed assuming a multiexponential decay function for the time-resolved fluorescence intensity, 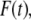 according to
according to
 |
(5) |
where αi and τi are the ith preexponential factor (amplitude) and the corresponding fluorescence lifetime, respectively, with 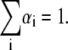 The fractional (steady-state) intensity is given by
The fractional (steady-state) intensity is given by 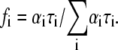 The data were best fit with a model of two components (i = 1, 2), in some cases including an additional scattered-light contribution with an arbitrary short-lifetime (0.055 ns) contribution. In general,
The data were best fit with a model of two components (i = 1, 2), in some cases including an additional scattered-light contribution with an arbitrary short-lifetime (0.055 ns) contribution. In general, 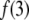 was <0.1. The remaining fractional intensities were renormalized. The goodness of the fits was assessed from the χ2 values, which were close to 1 for all samples, and the weighted residuals fluctuated about zero.
was <0.1. The remaining fractional intensities were renormalized. The goodness of the fits was assessed from the χ2 values, which were close to 1 for all samples, and the weighted residuals fluctuated about zero.
Time-resolved emission spectra (TRES) were derived from wavelength-dependent decays, acquired through an emission monochromator with a slit width of 8 nm. Fluorescence intensity decay curves were collected across the emission spectrum of the dyes in increments of 10 nm. At each wavelength, the decay fitted functions were rescaled by the corresponding steady-state corrected emission fluorescence intensity, 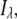 according to
according to
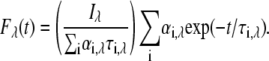 |
(6) |
Equation 6 was used to obtain the TRES profile at given time points after the excitation. The mean fluorescence decay, 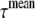 , is defined as
, is defined as 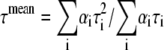 and the center of mass,
and the center of mass, 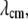 of the fluorescence spectra was calculated according to
of the fluorescence spectra was calculated according to 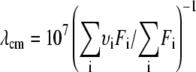 where
where 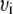 is the wavenumber (
is the wavenumber (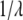 ) in cm−1 and
) in cm−1 and 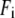 is the fluorescence intensity at that
is the fluorescence intensity at that 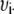 .
.
The time-resolved anisotropy decays were determined from the parallel and crossed polarized decays according to
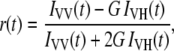 |
(7) |
where the denominator represents the total emission intensity 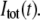 The limiting value of the anisotropy decay,
The limiting value of the anisotropy decay, 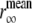 , was calculated by averaging over a window selected at a time longer than the fluorescence lifetime (25–40 ns for NAS derivatives and 8–16 ns for ThioT) according to
, was calculated by averaging over a window selected at a time longer than the fluorescence lifetime (25–40 ns for NAS derivatives and 8–16 ns for ThioT) according to 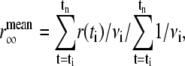 where the variance in anisotropy,
where the variance in anisotropy, 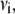 takes into account the uncertainty in the measured parameter due to photon statistics (46).
takes into account the uncertainty in the measured parameter due to photon statistics (46).
For cases in which perceptible time-dependent anisotropies were observed, the parallel and perpendicular polarized decays [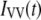 and
and 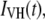 respectively] were analyzed by a procedure designed to obtain apparent rotational correlation times,
respectively] were analyzed by a procedure designed to obtain apparent rotational correlation times, 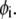 These were on the order of the width of the IRF, making it difficult to employ certain analysis routines incorporated into the DAS6 software. The following alternative strategy was developed and implemented with Mathematica (Wolfram Research, Champaign, IL). The IRF was fit to an analytical seven-parameter expression consisting of the sum of a Gaussian distribution function and a normal cumulative distribution function multiplied by an exponential function:
These were on the order of the width of the IRF, making it difficult to employ certain analysis routines incorporated into the DAS6 software. The following alternative strategy was developed and implemented with Mathematica (Wolfram Research, Champaign, IL). The IRF was fit to an analytical seven-parameter expression consisting of the sum of a Gaussian distribution function and a normal cumulative distribution function multiplied by an exponential function:
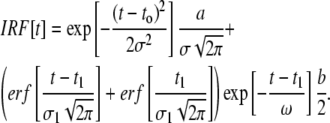 |
(8) |
Such a synthetic IRF can be convoluted analytically with an exponential decay function, the result of which can be fit directly to the time resolved data to derive the lifetime [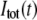 ] and anisotropy [global fit of
] and anisotropy [global fit of 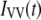 and
and 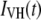 ] decay parameters. With this approach, the effect of noise in the IRF is minimized and the formulation and exploration of complex associated (linked) intensity and anisotropy decay models is greatly facilitated.
] decay parameters. With this approach, the effect of noise in the IRF is minimized and the formulation and exploration of complex associated (linked) intensity and anisotropy decay models is greatly facilitated.
Atomic force microscopy
AFM images were acquired on a Digital Instruments (Veeco) Multimode scanning probe microscope IIIa using a J-Scanner in air at a scan rate of 1 Hz with 512 lines/image. Imaging was performed in tapping mode with a DNP-S (nominal spring constant, 0.06 N/m) cantilever. An aliquot of the aggregated sample was diluted with water and spin-coated on a freshly cleaved mica surface.
RESULTS AND DISCUSSION
Induction of AS fibrillation
Fibrillation of AS in vitro is typically carried out by incubation of 70–100 μM protein solutions at 37°C, pH 7.0–7.5, under continuous stirring. Under these experimental conditions, the reaction was completed within 4 days (33), although there existed unpredictable variability between replicates in terms of the fraction of aggregated material and t1/2.
Elevated temperature and lowered pH lead to an enhanced rate of AS fibrillation (32,33), manifested by an increase in the population of partially folded intermediates with a significant content of β-structure (32). For the purpose of quickly generating significant amounts of fibrils, 300 μM AS protein solutions were incubated at 70°C with continuous shaking. We also took advantage of the acceleration provided by the decrease in the pH of the Tris-HCl buffer, from 7.2 at 37°C to 6.2 at 70°C (33). After 4 h of incubation, the solutions were visibly turbid and yielded a positive signal with ThioT, the reference dye used for detection of amyloid fibrils, the presence of which was confirmed by atomic force microscopy (AFM, Fig. 1). Fibrils with the characteristic twists of mature fibrils and a diameter of ∼9 nm (from the height) were clearly observed. Preliminary comparative solid-state NMR measurements of AS fibrils formed at 37°C and 70°C indicate that the major conformational features of the fibrillar core are unchanged at the higher temperature (H. Heise and M. Baldus, MPI for Biophysical Chemistry, personal communication, 2007). This material was used for all further experiments with purified fibrils.
FIGURE 1.

AFM images of AS fibrils formed at 70°C. (A) Fibrils showing characteristic twists and a diameter (height) of ∼9 nm. Image, 10 × 10 μm2. (B) Expanded view of the boxed section shown in A. Image, 3.5 × 3.5 μm2.
The kinetics of aggregation at 70°C was characterized by an aggregation halftime (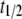 ) 1/10th that observed at 37°C, whereas
) 1/10th that observed at 37°C, whereas 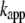 was increased by a modest factor of 2 (data not shown). These findings indicate that at 70°C, the rate of monomer addition was somewhat greater but that the major effect of the higher temperature and lower pH was to increase the efficiency of nucleation (32,33).
was increased by a modest factor of 2 (data not shown). These findings indicate that at 70°C, the rate of monomer addition was somewhat greater but that the major effect of the higher temperature and lower pH was to increase the efficiency of nucleation (32,33).
Fluorescence and binding properties of ANS/TNS derivatives interacting with AS
To apply the NAS probes for amyloid detection, a characterization of the spectroscopic properties of their protein complexes and their underlying structural basis was required. We compared the fluorescence properties of the ANS/TNS derivatives interacting with monomeric and fibrillar AS to those of ThioT. These determinations included the estimation of affinities for fibrillar AS in an effort to further elucidate the modes of interaction.
The steady-state emission of the probes in buffer or in the presence of monomeric AS was negligible or very low (Fig. 2), suggesting that the dyes either interact poorly with the monomer or that they are bound in a solvent-exposed mode. The anionic forms of the ANS/TNS derivatives may sense and interact directly with positively charged side chains of the protein. Ion pair formation, particularly external, of 1-anilinonaphthalene-8-sulfonate (1,8-ANS) with several proteins and peptides has been reported (47,48). The presumption that the binding of NAS is enhanced by electrostatic interactions is supported by the pH dependence of 2,6-TNS and bis-ANS fluorescence in the presence of Aβ(1–40) amyloid fibrils or the soluble form, in which the gain in signal correlates with the increasingly positively charged peptide at acidic pH (30,35). Bis-ANS also binds strongly to various soluble and aggregated (amyloid) species of transthyretin, albeit at very acidic pH (14). At neutral pH, AS possesses 15 positively charged Lys residues that may function as potential binding sites. Eleven are located in the amphipathic N-terminus (residues 1–60; net charge +6 including the N-terminal amino group) and one in the hydrophobic NAC region (residues 61–95); the remaining C-terminal region has a net charge of −13 including the C-terminal carboxyl group. The weak fluorescence signals observed in the presence of native AS may also arise from an interaction of the naphthalene dyes with transient highly-solvent-exposed hydrophobic patches on the disordered monomeric protein. This view is supported by NMR data indicating that native AS populates a dynamic ensemble of conformers ranging from highly unfolded to fairly compact structures (42).
FIGURE 2.

Fluorescence probes for aggregated states of AS. (A) Chemical structures of the dyes used in this work: (left to right) 2,6-ANS, 2,6-TNS, bis-ANS, bis-TNS, and ThioT. (B–F) Fluorescence emission spectra of (B) 2,6-ANS, (C) 2,6-TNS, (D) bis-ANS (E) bis-TNS and (F) ThioT, in buffer alone (black), or in the presence of monomeric (red) or purified fibrillar (blue) AS. The curves corresponding to the dye in buffer or with monomeric AS are multiplied fivefold. Dye, 1 μM; protein, 10 μM.
Upon mixing the NAS probes with AS fibrils, a pronounced increase in the fluorescence intensity, accompanied by a hyperchromic shift (not well defined for the probes that are virtually nonfluorescent in water), was observed (Fig. 2 and Table 1). In comparison with the free dyes, the spectra of 2,6-ANS and 2,6-TNS bound to fibrillar AS were shifted toward the blue edge by 54 nm and 38 nm, respectively (Table 1). These changes suggest that the bound probes experience a hydrophobic environment and/or occupy a polar region allowing only restricted motion (26,27).
TABLE 1.
Properties of different fluorescent probes upon binding to fibrillar AS
| 2,6-ANS | 2,6-TNS | bis-ANS | bis-TNS | ThioT | |
|---|---|---|---|---|---|
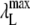 (nm)* (nm)*
|
471 | 475 | NA | NA | NA |
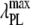 (nm) (nm) |
417 | 437 | 493 | 505 | 483 |
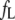 |
1.22 ± 0.01 | 0.35 ± 0.01 | 0 | 0 | 0 |
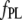 |
167 ± 2 | 169 ± 2 | 198 ± 3 | 124 ± 2 | 21 ± 1† |
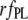 |
0.61 | 0.64 | 1 | 0.66 | — |
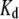 (μM) (μM) |
8.8 ± 0.5 | 11.7 ± 0.4 | 8.6 ± 0.5 | 11.6 ± 0.7 | 14.9 ± 0.4† |
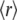 |
0.29 | 0.30 | 0.34 | 0.33 | 0.38 |
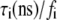 ‡ ‡
|
4.2/0.10 | 3.9/0.10 | 3.4/0.22 | 3.3/0.27 | 0.5/0.23 |
| 9.9/0.90 | 10.3/0.90 | 7.3/0.88 | 7.0/0.73 | 2.1/0.77 |
NA, not applicable.
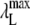 and
and 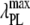 are the maximum wavelengths of fluorescence emission spectrum of the unbound and bound dye, respectively.
are the maximum wavelengths of fluorescence emission spectrum of the unbound and bound dye, respectively.
Values in the range 20–90 and 1–15 μM were obtained for 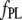 and
and 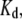 respectively, for different fibril preparations.
respectively, for different fibril preparations.
Standard deviation errors of fitted lifetimes were typically 0.1 ns for the fast component and 0.05 ns for the slow component.
The affinities of the probes for AS fibrils were determined by fluorimetric titration, in which small increments of concentrated probe solution were added to the protein. The intensities corresponding to the maximum of the emission band for each dye are plotted in Fig. 3 as a function of the dye concentrations. Control experiments (dyes injected into a buffer solution) are also shown. The data were corrected for inner filter effects and analyzed according to Eq. 4, yielding the intrinsic fluorescence of the unbound, 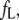 and bound,
and bound, 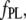 dye, and the apparent dissociation constant
dye, and the apparent dissociation constant 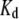 (Table 1). To minimize the number of adjustable parameters,
(Table 1). To minimize the number of adjustable parameters, 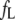 was obtained from the blank curves and was appreciable only for 2,6-ANS and 2,6-TNS.
was obtained from the blank curves and was appreciable only for 2,6-ANS and 2,6-TNS.
FIGURE 3.

Titration of fibrillar AS with NAS probes and ThioT. Purified AS fibrils with increasing amounts of (A) 2,6-ANS, (B) 2,6-TNS, (C) bis-ANS, (D) bis-TNS, and (E) ThioT. Data correspond to the emission at the indicated wavelengths, in the presence of fibrils (squares with dots) or in buffer (circles with dots). The initial protein concentration in monomeric units was 2 μM (A), 4 μM (B), and 8 μM (C–E). The lines represent the best fits obtained using Eq. 4 and the estimated parameters are given in Table 1. Data were corrected for inner filter effects.
A more informative comparison of the relative fluorescent signals generated by the bound NAS probes was obtained by correcting the 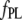 by multiplicative factors accounting for the wavelength-dependent differences in excitation intensities and detection efficiencies. Such estimates of relative fluorescence (
by multiplicative factors accounting for the wavelength-dependent differences in excitation intensities and detection efficiencies. Such estimates of relative fluorescence (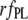 ), normalized by the value for bis-ANS, are given in Table 1. They follow the order bis-ANS > 2,6-TNS ≈ 2,6-ANS ≈ bis-TNS. We conclude that bis-ANS offers the highest sensitivity in the detection of amyloid protein. The corresponding apparent equilibrium dissociation constants (all in the micromolar range) were ranked differently: 2,6-ANS ≈ bis-ANS < 2,6-TNS ≈ bis-TNS (Table 1). It is interesting that the dissociation constants of the monomeric and dimeric forms of both ANS and TNS derivatives were similar, suggesting that only one naphthalene ring contributes to the binding, albeit manifesting somewhat distinct spectroscopic parameters. (In the event that the binding site accommodates two ANS or one bis-ANS, the derived value of
), normalized by the value for bis-ANS, are given in Table 1. They follow the order bis-ANS > 2,6-TNS ≈ 2,6-ANS ≈ bis-TNS. We conclude that bis-ANS offers the highest sensitivity in the detection of amyloid protein. The corresponding apparent equilibrium dissociation constants (all in the micromolar range) were ranked differently: 2,6-ANS ≈ bis-ANS < 2,6-TNS ≈ bis-TNS (Table 1). It is interesting that the dissociation constants of the monomeric and dimeric forms of both ANS and TNS derivatives were similar, suggesting that only one naphthalene ring contributes to the binding, albeit manifesting somewhat distinct spectroscopic parameters. (In the event that the binding site accommodates two ANS or one bis-ANS, the derived value of 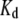 for ANS would increase by a factor of ∼2.) As discussed further below, we cannot exclude the influence of site heterogeneity nor probe structural features (methyl group substituent and/or substitution position) on the distinctive binding of the ANS and TNS moieties. Most probably, electrostatic (47,48) as well as predominant van der Waals and hydrophobic interactions are involved. By way of comparison, we note that the reported dissociation constants of ThioT for different amyloid fibrils vary from 0.03 to 10 μM (5). We obtained a somewhat higher value for AS fibrils (Table 1), but the methods of determination may not be directly comparable. In addition, we obtained
for ANS would increase by a factor of ∼2.) As discussed further below, we cannot exclude the influence of site heterogeneity nor probe structural features (methyl group substituent and/or substitution position) on the distinctive binding of the ANS and TNS moieties. Most probably, electrostatic (47,48) as well as predominant van der Waals and hydrophobic interactions are involved. By way of comparison, we note that the reported dissociation constants of ThioT for different amyloid fibrils vary from 0.03 to 10 μM (5). We obtained a somewhat higher value for AS fibrils (Table 1), but the methods of determination may not be directly comparable. In addition, we obtained 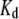 and
and 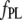 values for ThioT with different fibril preparations ranging from 1 to 15 μM and 20 to 90, respectively, reflecting the inherent irreproducibility associated with the use of this dye.
values for ThioT with different fibril preparations ranging from 1 to 15 μM and 20 to 90, respectively, reflecting the inherent irreproducibility associated with the use of this dye.
Steady-state fluorescence anisotropy, 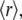 is a quantity that reflects the rate and extent of molecular motions and/or energy transfer during the excited-state lifetime of a fluorophore. The
is a quantity that reflects the rate and extent of molecular motions and/or energy transfer during the excited-state lifetime of a fluorophore. The 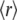 values determined for the probes bound to AS fibrils ranged from 0.29 to 0.38 (Table 1), indicating that the dyes were rigidly bound to the fibrillar structure. The values were very similar for 2,6-ANS and 2,6-TNS, and 10% higher for bis-ANS and bis-TNS (Table 1).
values determined for the probes bound to AS fibrils ranged from 0.29 to 0.38 (Table 1), indicating that the dyes were rigidly bound to the fibrillar structure. The values were very similar for 2,6-ANS and 2,6-TNS, and 10% higher for bis-ANS and bis-TNS (Table 1).
Time-resolved fluorescence spectroscopy provides information about binding-site heterogeneity, which is often difficult to obtain from steady-state measurements due to spectral overlap of the absorption and emission and reactions occurring during the excited state of the fluorophore. Determinations of intensity decays were performed on the complexes of the various probes with AS fibrils.
The experimental profiles, along with the corresponding fits according to a multiexponential decay model (Eq. 5) and the weighted residuals, are shown in Fig. 4 (fit parameters are given in Table 1). The probes 2,6-ANS and 2,6-TNS bound to AS fibrils had a short component of ∼4 ns and a long lifetime of ∼10 ns, accounting for 90% of the steady-state fluorescence intensity (Table 1). In the case of the dimeric probes, the short component was ∼3 ns and the long component of ∼7 ns represented ∼80% of the signal (Table 1). The fluorescence decays of NAS derivatives in aqueous solutions are generally monoexponential, with lifetimes of <0.25 ns (43), i.e., much shorter than the values obtained with the fibril complexes. The double-exponential decay of the latter indicates that the excited states experience at least two microenvironments with rather different polarities.
FIGURE 4.

Time-resolved fluorescence of the different probes bound to fibrillar AS. Fluorescence decay profiles of (A) 2,6-ANS, (B) 2,6-TNS, (C) bis-ANS, (D) bis-TNS, and (E) ThioT bound to AS fibrils. The lines represent the best fits obtained according to Materials and Methods, and the estimated parameters are given in Table 1. The weighted residuals are shown in the lower panels. Dye, 10 μM; protein, 100 μM.
Naphthalene derivatives generally exhibit multiexponential decays upon interaction with proteins. Examples are 2,6-TNS bound to apomyoglobin (49), bis-ANS interacting with aggregated states of the amyloid protein transthyretin (14) and native AS (37), and 1,8-ANS bound to proteins (50). For 1,8-ANS, the short lifetime (∼3 ns) was ascribed to probe molecules located at the protein-solvent interface and shown to be sensitive to protein association (50). Similar values were recently reported for polyamino acids employed as models for external binding sites of proteins (48). The long-lived decay has been attributed to buried binding sites excluded from water. These are presumed to be responsive to overall conformational changes, yet independent of the association state (50).
In accordance with these observations, we tentatively assign the short lifetimes of the probe-AS complexes to interactions with the N-terminal protein segments rich in Lys residues. Upon incorporation of AS into fibrils, the N-terminus is excluded from the fibrillar core yet exhibits a rigid conformation as of residue 22, albeit with some degree of static disorder (51). The long lifetime component would correspond to a binding site embedded in the fibril structure and protected from bulk water, as suggested by high-resolution solid-state NMR data showing that the central core of fibrillar AS consists of well-defined β-strands comprising at least residues 38–95 (51). A water-shielded core region extending from at least residue 65 to 89 has also been identified (H. Heise and M. Baldus, MPI for Biophysical Chemistry, personal communication, 2007). The existence of water-excluded cavities can be inferred from the dissociation of fibrils exposed to high pressure (52) and by numerous spectral features of pyrene-labeled cysteine mutants of AS (see (60)). These regions would be expected to bind hydrophobic compounds such as bis-ANS (52).
In the case of ThioT, two lifetime components were also observed, although both were short (0.5 ns and 2.1 ns, the latter accounting for 77% relative intensity) (Table 1). As discussed earlier, these results suggest the existence of different microenvironments for bound ThioT, as has also been inferred in studies of insulin and Aβ(1–40) amyloid fibrils (30). ThioT bound to aggregated transthyretin exhibits three lifetimes ranging from 0.1 to 3.9 ns (14).
The pronounced red shift and the decrease in quantum yield of NAS derivatives placed in a polar medium are usually interpreted in terms of solvent relaxation about the excited-state species (27). The greater dipole moment of the excited state relative to the ground state induces a greater degree of solvent relaxation (26,27). In the event that equilibrium about the excited-state dipole is not achieved, emission during the process of solvent relaxation leads to a spectrum reflecting the time-dependent ensemble of partially relaxed states. The phenomenon can be explored in the nanosecond timescale by measurement of TRES, as demonstrated by the environmental relaxation of 2,6-TNS bound to proteins and membranes (49,53). In this study, we examined the dynamic spectral relaxation of complexes of certain NAS and AS fibrils with TRES, as illustrated in Fig. 5 for 2,6-TNS.
FIGURE 5.

TRES of 2,6-TNS bound to AS fibrils. (A) Experimental fluorescence decays at selected emission wavelengths. (B) Normalized fits of fluorescence decays. (Inset) Mean fluorescence lifetime. (C) Steady-state normalized decays obtained with Eq. 6. (Inset) Corrected normalized steady-state emission spectrum of 2,6-TNS bound to AS fibrils. (D) Intensity normalized TRES at different time points after excitation, calculated according to Eq. 6. (Inset) Center of mass of the fluorescence spectra as a function of time.
Fluorescence decays were acquired at a number of wavelengths across the emission spectrum (Fig. 5 A). The decays were faster at the shorter wavelengths. In such cases, one presumes that deactivation at the blue edge is by both emission and relaxation, whereas the molecules selected at the red edge are relaxed before emission. The deconvoluted data analyzed in terms of the multiexponential model (Eq. 5) led to decay curves as a function of emission wavelength (Fig. 5 B). The mean lifetime increased with wavelength (Fig. 5 B, inset). A new set of intensity decays were scaled so as to equate their integrated intensities to the corresponding relative steady-state intensities at the respective emission wavelengths (Eq. 6) The normalized intensity decay profiles are shown in Fig. 5 C along with the corrected steady-state emission (inset) and the normalized TRES spectra at selected time points (Fig. 5 D). A distinct red shift with increasing time (3 nm in 20 ns) was observed, as represented by the time-dependent center of mass (Fig. 5 D, inset). As discussed above, the dye-binding sites in amyloids are probably heterogeneous, accounting at least in part for the observed TRES behavior of 2,6-TNS. In the case of two ground-state species with distinct emission spectra, either inherent to the probe or reflecting heterogeneous binding sites, one would expect two lifetimes with relative amplitudes (but not lifetimes) varying with wavelength. However, in the case of 2,6-TNS, both the lifetimes and relative intensities varied across the spectrum (data not shown). It has been reported that the intensity (quantum yield) of NAS derivatives decreases in environments leading to red shifts of the emission maximum (26,27). Our data, in contrast (Fig. 5 B, inset), are indicative of a time-dependent reorientation of the immediate environment of the bound dye as the most plausible basis for the observed behavior. One should be mindful that the concept of “environment” includes consideration of rigid polar residues of the protein in the binding pocket, as well as solvent molecules with properties differing from those of the bulk medium.
For 2,6-ANS, a dual wavelength-dependent behavior was observed. Initially, the decay rates decreased with wavelength but then gradually increased. This complex behavior requires further exploration. No time-dependent changes were observed with the other dyes, a finding that does not necessarily imply an absence of excited-state processes. They may simply have been very fast and thus unresolved on the experimentally available nanosecond timescale.
Monitoring AS aggregation with ANS/TNS derivatives
A major aim of our study was to demonstrate that NAS derivatives can serve as useful external optical probes for monitoring amyloid formation. The kinetics of AS aggregation was studied by both steady-state and time-resolved fluorescence spectroscopy, including anisotropy decay. Fig. 6 shows the fluorescence spectra of bis-TNS at different time points during protein fibrillation (Fig. 6 A) along with the normalized kinetic aggregation curves detected for all the probes (Fig. 6 B). The changes in time-resolved fluorescence parameters during the course of aggregation are presented in Fig. 7 and Table 2. Time-dependent anisotropy data are featured in Figs. 8 and 9.
FIGURE 6.

Aggregation kinetics of AS monitored by steady-state fluorescence. (A) Emission spectra of bis-TNS during AS aggregation. (B) Time course of AS aggregation sensed by NAS dyes and ThioT; blank (0 time value) subtracted intensities at 415 (2,6-ANS), 426 (2,6-TNS), 480 (bis-ANS), 485 (bis-TNS), and 483 nm (ThioT), respectively. Lines represent the fits obtained with Eq. 1. Dye, 10 μM; protein, ∼97 μM. Samples were run in duplicate.
FIGURE 7.

Aggregation kinetics of AS monitored by time-resolved fluorescence. Changes in fluorescence lifetimes (bars, left axis) and fractional intensity of the long-lived component (□, right axis) of (A) 2,6-ANS, (B) 2,6-TNS, (C) bis-ANS, (D) bis-TNS, and (E) ThioT during protein aggregation.
TABLE 2.
Time resolved fluorescence parameters of different probes during AS aggregation
| 2,6-ANS
|
2,6-TNS
|
bis-ANS
|
bis-TNS
|
ThioT
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) |
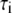 (ns) (ns) |
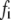 |
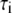 (ns) (ns) |
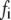 |
τi (ns) | 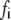 |
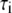 (ns) (ns) |
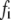 |
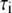 (ns) (ns) |
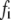 |
| 0 | 1.7 | 0.24 | 2.4 | 0.26 | 1.8 | 0.29 | 1.4 | 0.33 | ||
| 8.9 | 0.76 | 9.6 | 0.74 | 5.7 | 0.71 | 4.8 | 0.67 | |||
| 24 | 2.3 | 0.17 | 2.7 | 0.24 | 2.3 | 0.31 | 1.9 | 0.33 | 0.6 | 0.49 |
| 9.0 | 0.83 | 9.4 | 0.76 | 6.0 | 0.69 | 5.5 | 0.67 | 1.5 | 0.51 | |
| 48 | 3.0 | 0.15 | 3.3 | 0.20 | 2.9 | 0.28 | 2.5 | 0.25 | 0.5 | 0.33 |
| 9.4 | 0.85 | 9.7 | 0.80 | 6.5 | 0.72 | 6.5 | 0.75 | 1.3 | 0.65 | |
| 72 | 3.2 | 0.17 | 3.2 | 0.17 | 3.3 | 0.29 | 2.7 | 0.29 | 0.4 | 0.26 |
| 9.4 | 0.83 | 9.7 | 0.83 | 7.1 | 0.71 | 7.0 | 0.71 | 1.3 | 0.74 | |
| 96 | 3.3 | 0.13 | 3.0 | 0.16 | 3.1 | 0.25 | 2.8 | 0.27 | 0.4 | 0.29 |
| 9.5 | 0.87 | 9.5 | 0.84 | 6.7 | 0.75 | 6.8 | 0.73 | 1.2 | 0.71 | |
| 120 | 3.3 | 0.12 | 3.2 | 0.17 | 3.2 | 0.27 | 2.9 | 0.29 | 0.6 | 0.36 |
| 9.5 | 0.88 | 9.6 | 0.83 | 7.0 | 0.73 | 7.1 | 0.71 | 1.4 | 0.64 | |
Analyses were performed with the DAS6 software. Standard deviation errors of fitted lifetimes were typically 0.1 ns for the fast component and 0.05 ns for the slow component.
FIGURE 8.

Anisotropy decays during the course of AS aggregation. Anisotropy profiles for (A) 2,6-ANS, (B) 2,6-TNS, (C) bis-ANS, (D) bis-TNS, and (E) ThioT at 0 (black), 24 (red), 48 (blue), 72 (magenta), 96 (green), and 120 h (purple). The peak of the IRF was at ∼16 ns for NAS dyes and at ∼9 ns for ThioT.
FIGURE 9.

Fits to anisotropy data acquired during the course of AS aggregation. (A) Limiting anisotropy (mean value, see Materials and Methods) as a function of incubation time. (B) Time-resolved anisotropy for bis-TNS at 48 h incubation. Fit according to the analytical convolution technique (see text for parameters). (C) Anisotropy magnitude 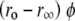 for bis-ANS and bis-TNS during aggregation (see text for details). Symbols are as defined in A; closed and open represent duplicate samples.
for bis-ANS and bis-TNS during aggregation (see text for details). Symbols are as defined in A; closed and open represent duplicate samples.
At the onset of the aggregation assay, a low but distinct fluorescence signal was detected with bis-TNS (Fig. 6 A) and the other NAS derivatives, but not with ThioT. We attribute these initial signals to interactions with monomeric AS but do not exclude a finite contribution from low amounts of preaggregated material that were not removed using the moderate centrifugation employed in these experiments.
As aggregation proceeded, all probes exhibited an increase in fluorescence intensity (Fig. 6 B) and, in some cases, spectral shifts, as featured for bis-TNS in Fig. 6 A. We propose that these phenomena reflect the progressive formation of hydrophobic binding sites and restructuring of preexistent binding sites, resulting in the state of reduced polarity inherent to the cross-β-amyloid conformation of prefibrillar intermediates and mature fibrils. All of the aggregation profiles monitored by intensity had the characteristic sigmoidal shapes expected for a nucleation-polymerization process (Fig. 6 B). According to this model, nucleation centers formed at the outset and during the lag phase spawn oligomeric species leading to the initiation and elongation of filamentous and fibrillar structures. However, a close comparison of the aggregation curves revealed significant differences in the aggregation kinetics. In the case of 2,6-ANS and 2,6-TNS, the increase in fluorescence intensity paralleled that of ThioT, with aggregation 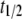 values of ∼46 ± 3 h. The
values of ∼46 ± 3 h. The 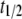 measured in the standard ThioT assay (dye excess), was very similar, but was shorter (38 ± 1 h) for bis-ANS and bis-TNS. It is unlikely that this result was due to differences in binding affinity, inasmuch as the hierarchy in the progression curves did not correspond to the order of dissociation constants (Table 1). It is more likely that the lower
measured in the standard ThioT assay (dye excess), was very similar, but was shorter (38 ± 1 h) for bis-ANS and bis-TNS. It is unlikely that this result was due to differences in binding affinity, inasmuch as the hierarchy in the progression curves did not correspond to the order of dissociation constants (Table 1). It is more likely that the lower 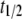 reflected interactions with prefibrillar species, as was further indicated by the steady-state and time-resolved anisotropy data (see below). Myriad conformational structures are present in the intermediate stages of aggregation, ranging from spherical oligomers to rodlike and wormlike protofibrils (54). In the case of AS, spheroids, chains of spheres, and rings resembling circularized protofibrils were identified by AFM (33,54). In addition, the β-sheet content of AS aggregates, assessed by Raman spectroscopy, is greater in protofibrils than in spherical oligomers ((54) and references therein).
reflected interactions with prefibrillar species, as was further indicated by the steady-state and time-resolved anisotropy data (see below). Myriad conformational structures are present in the intermediate stages of aggregation, ranging from spherical oligomers to rodlike and wormlike protofibrils (54). In the case of AS, spheroids, chains of spheres, and rings resembling circularized protofibrils were identified by AFM (33,54). In addition, the β-sheet content of AS aggregates, assessed by Raman spectroscopy, is greater in protofibrils than in spherical oligomers ((54) and references therein).
The time-resolved fluorescence parameters were also sensitive to protein aggregation. Already at the beginning of the aggregation, the fluorescence decays of the NAS probes exhibited a double-exponential character with a short-lived component of 1.4–2.4 ns and a longer dominant lifetime of 5–9 ns (Fig. 7 and Table 2). Both lifetimes exceeded values reported for the free dyes, indicating the existence of at least two distinct microenvironments of the bound probes. In accordance with our previous assignments, we attribute the short lifetimes to the interaction of the dyes with lysine-rich N-terminal domains and the longer-lifetime components to binding in the area of tertiary interactions within monomeric AS. Despite its predominantly disordered state, AS exhibits a compaction of the N- and C-termini and a foldback of the C-terminus over the NAC region (42). In a previous study of bis-ANS complexes with AS, a three-exponential decay was reported with lifetimes of 0.15, 2.6, and 7.6 ns (35). The authors assigned the short lifetime to the unbound probe, and the other two components to at least two types of hydrophobic surfaces on the protein. Using the second method of data analysis (Materials and Methods), we also resolved three components for the bis-ANS/AS complexes (initial values 0.4–0.5 ns, 2.5–2.8 ns, and 6.5–6.6 ns), the shortest of which accounted for only 7–8% of the steady-state fluorescence intensity. This value decreased during aggregation, as expected from the progressively decreasing concentration of free dye. The two-component fits to the bis-ANS decays sufficed for most purposes of data interpretation. The lifetimes for both the bis-ANS and bis-TNS complexes increased during aggregation, with little change in relative amplitude: the fast component almost doubled and the slow component increased from 4.8–5.7 ns to ∼7 ns at the end of fibrillation (Fig. 7 and Table 2).
The two lifetimes of 2,6-ANS, as well as the fractional intensity of the long-lived component, also increased as aggregation proceeded. In contrast, only the lifetime of the fast-decay component of 2,6-TNS increased with incubation time. These observations are in agreement with the steady-state fluorescence data, suggesting the formation of new hydrophobic cavities and/or a reorganization of the existing binding sites leading to a decreased polar character during fibril formation. According to our proposed mode of binding, the short lifetimes are modified due both to protein self-association (50) and loss of conformational flexibility of the N-terminus (51). On the other hand, the long-lived components change due to the formation of a well-defined cross-β-structure in the NAC region, which limits the mobility of the dyes and protects them from bulk solvent molecules.
In the case of ThioT, the steady-state fluorescence intensity began to rise after 24 h of incubation (Fig. 6 B) and the two lifetimes remained constant thereafter (Fig. 7 and Table 2). These observations confirm that binding of ThioT is restricted to the mature amyloid fibrils as a consequence of their specific secondary, tertiary, and quaternary structural features (5–7).
The time-resolved anisotropies of the various dyes showed a distinctive dependence on the elapsed time of the aggregation reaction (Fig. 8). With the exception of ThioT, all curves increased from an initial value of anisotropy (not fully resolved in Fig. 8 due to truncation). The limiting values likewise increased and plateaued after 72 h (Figs. 8 and 9 A). The intensity-weighted mean values 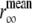 differed for the three classes of molecules: monomeric NAS, 0.28; dimeric NAS, 0.26; ThioT, 0.33. The progressive increase of NAS dye anisotropy during aggregation indicates that these compounds sense the presence and properties of oligomeric prefibrillar intermediates. In particular, the single NAS dyes appear to detect a compact conformation of the native protein as well as oligomeric species, whereas the dimeric NAS dyes exhibit a higher sensitivity for the early steps of aggregation. ThioT exclusively detects fibrils and as a consequence the anisotropy of its complexes is high and constant after the onset of finite emission intensities (Fig. 9).
differed for the three classes of molecules: monomeric NAS, 0.28; dimeric NAS, 0.26; ThioT, 0.33. The progressive increase of NAS dye anisotropy during aggregation indicates that these compounds sense the presence and properties of oligomeric prefibrillar intermediates. In particular, the single NAS dyes appear to detect a compact conformation of the native protein as well as oligomeric species, whereas the dimeric NAS dyes exhibit a higher sensitivity for the early steps of aggregation. ThioT exclusively detects fibrils and as a consequence the anisotropy of its complexes is high and constant after the onset of finite emission intensities (Fig. 9).
We subjected the anisotropy decay data of bis-ANS and bis-TNS to further analysis using the analytical convolution technique described in Material and Methods (see Eq. 8). Numerous models of associated intensity-anisotropy decay modes were explored, the simplest of which was most successful in fitting the data. The model in question assumes an exponential decay of anisotropy from an initial (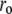 ) to a terminal (
) to a terminal (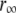 ) limiting value, which then remains constant. These parameters and the rotational correlation times, φ, are common to both intensity lifetime components, such that
) limiting value, which then remains constant. These parameters and the rotational correlation times, φ, are common to both intensity lifetime components, such that
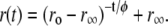 |
(9) |
In view of the interpretation of the two lifetime components in the context of binding site heterogeneity, this interpretation is clearly a simplification, one that would be biased to the properties of the dominant slower-decaying species. Nonetheless, the fits provided good representations of the experimental time-resolved anisotropy, including the initial period extending into the IRF (Fig. 9 B). For both dyes, 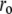 and
and 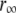 increased during aggregation, but the apparent rotational correlation times clustered in the range of 0.4–1 ns. This low value is consistent with other determinations based on covalent adducts of short-lifetime organic dyes with monomeric AS (C. W. Bertoncini, E. A. Jares-Erijman, and T. M. Jovin, unpublished) and is much smaller than that predicted for the global rotational relaxation of a protein molecule with the molecular mass of AS, either in a compact or extended (41) conformation. We conclude that the observed relaxation reflected local motion of the dye docked to its binding site(s) and that its amplitude decreased as the oligomeric-prefibrillar-fibrillar forms increased in size and rigidity. This phenomenon, expressed quantitatively as an “anisotropy magnitude”, the product of (ro−r∞) φ, appears to distinguish distinct initial and terminal phases of aggregation (Fig. 9 C), an interesting observation that requires further investigation.
increased during aggregation, but the apparent rotational correlation times clustered in the range of 0.4–1 ns. This low value is consistent with other determinations based on covalent adducts of short-lifetime organic dyes with monomeric AS (C. W. Bertoncini, E. A. Jares-Erijman, and T. M. Jovin, unpublished) and is much smaller than that predicted for the global rotational relaxation of a protein molecule with the molecular mass of AS, either in a compact or extended (41) conformation. We conclude that the observed relaxation reflected local motion of the dye docked to its binding site(s) and that its amplitude decreased as the oligomeric-prefibrillar-fibrillar forms increased in size and rigidity. This phenomenon, expressed quantitatively as an “anisotropy magnitude”, the product of (ro−r∞) φ, appears to distinguish distinct initial and terminal phases of aggregation (Fig. 9 C), an interesting observation that requires further investigation.
In studies based on fluorescence anisotropy, it is important to consider mechanisms for depolarization other than rotational diffusion. In particular, rapid decays of anisotropy can occur due to energy transfer between excited- and ground-state molecules in close proximity, in which case the perceived time constant is not a rotational correlation time but rather is related to the rate of energy transfer. We estimated a homotransfer Förster radius (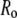 ) for bis-ANS of 2.2 nm (using a value of 2/3 for κ2). Under the condition of our experiments, and assuming a finite degree of binding of the dyes to monomeric AS, the estimated ratio of bound ligand to aggregated protein would probably not exceed a value of ∼0.1. We also note that the measured time constants did not display the systematic increase expected for homotransfer occurring during a progressive conversion of the protein. We conclude tentatively that homotransfer was not significant, although it cannot be excluded.
) for bis-ANS of 2.2 nm (using a value of 2/3 for κ2). Under the condition of our experiments, and assuming a finite degree of binding of the dyes to monomeric AS, the estimated ratio of bound ligand to aggregated protein would probably not exceed a value of ∼0.1. We also note that the measured time constants did not display the systematic increase expected for homotransfer occurring during a progressive conversion of the protein. We conclude tentatively that homotransfer was not significant, although it cannot be excluded.
The rotational correlation times corresponding to global tumbling of structures the size of protofibrils and fibrils are expected to largely exceed the lifetime of the molecular probes used in this work. In this regard, fluorescence probes having longer decay times are more appropriate for the determination of the size distribution of oligomeric species. In an parallel effort conducted in our laboratory, we exploited the long fluorescent lifetime as well as the spectral and polarization properties of residue-specific covalent adducts of pyrene with AS for detecting the multiple stages of AS aggregation (see (60)).
Structural interpretations
The interest in protein aggregation has increased tremendously in the last decades due to its relevance to protein misfolding and deposition diseases (1). Historically, the presence of amyloid fibrils ex vivo and insights into the mechanisms and kinetics of amyloid formation in vitro have been obtained by use of fluorescence dyes such as Congo red and ThioT (3). However, the specificity and sensitivity of these two dyes have been questioned (3,5,13). In addition, the need for periodic manual sampling, and the dependence of the signal on fibril morphology and solution conditions are deficiencies inherent to these in vitro assays. Furthermore, the dyes are believed to be insensitive to oligomeric intermediates, although some analogs appear to detect soluble Aβ oligomers (12).
NAS derivatives have been used extensively to explore particular aspects of protein misfolding and self-assembly. We present here a systematic and comparative evaluation of the interactions of selected members of the family (2,6-ANS, 2,6-TNS, bis-ANS, and bis-TNS) with AS in an effort dedicated to the expansion of the available set of fluorescent tools for studying mechanisms of amyloid formation. The sensitivity of these extrinsic reporter molecules to polarity and to the mobility of surrounding dipoles provides additional information related to structural aspects not revealed by ThioT. This circumstance is of particular interest due to the increasing number of high-resolution structural models of fibrillar aggregates from different sources.
We have shown that NAS derivatives exhibit a low fluorescence in the presence of native AS but become highly fluorescent when bound to AS fibrils. Hyperchromic shifts, high steady-state anisotropy values, increased fluorescence lifetimes, and solvent relaxation in the nanosecond timescale observed upon interaction with fibrillar structures suggest that the bound probes experience a hydrophobic environment and/or a polar surrounding with restricted motion. In addition, both steady-state and time-resolved fluorescence parameters change during the course of amyloid formation. The side chains on each side of the β-sheet form rows (so-called channels) running along the β-sheet (and therefore the fibril), perpendicular to the strands (55). Taking into account the volume occupied by the side chains, the width of the channels would be <6.5 Å (55). It has been proposed that the long and thin ThioT molecule (molecular dimensions 15.2 Å, 6.1 Å, and 4.3 Å, corresponding to the x, y, and z axes, respectively) inserts itself into these channels with its shortest axis perpendicular to the fibril axis, closely surrounded by the side chains (6). Since NAS probes have comparable molecular dimensions, one expects them to bind to AS fibrils in a similar fashion. The physical confinement conferred by the close proximity of the side chains would result in restricted motions of the probes (and their molecular environment), along with a shielding from bulk solvent molecules, thereby leading to the observed spectroscopic characteristics.
NAS probes bind to AS fibrils with apparent dissociation constants in the micromolar range. It has been shown that bis-ANS can interfere with protein-protein interactions, including those in thermally or chemically induced aggregation processes (56), as well as in the assembly of microtubules in vitro (57), bacteriophage P22 capsids (58), vesicular stomatitis virus (59), and amyloid formation of the prion protein (38,39). However, to abolish protein association, a high dye/protein molar ratio is required. According to our data, substoichiometric quantities should suffice for biosensing purposes, and we are exploring the requirements for establishing sensitive and quantitative continuous aggregation assays based on the NAS dyes.
Acknowledgments
We are very grateful to A. Pelah for AFM measurements and C. Bertoncini for suggestions regarding the conditions for protein aggregation and data interpretation. C. Bertoncini and S. Thirunavukkuarasu provided helpful comments on the manuscript.
This work was supported in part by the Universidad de Buenos Aires, Consejo Nacional de Investigaciones Cientificas y Técnicas, and Agencia Nacional de Promoción Científica y Tecnológica. M.S.C. is the recipient of a postdoctoral fellowship from the Alexander von Humboldt Foundation.
Editor: G. Barisas.
References
- 1.Hamley, I. W. 2007. Peptide fibrillization. Angew. Chem. Int. Ed. 46:8128–8147. [DOI] [PubMed] [Google Scholar]
- 2.Wetzel, R. 2006. Kinetics and thermodynamics of amyloid fibril assembly. Acc. Chem. Res. 39:671–679. [DOI] [PubMed] [Google Scholar]
- 3.Nilsson, M. R. 2004. Techniques to study amyloid fibril formation in vitro. Methods. 34:151–160. [DOI] [PubMed] [Google Scholar]
- 4.Klunk, W. E., J. W. Pettegrew, and D. J. Abraham. 1989. Quantitative evaluation of congo red binding to amyloid-like proteins with a β-pleated sheet conformation. J. Histochem. Cytochem. 37:1273–1281. [DOI] [PubMed] [Google Scholar]
- 5.LeVine 3rd, H. 1995. Thioflavine T interaction with amyloid β-sheet structures. Amyloid: Int. J. Exp. Clin. Invest. 2:1–6. [Google Scholar]
- 6.Krebs, M. R., E. H. Bromley, and A. M. Donald. 2005. The binding of thioflavin-T to amyloid fibrils: localisation and implications. J. Struct. Biol. 149:30–37. [DOI] [PubMed] [Google Scholar]
- 7.Groenning, M., L. Olsen, M. van de Weert, J. M. Flink, S. Frokjaer, and F. S. Jorgensen. 2007. Study on the binding of Thioflavin T to β-sheet-rich and non-β-sheet cavities. J. Struct. Biol. 158:358–369. [DOI] [PubMed] [Google Scholar]
- 8.Klunk, W. E., M. L. Debnath, and J. W. Pettegrew. 1995. Chrysamine-G binding to Alzheimer and control brain: autopsy study of a new amyloid probe. Neurobiol. Aging. 16:541–548. [DOI] [PubMed] [Google Scholar]
- 9.Styren, S. D., R. L. Hamilton, G. C. Styren, and W. E. Klunk. 2000. X-34, a fluorescent derivative of Congo red: a novel histochemical stain for Alzheimer's disease pathology. J. Histochem. Cytochem. 48:1223–1232. [DOI] [PubMed] [Google Scholar]
- 10.Crystal, A. S., B. I. Giasson, A. Crowe, M. P. Kung, Z. P. Zhuang, J. Q. Trojanowski, and V. M. Lee. 2003. A comparison of amyloid fibrillogenesis using the novel fluorescent compound K114. J. Neurochem. 86:1359–1368. [DOI] [PubMed] [Google Scholar]
- 11.Klunk, W. E., Y. Wang, G. F. Huang, M. L. Debnath, D. P. Holt, and C. A. Mathis. 2001. Uncharged thioflavin-T derivatives bind to amyloid-β protein with high affinity and readily enter the brain. Life Sci. 69:1471–1484. [DOI] [PubMed] [Google Scholar]
- 12.Maezawa, I., H. S. Hong, R. Liu, C. Y. Wu, R. H. Cheng, M. P. Kung, H. F. Kung, K. S. Lam, S. Oddo, F. M. Laferla, and L. W. Jin. 2008. Congo red and thioflavin-T analogs detect Aβ oligomers. J. Neurochem. 104:457–468. [DOI] [PubMed] [Google Scholar]
- 13.Khurana, R., V. N. Uversky, L. Nielsen, and A. L. Fink. 2001. Is Congo red an amyloid-specific dye? J. Biol. Chem. 276:22715–22721. [DOI] [PubMed] [Google Scholar]
- 14.Lindgren, M., K. Sorgjerd, and P. Hammarstrom. 2005. Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy. Biophys. J. 88:4200–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Volkova, K. D., V. B. Kovalska, A. O. Balanda, R. J. Vermeij, V. Subramaniam, Y. L. Slominskii, and S. M. Yarmoluk. 2007. Cyanine dye-protein interactions: looking for fluorescent probes for amyloid structures. J. Biochem. Biophys. Methods. 5:722–733. [DOI] [PubMed] [Google Scholar]
- 16.Roberti, M. J., C. W. Bertoncini, R. Klement, E. A. Jares-Erijman, and T. M. Jovin. 2007. Fluorescence imaging of amyloid formation in living cells by a functional, tetracysteine-tagged α-synuclein. Nat. Methods. 4:345–351. [DOI] [PubMed] [Google Scholar]
- 17.Krishnan, R., and S. L. Lindquist. 2005. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature. 435:765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaylor, J., N. Bodner, S. Edridge, G. Yamin, D. P. Hong, and A. L. Fink. 2005. Characterization of oligomeric intermediates in α-synuclein fibrillation: FRET studies of Y125W/Y133F/Y136F α-synuclein. J. Mol. Biol. 353:357–372. [DOI] [PubMed] [Google Scholar]
- 19.Mukhopadhyay, S., R. Krishnan, E. A. Lemke, S. Lindquist, and A. A. Deniz. 2007. A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc. Natl. Acad. Sci. USA. 104:2649–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giese, A., B. Bader, J. Bieschke, G. Schaffar, S. Odoy, P. J. Kahle, C. Haass, and H. Kretzschmar. 2005. Single particle detection and characterization of synuclein co-aggregation. Biochem. Biophys. Res. Commun. 333:1202–1210. [DOI] [PubMed] [Google Scholar]
- 21.Allsop, D., L. Swanson, S. Moore, Y. Davies, A. York, O. M. El-Agnaf, and I. Soutar. 2001. Fluorescence anisotropy: a method for early detection of Alzheimer β−peptide (Aβ) aggregation. Biochem. Biophys. Res. Commun. 285:58–63. [DOI] [PubMed] [Google Scholar]
- 22.Padrick, S. B., and A. D. Miranker. 2002. Islet amyloid: phase partitioning and secondary nucleation are central to the mechanism of fibrillogenesis. Biochemistry. 41:4694–4703. [DOI] [PubMed] [Google Scholar]
- 23.Koo, B. W., and A. D. Miranker. 2005. Contribution of the intrinsic disulfide to the assembly mechanism of islet amyloid. Protein Sci. 14:231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mukhopadhyay, S., P. K. Nayak, J. B. Udgaonkar, and G. Krishnamoorthy. 2006. Characterization of the formation of amyloid protofibrils from barstar by mapping residue-specific fluorescence dynamics. J. Mol. Biol. 358:935–942. [DOI] [PubMed] [Google Scholar]
- 25.Luk, K. C., E. G. Hyde, J. Q. Trojanowski, and V. M. Lee. 2007. Sensitive fluorescence polarization technique for rapid screening of α-synuclein oligomerization/fibrillization inhibitors. Biochemistry. 46:12522–12529. [DOI] [PubMed] [Google Scholar]
- 26.Brand, L., and J. R. Gohlke. 1972. Fluorescence probes for structure. Annu. Rev. Biochem. 41:843–868. [DOI] [PubMed] [Google Scholar]
- 27.Slavik, J. 1982. Anilinonaphthalene sulfonate as a probe of membrane composition and function. Biochim. Biophys. Acta. 694:1–25. [DOI] [PubMed] [Google Scholar]
- 28.Condie, C. C., and S. C. Quay. 1983. Conformational studies of aqueous melittin. Characteristics of a fluorescent probe binding site. J. Biol. Chem. 258:8231–8234. [PubMed] [Google Scholar]
- 29.Horowitz, P. M., S. Hua, and D. L. Gibbons. 1995. Hydrophobic surfaces that are hidden in chaperonin Cpn60 can be exposed by formation of assembly-competent monomers or by ionic perturbation of the oligomer. J. Biol. Chem. 270:1535–1542. [PubMed] [Google Scholar]
- 30.LeVine 3rd, H. 1997. Stopped-flow kinetics reveal multiple phases of thioflavin T binding to Alzheimer β (1–40) amyloid fibrils. Arch. Biochem. Biophys. 342:306–316. [DOI] [PubMed] [Google Scholar]
- 31.Kayed, R., J. Bernhagen, N. Greenfield, K. Sweimeh, H. Brunner, W. Voelter, and A. Kapurniotu. 1999. Conformational transitions of islet amyloid polypeptide (IAPP) in amyloid formation in vitro. J. Mol. Biol. 287:781–796. [DOI] [PubMed] [Google Scholar]
- 32.Uversky, V. N., J. Li, and A. L. Fink. 2001. Evidence for a partially folded intermediate in α-synuclein fibril formation. J. Biol. Chem. 276:10737–10744. [DOI] [PubMed] [Google Scholar]
- 33.Hoyer, W., T. Antony, D. Cherny, G. Heim, T. M. Jovin, and V. Subramaniam. 2002. Dependence of α-synuclein aggregate morphology on solution conditions. J. Mol. Biol. 322:383–393. [DOI] [PubMed] [Google Scholar]
- 34.Baskakov, I. V., G. Legname, M. A. Baldwin, S. B. Prusiner, and F. E. Cohen. 2002. Pathway complexity of prion protein assembly into amyloid. J. Biol. Chem. 277:21140–21148. [DOI] [PubMed] [Google Scholar]
- 35.LeVine 3rd, H. 2002. 4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonate: report on non-β-sheet conformers of Alzheimer's peptide β(1–40). Arch. Biochem. Biophys. 404:106–115. [DOI] [PubMed] [Google Scholar]
- 36.Martins, S. M., A. Chapeaurouge, and S. T. Ferreira. 2003. Folding intermediates of the prion protein stabilized by hydrostatic pressure and low temperature. J. Biol. Chem. 278:50449–50455. [DOI] [PubMed] [Google Scholar]
- 37.Ahmad, M. F., T. Ramakrishna, B. Raman, and M. Rao Ch. 2006. Fibrillogenic and non-fibrillogenic ensembles of SDS-bound human α-synuclein. J. Mol. Biol. 364:1061–1072. [DOI] [PubMed] [Google Scholar]
- 38.Cordeiro, Y., L. M. Lima, M. P. Gomes, D. Foguel, and J. L. Silva. 2004. Modulation of prion protein oligomerization, aggregation, and β-sheet conversion by 4,4′-dianilino-1,1′-binaphthyl-5,5′-sulfonate (bis-ANS). J. Biol. Chem. 279:5346–5352. [DOI] [PubMed] [Google Scholar]
- 39.Ferrao-Gonzales, A. D., B. K. Robbs, V. H. Moreau, A. Ferreira, L. Juliano, A. P. Valente, F. C. Almeida, J. L. Silva, and D. Foguel. 2005. Controlling β-amyloid oligomerization by the use of naphthalene sulfonates: trapping low molecular weight oligomeric species. J. Biol. Chem. 280:34747–34754. [DOI] [PubMed] [Google Scholar]
- 40.Shults, C. W. 2006. Lewy bodies. Proc. Natl. Acad. Sci. USA. 103:1661–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weinreb, P. H., W. Zhen, A. W. Poon, K. A. Conway, and P. T. Lansbury, Jr. 1996. NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 35:13709–13715. [DOI] [PubMed] [Google Scholar]
- 42.Bertoncini, C. W., Y. S. Jung, C. O. Fernandez, W. Hoyer, C. Griesinger, T. M. Jovin, and M. Zweckstetter. 2005. Release of long-range tertiary interactions potentiates aggregation of natively unstructured α-synuclein. Proc. Natl. Acad. Sci. USA. 102:1430–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Farris, F. J., G. Weber, C. C. Chiang, and I. C. Paul. 1978. Preparation, crystalline structure, and spectral properties of the fluorescent probe 4,4′-bis-1-phenylamino-8-naphthalenesulfonate. J. Am. Chem. Soc. 100:4469–4474. [Google Scholar]
- 44.Haugland, R. P. 2002. Probes for lipids and membranes. In Handbook of Fluorescent Probes and Research Products, 9th ed. J. Gregory, editor. Molecular Probes, Eugene, OR. 503–542.
- 45.Fernandez, C. O., W. Hoyer, M. Zweckstetter, E. A. Jares-Erijman, V. Subramaniam, C. Griesinger, and T. M. Jovin. 2004. NMR of α-synuclein-polyamine complexes elucidates the mechanism and kinetics of induced aggregation. EMBO J. 23:2039–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lidke, K. A., B. Rieger, D. S. Lidke, and T. M. Jovin. 2005. The role of photon statistics in fluorescence anisotropy imaging. IEEE Trans. Image Process. 14:1237–1245. [DOI] [PubMed] [Google Scholar]
- 47.Matulis, D., and R. Lovrien. 1998. 1-anilino-8-naphthalene sulfonate anion-protein binding depends primarily on ion pair formation. Biophys. J. 74:422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gasymov, O. K., and B. J. Glasgow. 2007. ANS fluorescence: potential to augment the identification of the external binding sites of proteins. Biochim. Biophys. Acta. 1774:403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gafni, A., R. P. DeToma, R. E. Manrow, and L. Brand. 1977. Nanosecond decay studies of a fluorescence probe bound to apomyoglobin. Biophys. J. 17:155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Uversky, V. N., S. Winter, and G. Lober. 1996. Use of fluorescence decay times of 8-ANS-protein complexes to study the conformational transitions in proteins which unfold through the molten globule state. Biophys. Chem. 60:79–88. [DOI] [PubMed] [Google Scholar]
- 51.Heise, H., W. Hoyer, S. Becker, O. C. Andronesi, D. Riedel, and M. Baldus. 2005. Molecular-level secondary structure, polymorphism, and dynamics of full-length α-synuclein fibrils studied by solid-state NMR. Proc. Natl. Acad. Sci. USA. 102:15871–15876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foguel, D., M. C. Suarez, A. D. Ferrao-Gonzales, T. C. Porto, L. Palmieri, C. M. Einsiedler, L. R. Andrade, H. A. Lashuel, P. T. Lansbury, J. W. Kelly, and J. L. Silva. 2003. Dissociation of amyloid fibrils of α-synuclein and transthyretin by pressure reveals their reversible nature and the formation of water-excluded cavities. Proc. Natl. Acad. Sci. USA. 100:9831–9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brand, L., and J. R. Gohlke. 1971. Nanosecond time-resolved fluorescence spectra of a protein-dye complex. J. Biol. Chem. 246:2317–2319. [PubMed] [Google Scholar]
- 54.Kodali, R., and R. Wetzel. 2007. Polymorphism in the intermediates and products of amyloid assembly. Curr. Opin. Struct. Biol. 17:48–57. [DOI] [PubMed] [Google Scholar]
- 55.Salemme, F. R. 1983. Structural properties of protein β-sheets. Prog. Biophys. Mol. Biol. 42:95–133. [DOI] [PubMed] [Google Scholar]
- 56.Fu, X., X. Zhang, and Z. Chang. 2005. 4,4′-dianilino-1,1′-binaphthyl-5,5′-sulfonate, a novel molecule having chaperone-like activity. Biochem. Biophys. Res. Commun. 329:1087–1093. [DOI] [PubMed] [Google Scholar]
- 57.Horowitz, P., V. Prasad, and R. F. Luduena. 1984. Bis(1,8-anilinonaphthalenesulfonate). A novel and potent inhibitor of microtubule assembly. J. Biol. Chem. 259:14647–14650. [PubMed] [Google Scholar]
- 58.Teschke, C. M., J. King, and P. E. Prevelige, Jr. 1993. Inhibition of viral capsid assembly by 1,1′-bi(4-anilinonaphthalene-5-sulfonic acid). Biochemistry. 32:10658–10665. [DOI] [PubMed] [Google Scholar]
- 59.Bonafe, C. F., M. Glaser, E. W. Voss, G. Weber, and J. L. Silva. 2000. Virus inactivation by anilinonaphthalene sulfonate compounds and comparison with other ligands. Biochem. Biophys. Res. Commun. 275:955–961. [DOI] [PubMed] [Google Scholar]
- 60.Thirunavukkuarasu, S., E. A. Jares-Erijman, and T. M. Jovin. 2008. Multiparametric fluorescence detection of early stages in the amyloid protein aggregation of pyrene-labeled α-synuclein. J. Mol. Biol. 378:1064–1073. [DOI] [PubMed] [Google Scholar]