

Abstract
The previously identified membrane-active regions of the hepatitis C virus (HCV) E1 and E2 envelope glycoproteins led us to identify different segments that might be implicated in viral membrane fusion, membrane interaction, and/or protein-protein binding. HCV E2 glycoprotein contains one of the most membranotropic segments, segment 603–634, which has been implicated in CD81 binding, E1/E2 and E2/E2 dimerization, and membrane interaction. Through a series of complementary experiments, we have carried out a study of the binding and interaction with the lipid bilayer of a peptide corresponding to segment 603–634, peptide E2FP, as well as the structural changes induced by membrane binding that take place in both the peptide and the phospholipid molecules. Here, we demonstrate that peptide E2FP binds to and interacts with phospholipid model membranes, modulates the polymorphic phase behavior of membrane phospholipids, is localized in a shallow position in the membrane, and is probably oligomerized in the presence of membranes. These data support the role of E2FP in HCV-mediated membrane fusion, and sustain the notion that this segment of the E2 envelope glycoprotein, together with other segments of E2 and E1 glycoproteins, provides the driving force for the merging of the viral and target cell membranes.
INTRODUCTION
Hepatitis C virus (HCV), an enveloped positive single-stranded RNA virus of the Flaviviridae family, has an important impact on public health since it is the leading cause of acute and chronic liver disease in humans, including chronic hepatitis, cirrhosis, and hepatocellular carcinoma (1–3). At the moment, there exists no vaccine to prevent HCV infection, and current therapeutic agents have limited success against HCV (4). The HCV genome consists of one translational open reading frame encoding a polyprotein precursor of ∼3010 amino acids in length, including structural and nonstructural proteins, that is cleaved by host and viral proteases (5). The polyprotein precursor is cotranslationally and posttranslationally processed by both cellular and viral proteases at the level of the endoplasmic reticulum membrane to yield 10 mature structural and nonstructural proteins. The structural proteins include the core, which forms the viral nucleocapsid, and the envelope transmembrane glycoproteins E1 and E2. The structural proteins are separated from the nonstructural proteins by the short membrane protein p7. HCV entry into the cell is achieved by the fusion of viral and cellular membranes, and morphogenesis and budding has been suggested to take place in the endoplasmic reticulum (6). Therefore, the protein regions implicated in fusion and/or budding must interact with the biological membrane and should be conservative membranotropic sequences. The variability of the HCV proteins gives the virus the ability to escape the host's immune surveillance system and the development of a vaccine proves to be a difficult task (7). Furthermore, HCV proteins are very sensitive to folding, assembly, mutations, and deletions. Finding protein-membrane and protein-protein interaction inhibitors could be a good strategy against HCV infection, since they might prove to be potential therapeutic agents.
The viral structure of HCV is thought to adopt a classical icosahedral scaffold in which the E1 and E2 envelope glycoproteins are anchored to the host-cell-derived double-layer lipid envelope. Both of them are essential for host-cell entry, binding to receptor(s), and fusion with the host cell membrane, as well as in viral particle assembly (8). E1 and E2 are type I transmembrane glycoproteins, with an N-terminal ectodomain and a short C-terminal transmembrane domain. They interact with each other and assemble as noncovalent heterodimers. It is significant that their transmembrane domains play a major role in the E1/E2 heterodimer formation, membrane anchoring, and endoplasmic reticulum retention (9–12). The HCV envelope glycoproteins E1 and E2 are included in the class II fusion proteins, because the putative fusion peptide is localized in an internal sequence linked by antiparallel β-sheets; moreover, proteomics computational analyses suggest that HCV envelope glycoprotein E1 is a truncated class II fusion protein (13). The HCV membrane fusion process is pH-dependent, and low endosomal pH promotes the arrangement of E1/E2 to its active form (14). Many aspects regarding the function and properties of both HCV E1 and E2 glycoproteins still remain unresolved; even so, the location of the fusion peptide is controversial, since several data suggest that it could be located in either E1 or E2. Although previous data have proposed a direct role of HCV E1 in membrane fusion, whereas HCV E2 should mediate the binding with receptor CD81, and should also be responsible for heterodimerization with E1, recent data suggest that indeed both E1 and E2 glycoproteins participate in the membrane fusion mechanism (14–18).
We have recently identified the membrane-active regions of the HCV E1 and E2 glycoproteins by observing the effect of E1 and E2 glycoprotein-derived peptide libraries on model membrane integrity, and have found different segments that present high positive values of hydrophobic moment, hydrophobicity, and interfaciality (19). These results permit us to suggest the possible location of different segments in these proteins that might be implicated in protein-lipid and protein-protein interactions, helping us to understand the processes that give rise to the interaction between the different proteins and the membrane. The most membranotropic regions of E2 are made up of segments comprising residues 455–489, which include the hypervariable HVR-2 region, 525–565 and 603–634 (19). Region 603–634 shows a very high hydrophobicity and interfaciality, as well as significant leakage, hemifusion, and fusion effects in membrane model systems (19). Of significance is the demonstration, using infectious HCV pseudoparticles, that mutations in this region abolished infectivity and membrane fusion, demonstrating that this segment of the E2 glycoprotein participates in the viral fusion process (15). It is known that the mechanism by which proteins facilitate the formation of fusion intermediates is a complex process involving several segments of fusion proteins (20,21), and at the same time, there are still many questions to be answered regarding the E1 and E2 mode of action in HCV and cell host membrane fusion. Since the 603–634 region of the HCV E2 envelope glycoprotein might be involved in membrane destabilization and at the same time take part in the fusion events as a helper for the fusion peptide and/or the pretransmembrane regions, as well as other segments, we have made an in-depth study of the 603–634 HCV E2 region, peptide E2FP, patterned after peptides 603-LTPRCLVDYPYRLWHYPC-620, 610-DYPYRLWHYPCTLNFSIF-627, and 617-HYPCTLNFSIFKVRMYVG-634 (peptides 34–36 from the original HCV E1/E2 peptide library, respectively (19)). We have studied the binding and interaction of E2FP with membrane model systems, as well as the structural changes that take place in both the peptide and phospholipid molecules induced by membrane binding through a series of complementary experiments. In this work, we show that peptide E2FP strongly partitions and buries into phospholipid membranes, interacts with negatively charged phospholipids, and modulates the phase polymorphic behavior of model membranes.
MATERIALS AND METHODS
Materials and reagents
The peptide E2FP, corresponding to the sequence 603-LTPRCLVDYPYRLWHYPCTLNFSIFKVRMYVG-634 from HCV strain 1B4J (with N-terminal acetylation and C-terminal amidation) was obtained from Genemed Synthesis, San Francisco, CA. The peptide E2FP was purified by reverse-phase high-performance liquid chromatography (C-8 column (Vydac, Hesperia, CA), 250 × 4.6 mm, flow rate 1 ml/min; solvent A, 0.1% trifluoroacetic acid; solvent B, 99.9% acetonitrile, and 0.1% trifluoroacetic acid) to >95% purity, and its composition and molecular mass were confirmed by amino acid analysis and mass spectroscopy. Since trifluoroacetate has a strong infrared absorbance at ∼1673 cm−1, which interferes with the characterization of the peptide amide I band (22), residual trifluoroacetic acid, used both in peptide synthesis and in the high-performance liquid chromatography mobile phase, was removed by several lyophilization/solubilization cycles in 10 mM HCl (23). Egg L-α-phosphatidylcholine (EPC), egg L-α-phosphatidic acid (EPA), egg sphingomyelin (SM), bovine brain phosphatidylserine (BPS), egg transsterified L-α-phosphatidylethanolamine (TPE), lyso-α-phosphatidylcholine (LPC), 1,2-dimyristoylphosphatidylcholine (DMPC), 1,2-dimyristoylphosphatidylglycerol (DMPG), 1,2-dimyristoylphosphatidylserine, 1,2-dimyristoylphosphatidic acid (DMPA), 1,2-dielaidoyl-sn-glycero-3-phosphatidylethanolamine (DEPE), 1-palmitoyl, 2-oleoyl-sn-glycero-3-phosphatidylethanolamine, liver lipid extract, cholesterol (Chol), Lissamine rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, and N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphatidylethanolamine (NBD-PE) were obtained from Avanti Polar Lipids (Alabaster, AL). 5-Carboxyfluorescein (CF) (>95% by high-performance liquid chromatography), 5-doxyl-stearic acid (5NS), 16-doxyl-stearic acid (16NS), dehydroergosterol (ergosta-5,7,9(11),22-tetraen-3β-ol), sodium dithionite, deuterium oxide (99.9% by atom), Triton X-100, EDTA, and HEPES were purchased from Sigma-Aldrich (Madrid, Spain). 1,6-Diphenyl-1,3,5-hexatriene (DPH), 1-(4-trimethylammoniumphenyl)-6-phenyl-1,3,5-hexatriene (TMA-DPH), and 4-(2-(6-(dioctylamino)-2-naphthalenyl) (ethenyl)-1-(3-sulfopropyl)-pyridinium inner salt (di-8-ANEPPS) were obtained from Molecular Probes (Eugene, OR). All other reagents used were of analytical grade from Sigma-Aldrich. Water was deionized, twice distilled, and passed through a Milli-Q equipment (Millipore Ibérica, Madrid, Spain) to a resistivity better than 18 MΩ cm.
Vesicle preparation
Aliquots containing the appropriate amount of lipid in chloroform/methanol (2:1, v/v) were placed in a test tube, the solvents removed by evaporation under a stream of O2-free nitrogen, and finally, traces of solvents were eliminated under vacuum in the dark for >3 h. The lipid films were resuspended in an appropriate buffer and incubated either at 25°C or 10°C above the gel-to-liquid-crystal-phase transition temperature (Tm) with intermittent vortexing for 30 min to hydrate the samples and obtain multilamellar vesicles (MLV). The samples were frozen and thawed five times to ensure complete homogenization and maximization of peptide/lipid contacts, with occasional vortexing. Large unilamellar vesicles (LUV) with mean diameter 0.1 and 0.2 μm for leakage or hemifusion and fusion experiments, respectively, were prepared from multilamellar vesicles by the extrusion method (24), using polycarbonate filters with a pore size of 0.1 and 0.2 μm (Nuclepore Corp., Cambridge, CA). The phospholipid and peptide concentration were measured by methods described previously (25,26).
Membrane leakage measurement
LUVs with a mean diameter of 0.1 μm were prepared as indicated above in buffer containing 10 mM Tris, 20 mM NaCl, pH 7.4, and CF at a concentration of 40 mM. Nonencapsulated CF was separated from the vesicle suspension through a Sephadex G-75 filtration column (Pharmacia, Uppsala, Sweden) eluted with buffer containing 10 mM TRIS, 100 mM NaCl, 1 mM EDTA, pH 7.4. Membrane rupture (leakage) of intraliposomal CF was assayed by treating the probe-loaded liposomes (final lipid concentration, 0.125 mM) with the appropriate amounts of peptide using a 5 × 5 mm fluorescence cuvette on a Cary Eclipse spectrofluorometer (Varian, San Carlos, CA), stabilized at 25°C with the appropriate amounts of peptide, each well containing a final volume of 400 μl. The medium in the cuvettes was continuously stirred to allow the rapid mixing of peptide and vesicles. Leakage was assayed until no more change in fluorescence was obtained. The fluorescence was measured using a Varian Cary Eclipse spectrofluorometer . Changes in fluorescence intensity were recorded with excitation and emission wavelengths set at 492 and 517 nm, respectively. Excitation and emission slits were set at 5 nm. One hundred percent release was achieved by adding Triton X-100 to the cuvette to a final concentration of 0.5% (w/w). Fluorescence measurements were made initially with probe-loaded liposomes, afterward by adding peptide solution, and finally by adding Triton X-100 to obtain 100% leakage. Leakage was quantified on a percentage basis according to the equation, 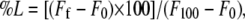 where Ff is the equilibrium value of fluorescence after peptide addition, F0 is the initial fluorescence of the vesicle suspension, and F100 is the fluorescence value after the addition of Triton X-100.
where Ff is the equilibrium value of fluorescence after peptide addition, F0 is the initial fluorescence of the vesicle suspension, and F100 is the fluorescence value after the addition of Triton X-100.
Phospholipid-mixing measurement
Peptide-induced vesicle lipid mixing was measured by resonance energy transfer (27). This assay is based on the decrease in resonance energy transfer between two probes (NBD-PE and Lissamine rhodamine B 1,2-dihexadecanoyl-sn-glycero-3- phosphoethanolamine) when the lipids of the probe-containing vesicles are allowed to mix with lipids from vesicles lacking the probes. The concentration of each of the fluorescent probes within the liposome membrane was 0.6% mol. LUVs with a mean diameter of 0.2 μm were prepared as described above. Labeled and unlabeled vesicles in the proportion 1:4 were placed in a 5 × 5-mm fluorescence cuvette at a final lipid concentration of 100 μM in a final volume of 400 μl, stabilized at 25°C under constant stirring. The fluorescence was measured using a Varian Cary Eclipse fluorescence spectrometer using 467 nm and 530 nm for excitation and emission, respectively. Excitation and emission slits were set at 10 nm. Since labeled/unlabeled vesicles were mixed in a proportion of 1:4, 100% phospholipid mixing was estimated with a liposome preparation in which the membrane concentration of each probe was 0.12%. Phospholipid mixing was quantified on a percentage basis according to the equation, 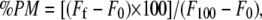 where Ff is the value of fluorescence obtained 15 min after peptide addition to a liposome mixture containing liposomes with 0.6% of each probe plus liposomes without any fluorescent probe, F0 is the initial fluorescence of the vesicles, and F100 is the fluorescence value of the liposomes containing 0.12% of each probe.
where Ff is the value of fluorescence obtained 15 min after peptide addition to a liposome mixture containing liposomes with 0.6% of each probe plus liposomes without any fluorescent probe, F0 is the initial fluorescence of the vesicles, and F100 is the fluorescence value of the liposomes containing 0.12% of each probe.
Inner-monolayer phospholipid-mixing (fusion) measurement
Peptide-induced phospholipid-mixing of the inner monolayer was measured by a modification of the phospholipid-mixing measurement stated above (28). This assay is based on the decrease in resonance energy transfer between two probes (NBD-PE and Lissamine rhodamine B 1,2-dihexadecanoyl-sn-glycero-3- phosphoethanolamine) when the lipids of the probe-containing vesicles are allowed to mix with lipids from vesicles lacking the probes. The concentration of each of the fluorescent probes within the liposome membrane was 0.6 mol %. LUVs with a mean diameter of 0.2 μm were prepared as described above. LUVs were treated with sodium dithionite to completely reduce the NBD-labeled phospholipid located at the outer monolayer of the membrane. Final concentration of sodium dithionite was 100 mM (from a stock solution of 1 M dithionite in 1 M TRIS, pH 10.0) and incubated for ∼1 h on ice in the dark. Sodium dithionite was then removed by size-exclusion chromatography through a Sephadex G-75 filtration column (Pharmacia) eluted with buffer containing 10 mM TRIS, 100 mM NaCl, 1 mM EDTA, pH 7.4. The proportion of labeled and unlabeled vesicles, lipid concentration, and other experimental and measurement conditions were the same as indicated above for the phospholipid mixing assay.
Peptide binding to vesicles
The partitioning of the peptide into the phospholipid bilayer was monitored by the fluorescence enhancement of tryptophan. Fluorescence spectra were recorded in an SLM Aminco 8000C spectrofluorometer with excitation and emission wavelengths of 290 and 348 nm, respectively, and 4-nm spectral bandwidths. Measurements were carried out in 20 mM HEPES, 50 mM NaCl, EDTA 0.1 mM, pH 7.4. Intensity values were corrected for dilution, and the scatter contribution was derived from lipid titration of a vesicle blank. Partitioning coefficients were obtained using (29)
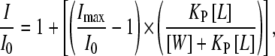 |
where I and I0 are the final and the initial intensities respectively, kP is a mole fraction partition coefficient that represents the amount of peptide in the bilayers as a fraction of the total peptide present in the system, Imax is a variable value for the fluorescence enhancement at complete partitioning determined by fitting the equation to the experimental data, [L] is the lipid concentration, and [W] is the concentration of water (55.3 M). The peptide concentration in the assays was 30 μM.
Steady-state fluorescence anisotropy
MLVs were formed in 100 mM NaCl, 0.05 mM EDTA, 25 mM HEPES, pH 7.4. Aliquots of TMA-DPH or DPH in N,N′-dimethylformamide (2 × 10−4 M) were directly added into the lipid dispersion to obtain a probe/lipid molar ratio of 1:500. Samples were incubated for 15 or 60 min for TMA-DPH and DPH, respectively, 10°C above the Tm of the phospholipid mixture. Afterward, the peptides were added to obtain a peptide/lipid molar ratio of 1:15 and incubated at 10°C above the Tm of each lipid for 1 h, with occasional vortexing. All fluorescence studies were carried using 5 × 5-mm quartz cuvettes in a final volume of 400 μl (315 μM lipid concentration). All the data were corrected for background intensities and progressive dilution. The steady-state fluorescence anisotropy, 〈r〉, was measured with an automated polarization accessory using a Varian Cary Eclipse fluorescence spectrometer, coupled to a Peltier device (Varian) for automatic temperature change. The vertically and horizontally polarized emission intensities, elicited by vertically polarized excitation, were corrected for background scattering by subtracting the corresponding polarized intensities of a phospholipid preparation lacking probes. The G-factor, accounting for differential polarization sensitivity, was determined by measuring the polarized components of the fluorescence of the probe with horizontally polarized excitation (G = IHV/IHH). Samples were excited at 360 nm (slit width, 5 nm) and fluorescence emission was recorded at 430 nm (slit width, 5 nm). The values were calculated from the equation (30). The steady-state anisotropy was defined by equation 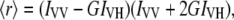 where IVV and IVH are the measured fluorescence intensities (after appropriate background subtraction) with the excitation polarizer vertically oriented and the emission polarizer vertically and horizontally oriented, respectively.
where IVV and IVH are the measured fluorescence intensities (after appropriate background subtraction) with the excitation polarizer vertically oriented and the emission polarizer vertically and horizontally oriented, respectively.
Fluorescence quenching of Trp emission by water-soluble and lipophylic probes
For acrylamide quenching assays, aliquots from a 4 M solution of the water-soluble quencher were added to the solution-containing peptide in the presence and absence of liposomes at a peptide/lipid molar ratio of 1:100. The results obtained were corrected for dilution and the scatter contribution was derived from acrylamide titration of a vesicle blank. The data were analyzed according to the Stern-Volmer equation (31), 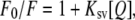 where I0 and I represent the fluorescence intensities in the absence and presence of the quencher [Q], respectively, and Ksv is the Stern-Volmer quenching constant, which is a measure of the accessibility of Trp to acrylamide. Quenching studies with lipophylic probes were performed by successive addition of small amounts of 5NS or 16NS in ethanol to the samples of the peptide incubated with LUV. The final concentration of ethanol was kept below 2.5% (v/v) to avoid any significant bilayer alterations. After each addition, there was an incubation period of 15 min before the measurement. The effective quencher concentration in the membrane, [Q]L, was calculated using the relationship (32)
where I0 and I represent the fluorescence intensities in the absence and presence of the quencher [Q], respectively, and Ksv is the Stern-Volmer quenching constant, which is a measure of the accessibility of Trp to acrylamide. Quenching studies with lipophylic probes were performed by successive addition of small amounts of 5NS or 16NS in ethanol to the samples of the peptide incubated with LUV. The final concentration of ethanol was kept below 2.5% (v/v) to avoid any significant bilayer alterations. After each addition, there was an incubation period of 15 min before the measurement. The effective quencher concentration in the membrane, [Q]L, was calculated using the relationship (32)
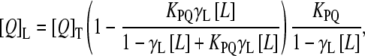 |
where γL is the phospholipid molar volume, [Q]T is the total quencher concentration, [L] is the lipid molar concentration, and KPQ is the partition coefficient of the quencher. The KPQ values for 5NS and 16NS were as reported previously (33). The fluorescence data were analyzed using a direct fit according to the relationship (34)
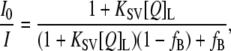 |
where I0 is the fluorescence intensity in the absence of quencher, KSV is the Stern-Volmer quenching constant, and fB = I0,B/I0, where I0,B is the fluorescence intensity of the fluorophore population accessible to the quencher. The excitation and emission wavelengths were 290 and 348 nm, respectively.
Fluorescence measurements using FPE-labeled membranes
LUVs with a mean diameter of 0.1 μm were prepared in buffer containing 10 mM TRIS-HCl, pH 7.4. The vesicles were labeled exclusively in the outer bilayer leaflet with fluoresceinphosphatidylethanolamine (FPE), as described previously (35). Briefly, LUVs were incubated with 0.1 mol % FPE dissolved in ethanol (never more than 0.1% of the total aqueous volume) at 37°C for 1 h in the dark. Any remaining unincorporated FPE was removed by gel filtration on a Sephadex G-25 column equilibrated with the appropriate buffer. FPE-vesicles were stored at 4°C until use in an oxygen-free atmosphere. Fluorescence time courses of FPE-labeled vesicles were measured after the desired amount of peptide was added into 400 μl of lipid suspensions (200 μM lipid) using a Varian Cary Eclipse fluorescence spectrometer. Excitation and emission wavelengths were set at 490 and 520 nm, respectively, using excitation and emission slits set at 5 nm. Temperature was controlled with a thermostatic bath at 25°C. The contribution of light scattering to the fluorescence signals was measured in experiments without the dye and was subtracted from the fluorescence traces. Data were fitted to a hyperbolic binding model (36) using the equation 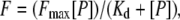 where F is the fluorescence variation, Fmax the maximum fluorescence variation, [P] the peptide concentration, and Kd the dissociation constant of the membrane binding process.
where F is the fluorescence variation, Fmax the maximum fluorescence variation, [P] the peptide concentration, and Kd the dissociation constant of the membrane binding process.
Measurement of the membrane dipole potential using di-8-ANEPPS-labeled membranes
Aliquots containing the appropriate amount of lipid in chloroform-methanol (2:1 v/v) and di-8-ANEPPS were placed in a test tube to obtain a probe/lipid molar ratio of 1:100 and LUVs, with a mean diameter of 90 nm, were prepared as described previously. Steady-state fluorescence measurements were recorded with a Varian Cary Eclipse spectrofluorometer. Dual wavelength recordings with the dye di-8-ANEPPS were obtained by exciting the samples at two different wavelengths (450 and 520 nm) and measuring their intensity ratio, R450/520, at an emission wavelength of 620 nm (37,38). By exciting the membrane suspensions at two different wavelengths corresponding to the maximum and the minimum of the difference spectrum, a fluorescence intensity ratio R can be calculated, which can be used as a measure of the relative changes in the magnitude of the dipole potential. The fluorescence ratio R is defined as the ratio of the fluorescence intensity at an excitation wavelength of 450 nm divided by the intensity at 520 nm. The lipid concentration was 200 μM, and all experiments were performed at 25°C.
Infrared spectroscopy
For IR spectroscopy, the samples were prepared as above but in D2O buffer; ∼25 μl of a pelleted sample in D2O was placed between two CaF2 windows separated by 50-μm thick Teflon spacers in a liquid demountable cell (Harrick, Ossining, NY). The spectra were obtained in a Bruker IFS55 spectrometer (Bruker Biospin, Rheinstetten, Germany) using a deuterated triglycine sulfate detector. Each spectrum was obtained by collecting 200 interferograms with a nominal resolution of 2 cm−1, transformed using triangular apodization, and a sample shuttle accessory was used to obtain sample and background spectra to average background spectra between sample spectra over the same time period. The spectrometer was continuously purged with dry air at a dew point of −40°C. All samples were equilibrated at the lowest temperature for 20 min before acquisition. An external bath circulator, connected to the infrared spectrometer, controlled the sample temperature. For temperature studies, samples were scanned using 2°C intervals with a 2-min delay between consecutive scans. Subtraction of buffer spectra taken at the same temperature as the samples, center-of-gravity frequencies, and band-narrowing methods were performed interactively using either GRAMS/32 or Spectra-Calc (Galactic Industries, Salem, MA), as described previously (39,40).
Differential scanning calorimetry
MLVs were formed as described above in 100 mM NaCl, 0.05 mM EDTA and 25mM HEPES, pH 7.4. The peptides were added to obtain a peptide/lipid molar ratio of 1:15. The final volume was 1.2 ml (500 μM lipid concentration), and was incubated 10°C above the Tm of each phospholipid for 1 h, with occasional vortexing. Samples were degassed under vacuum for 10–15 min with gentle stirring, before being loaded into the calorimetric cell. Differential scanning calorimetry (DSC) experiments were performed in a VP-DSC differential scanning calorimeter (MicroCal) under a constant external pressure of 30 psi to avoid bubble formation, and samples were heated at a constant scan rate of 60°C/h. Experimental data were corrected from small mismatches between the two cells by subtracting a buffer baseline before data analysis. The excess heat capacity functions were analyzed using Origin 7.0 (Microcal). The errors in determination of Tc and ΔH were 0.2°C and 0.5 kJ/mol, respectively. The thermograms were defined by the onset and completion temperatures of the transition peaks obtained from heating scans. To avoid artifacts due to the thermal history of the sample, the first scan was never considered; second and further scans were carried out until a reproducible and reversible pattern was obtained.
31P NMR
Samples were prepared as described above at a lipid/peptide molar ratio of 50:1 and concentrated by centrifugation (14,000 rpm for 15 min). 31P NMR spectra were recorded at different temperatures in the Fourier transform mode in a Bruker Avance 500 MHz NMR (Bruker BioSpin) spectrometer operating at a resonance frequency of 202.38 MHz for 31P-nuclei and using a 5 mm probe BBO BB-1H. Probe temperature was maintained at ±0.2°C by a Bruker BVT 3000 variable digital temperature unit. Measurements, spectra acquisition, and calibration were essentially performed as previously described (41). Typical acquisition parameters: spectral width of 30 kHz in 16 K data points, π/2 pulse widths of typically 6.5 μs, and relaxation delays of 5 s. Typically, 1600 scans were recorded with deuterium (D2O) lock.
Small-angle x-ray scattering experiments
MLVs at a concentration of 5% (w/w) prepared without or with the peptide at a lipid/peptide molar ratio of 50:1 were prepared as stated above and submitted to 15 temperature cycles (heating at 45°C and cooling at –20°C). Small-angle x-ray diffraction (SAXD) measurements were carried out using a Hecus SWAX-camera (Hecus X-ray Systems, Graz, Austria), as described previously (42), with Ni-filtered Cu-Ka radiation (λ = 1542 Å) originating from a sealed-tube x-ray generator (GE-Seifert, Ahrensburg, Germany) operating at a power of 2 kW (50 kV, 4 mA). Sample-to-detector distance was 27.8 cm. A linear-position-sensitive detector was used with 1024-channel resolution. SAXD angle calibration was done with silver stearate. The measurements were performed with the sample placed in a thin-walled 1-mm diameter quartz capillary held in a steel cuvette holder at different temperatures with an exposure time of 1 h. The SAXD curves were analyzed after background subtraction and normalization in terms of a full q-range model using the program GAP (43).
RESULTS
The HCV E1 envelope glycoprotein is thought to be responsible for the membrane fusion process, whereas the HCV E2 envelope glycoprotein is thought to mediate the binding to the host cell, although other roles could not be ruled out (14,15,44). It is interesting that several hydrophobic patches have been identified in the E2 protein that might be important for modulating membrane binding and interaction (17,19). In Fig. 1, we present the analysis of the hydrophobic moment, hydrophobicity, and interfaciality distribution along the E2 envelope glycoprotein sequence of the HCV_1B4J strain, assuming it forms an α-helical wheel along the whole sequence (19). Although the E1 and E2 HCV envelope glycoproteins are supposed to be class II membrane fusion proteins and therefore their α-helix content should not be as high as that of class I membrane fusion proteins, these data give us a depiction of the potential surface zones that could be implicated in the modulation of membrane binding. As we have shown previously, the sequence comprising residues 603–634 is one of the most membranotropic regions of HCV E2 (19). Furthermore, it has been shown recently that this segment participates in the viral fusion process (15). Because of that, we present here the results of the study of the interaction with model membranes of a peptide derived from this region, the E2FP peptide, comprising residues 603–634 (see Fig. 1). Peptide E2FP has been patterned after peptides 34 (603-LTPRCLVDYPYRLWHYPC-620), 35 (610-DYPYRLWHYPCTLNFSIF-627), and 36 (617-HYPCTLNFSIFKVRMYVG-634), from the original HCV E2 peptide library (19).
FIGURE 1.

Hydrophobic moment (HM), hydrophobicity (H), and interfaciality (I) distributions for HCV E2 envelope glycoprotein (19), assuming it forms an α-helical wheel (71). Only positive bilayer-to-water transfer free-energy values are shown (the darker, the greater). The residue numbers are indicated at the left. The sequence of the E2FP peptide studied in this work is shown and highlighted in the figure with dotted lines.
The ability of the E2FP peptide to interact with membranes was determined from the increase in fluorescence emission intensity of its Trp residues in the presence of model membranes (45) containing different phospholipid compositions at different lipid/peptide ratios (Fig. 2 A). It should be recalled that HCV pseudoparticle membrane fusion does not require any protein or receptor at the membrane surface, and it has been shown as well that the presence of Chol enhances it (14). The fluorescence emission of the peptide in buffer had a maximum at 344 nm, typical for Trp in a polar environment, whereas in the presence of liposomes it decreased to 333 nm, implying binding of the peptide to the membrane bilayer (a low-polarity environment was sensed by Trp). These results were further corroborated when the Trp fluorescence intensity of the E2FP peptide increased when the lipid/peptide ratio was increased, indicating a significant change in the environment of the Trp moiety of the peptide (Fig. 2 A). From fitting, Kp values in the range 105–106 were obtained for different phospholipid compositions (Table 1), indicating that the peptide was bound to the membrane surface with very high affinity (39,45–47). Slightly higher Kp values were obtained for negatively charged phospholipid-containing bilayers; the E2FP peptide has a positive net formal charge, so that an electrostatic effect might be the reason to observe higher-Kp-value compositions containing negatively charged phospholipids. Furthermore, in the presence of model membranes, the anisotropy values of the peptide Trp increased upon increasing the lipid/peptide ratio, with the limiting value being ∼0.125 for liver membranes (not shown), indicating a significant motional restriction of the Trp moieties of the peptide at a relatively high lipid/protein ratio (30). We have also used the electrostatic surface potential probe FPE (48) to monitor the binding of the E2FP peptide to model membranes composed of different lipid compositions at different lipid/peptide ratios (not shown). Supporting the previous data, E2FP had a high affinity for model membranes, higher for liposomes composed of the liver lipid extract than for the others. Changes in the membrane dipole potential magnitude elicited by E2FP were monitored by means of the spectral shift of the fluorescence probe di-8-ANEPPS (37,49,50). The variation of the fluorescence intensity ratio R450/520 normalized as a function of the peptide concentration for different membrane compositions is shown in Fig. 2 B. In the presence of the peptide, the greater decrease in R450/520 value was measured in the presence of negatively charged lipid compositions, confirming again the presence of a specific interaction of the peptide with vesicles bearing negatively charged phospholipids.
FIGURE 2.

(A) Determination of the partition constant, Kp, of E2FP through the change of the intrinsic tryptophan fluorescence in the presence of increasing lipid concentrations. (B) Effect of E2FP on the membrane dipole potential monitored through the fluorescence ratio (R) of di-8-ANEPPS. (C) Stern-Volmer plots of the quenching of the Trp fluorescence emission of E2FP by acrylamide. (D) Depth-dependent quenching of the Trp fluorescence emission of E2FP by 5NS (solid symbols) and 16NS (open symbols) in LUVs. Peptide in buffer is represented in B as an open pentagon. LUVs were composed of EPC/Chol at a molar ratio of 5:1 (○), BPS/Chol at a molar ratio of 5:1 (⋄), EPG/Chol at a molar ratio of 5:1 (□), EPA/Chol at a molar ratio of 5:1 (▵) and liver extract lipids (∇). In C and D, the lipid/peptide ratio was 100:1, whereas the lipid concentration was 200 μM in B–D. Vertical bars indicate standard deviations of the mean of triplicate samples.
TABLE 1.
Partition coefficients, Stern-Volmer quenching constants, and maximal leakage, hemifusion, and fusion values at a phospholipid/peptide ratio of 15:1 for the E2FP peptide incorporated in LUVs of different compositions
| LUV compositions | KP (Trp) | Ksv (M−1) Acrylamide | Ksv (M−1) 5NS | Ksv (M−1) 16NS | Leakage (max %) | Hemifusion (max %) | Fusion (max %) |
|---|---|---|---|---|---|---|---|
| EPG/Chol, 5:1 | 4.85 × 105 | 1.124 | 4.44435 | 3.119 | 100 | 100 | 59 |
| EPA/Chol, 5:1 | 1.24 × 106 | 2.006 | 2.69487 | 1.708 | 100 | 100 | 55 |
| BPS/Chol, 5:1 | 8.6 × 105 | 3.826 | 5.65617 | 1.979 | 100 | 100 | 56 |
| EPC/Chol, 5:1 | 2.49 × 105 | 3.418 | 2.16296 | 0.550 | 78 | 69 | 50 |
| Liver extract | 2.57 × 105 | 2.782 | 3.84889 | 0.131 | 46 | 46 | 40 |
| E2FP in buffer | — | 5.456 | — | — | — | — | — |
We also studied the accessibility of the Trp residue of membrane-bound E2FP peptide toward acrylamide, a neutral, water-soluble, highly efficient quenching molecule. The quenching data is presented in Fig. 2 C and the lower KSV values obtained in the presence of lipids, compared with the measurements in their absence (Table 1), suggest that the E2FP peptide is buried in the membrane, becoming less accessible for quenching by acrylamide. Linear Stern-Volmer plots are indicative of the Trp residue being accessible to acrylamide, and in all cases, the quenching of the peptide Trp residues showed acrylamide-dependent concentration behavior. It is interesting that the KSV values in the presence of EPG- and EPA-containing vesicles were slightly lower than in the presence of the other vesicles, indicating that the peptide was slightly more buried inside the membrane in the presence of those phospholipids. The transverse location (penetration) of the E2FP peptide into the lipid bilayer was evaluated by monitoring the relative quenching of the fluorescence of the Trp residues by the lipophylic spin probes 5NS and 16NS when the peptide was incorporated into vesicles with different phospholipid compositions (Fig. 2 D and Table 1). It can be seen that, in general and for each one of the different membrane compositions studied, the E2FP peptide was quenched more efficiently by 5NS, a quencher for molecules near or at the interface, than by 16NS, a quencher for molecules buried deep in the membrane, suggesting that E2FP remained close to the lipid/water interface.
To further explore the effect of the E2FP peptide in the destabilization of membrane vesicles, we studied their effect on the release of encapsulated fluorophores in model membranes of various compositions. The extent of leakage observed at different peptide/lipid molar ratios and the effect on different phospholipid compositions is shown in Fig. 3 A (see also Table 1). The E2FP peptide induced the highest percentage of leakage (leakage values between 90 and 100%), even at lipid/peptide ratios as high as 15:1, for liposomes containing negatively charged phospholipids (Fig. 3 A). Lower, but significant, leakage values were obtained for liposomes composed of EPC alone, EPC plus Chol, EPC plus TPE, and EPC/SM/Chol at a molar ratio of 52:18:30, since at a lipid/peptide ratio of 30:1, leakage values >60% were observed. Liposomes composed of lipid liver extract and liposomes composed of EPC/SM/Chol at a molar ratio of 5:1:1 were the ones that elicited the lowest leakage values, i.e., ∼50% (Fig. 3 A). The induction of outer-monolayer lipid mixing (hemifusion) by the E2FP peptide was tested with several types of vesicles utilizing the probe dilution assay (27,28), and the results are shown in Fig. 3 B. The higher hemifusion values were found for liposomes containing negatively charged phospholipids (liposomes containing EPG, EPA, and BPS), as well as liposomes containing TPE, which showed high hemifusion values near 100% at a lipid/peptide molar ratio of 5:1. Lower hemifusion values were observed for liposomes containing either EPC or EPC plus Chol (∼78% leakage). Liposomes containing complex mixtures of lipids, i.e., the liver extract and EPC/SM/Chol, showed ∼40% leakage at the lowest lipid/peptide molar ratio used. It is interesting that liposomes containing LPC displayed the least leakage values (∼20%). Inner-monolayer lipid-mixing (fusion) results induced by the E2FP peptide in liposomes of different compositions are shown in Fig. 3 C. In contrast to the results shown above, the highest fusion values were found for the complex mixture containing EPC/SM/Chol at a molar ratio of 52:18:30 (leakage values of ∼70% at a lipid/peptide molar ratio of 5:1) Leakage values between 60% and 50% were found for liposomes containing negatively charged phospholipids, as well as for liposomes containing TPE, whereas leakage values between 50% and 40% were found for liposomes composed of EPC, either pure or with added Chol, EPC/SM/Chol at a molar ratio of 5:1:1, and liposomes composed of the liver lipid extract.
FIGURE 3.

Effect of the E2FP peptide on (A) membrane rupture, i.e., leakage, (B) membrane phospholipid mixing of the outer monolayer, i.e., hemifusion, and (C) membrane phospholipid mixing of the inner monolayer, i.e., fusion, of fluorescent probes encapsulated in LUVs containing different lipid compositions at different lipid/peptide molar ratios. The lipid compositions used were EPC/Chol at a molar ratio of 5:1 (○), BPS/Chol at a molar ratio of 5:1 (⋄), EPG/Chol at a molar ratio of 5:1 (□), EPA/Chol at a molar ratio of 5:1 (▵), liver extract lipids (∇), EPC/SM/Chol at a molar ratio of 5:1:1 (◃), EPC/SM/Chol at a molar ratio of 52.18.30 (◃), EPC (•), EPC/LPC at a molar ratio of 5:1 (▪), and EPC/TPE at a molar ratio of 5:1 (▾).
The effect of the E2FP peptide on the structural and thermotropic properties of phospholipid membranes was investigated by measuring the steady-state fluorescence anisotropy of the fluorescent probes DPH and TMA-DPH (51–53) incorporated into model membranes composed of saturated synthetic phospholipids as a function of temperature (Fig. 4). In general, E2FP was capable of decreasing the cooperativity of the transition Tm for all phospholipids, as observed by both types of probes. With respect to the anisotropy, at temperatures above, but not below, the Tm for liposomes composed of DMPG, E2FP induced a greater increase in the anisotropy values for DPH (Fig. 4 B) than for TMA-DPH (Fig. 4 A). This effect was similar to that found for DMPA (Fig. 4, C and D), DMPC (Fig. 4, E and F) and the complex mixture EPC/SM/Chol at a molar ratio of 5:1:1 (Fig. 4, G and H). In contrast, there was no significant change in the anisotropy either below or above Tm for DEPE liposomes (Fig. 4, I and J). These data would suggest that E2FP was capable of decreasing the mobility of the phospholipid acyl chains when compared to the pure phospholipid (except in the case of DEPE). These results suggest that the E2FP peptide, although interacting with the membrane, should be located at the lipid-water interface, and that its incorporation should not be affected significantly by differences in headgroup charge (41). It is of interest that E2FP is capable of decreasing significantly the cooperativity of DEPE (Fig. 4, I and J; see below). We have also studied the intrinsic anisotropy of the peptide Trp residues in the presence of liposomes composed of pure synthetic phospholipids (Fig. 4 K). In solution, the anisotropy value was ∼0.3 along the whole range of temperatures studied. However, in the presence of the phospholipids, the anisotropy values differed depending on temperature: they were higher below the Tm of each specific phospholipid, but lower above it (Fig. 4 K). More important, the change in anisotropy occurred coincidentally when the main gel-to-liquid phase transition of the phospholipids took place; these data implied, first, that the peptide was effectively incorporated into the membrane palisade, and, second, that the peptide was capable of sensing the phase transition of each one of the phospholipids.
FIGURE 4.

Steady-state anisotropy, 〈r〉, as a function of temperature of TMA-DPH (A, C, E, G, and I) and DPH (B, D, F, H, and J) incorporated into MLVs composed of DMPG (A and B), DMPA (C and D), DMPC (E and F), EPC/SM/Chol at a lipid molar ratio of 5:1:1 (G and H), and DEPE (I and J). Data correspond to vesicles in the absence (•) and presence (○) of the E2FP peptide. (K) Intrinsic steady-state anisotropy of the E2FP peptide as a function of temperature in solution (•) and incorporated into MLVs composed of DMPC (○), DMPA (▵), DMPG (□), and DEPE (∇). The peptide/lipid molar ratio in all cases was 1:15.
It is well known that the capability of phosphatidylethanolamines in general, and DEPE in particular, to display, apart from the main gel-to-liquid crystalline phase transition, Lβ-Lα, a liquid-to-hexagonal phase transition, Lα-HII (54). To observe any effect of E2FP on the phase polymorphism behavior of DEPE, we have assayed the thermotropic phase behavior of DEPE by differential scanning calorimetry (DSC) (Fig. 5 A). As observed in Fig. 5 A, pure DEPE displays the main gel-to-liquid crystalline phase transition at ∼38°C, whereas the lamellar-to-hexagonal phase transition is observed at ∼64°C (54). In the presence of the peptide at a lipid/peptide molar ratio of 50:1, the main gel-to-liquid crystalline transition was broadened and slightly shifted to lower temperatures, whereas the liquid-to-hexagonal phase transition was completely abolished (Fig. 5 A). 31P NMR spectrometry, sensitive to local motion and orientation of the phosphate group in membrane phospholipids (55), is suitable for following structural changes in membranes. DEPE and other phosphatidylethanolamines, when organized in bilayer structures, give rise to an asymmetrical 31P NMR line shape with a high-field peak and a low-field shoulder, presenting a residual chemical shift anisotropy, Δσ, of 36–40 ppm in the gel state and 27–30 ppm in the liquid-crystalline state (Fig. 5 B). In the HII phase, the chemical shift anisotropy is further averaged due to rapid lateral diffusion of the phospholipid around the tubes of which this phase is composed, resulting in a line-shape with reverse symmetry, i.e., a high-field shoulder and a low-field peak, accompanied by a twofold reduction in the absolute value of Δσ (Fig. 5 B). This is the behavior we found for pure DEPE as expected (Fig. 5 B). However, when E2FP was added to attain a lipid/peptide molar ratio of 50:1, the NMR profile of DEPE was completely different, since, beginning at ∼60°C, an isotropic peak at 0 ppm was apparent. This peak, which should correspond to another phase induced by the presence of the E2FP peptide (see below), increased in intensity at increasing temperatures, being the only component present at high temperatures (Fig. 5 C).
FIGURE 5.

(A) DSC heating thermograms of DEPE in the absence (solid line) and presence (dashed line) of the E2FP peptide. (B and C) 31P NMR spectra of DEPE phospholipid dispersions in the absence (B) and presence (C) of the E2FP peptide at a phospholipid/peptide molar ratio of 15:1 at different temperatures, as stated. The 31P NMR spectra have been normalized.
Information on the structural organization of DEPE and DEPE in the presence of E2FP was obtained by the use of SAXD. This technique defines the nanoscopic structure and provides the interlamellar repeat distance in the lamellar phase, which comprises both the bilayer and the water layer thickness. The diffraction patterns of pure DEPE and DEPE in the presence of E2FP are shown in Fig. 6. The figure illustrates the diffraction patterns collected at 25°C, 45°C, and 70°C, i.e., in the Lβ, in the Lα, and in the HII phases, respectively, for pure DEPE (Fig. 6, A–C, respectively). In the presence of the peptide, the diffraction patterns showed an increase in membrane disorder, and also indicated that several bilayers had become positionally uncorrelated due to the peptide's presence; at the same time, the scattering and the fluctuation increased, and the membranes showed a slight increase in membrane thickness (from 66 Å and 54 Å in the absence of peptide to 69 Å and 59 Å in its presence, at 25°C and 45°C, respectively). Pure DEPE, when organized in the hexagonal HII structure, showed a first-order diffraction at 66 Å; however, the presence of the E2FP peptide induced the disappearance of the reflections characteristic of the phospholipid in the hexagonal phase (Fig. 6 C). The new pattern observed in the presence of the peptide showed the occurrence of a lamellar structure with a spacing and membrane thickness similar to that found in the Lα phase at 45°C.
FIGURE 6.

Small angle x-ray scattering of DEPE MLV suspension in the absence (○) and presence (•) of the E2FP peptide in (A) the gel phase at 25°C, (B) the crystalline liquid phase at 45°, and (C) the hexagonal phase at 70°. Solid lines represent the best fit to the SAXD data applying a global analysis technique. The insets (D–F) display the one-dimensional electron density profiles along the bilayer normal calculated from the SAXD diffraction patterns in the absence (dotted lines) and presence (solid lines) of the peptide.
The existence of structural changes on the E2FP peptide induced by membrane binding was studied by analyzing the infrared amide I′ band located between 1700 and 1600 cm−1 in membranes by Fourier transformed infrared spectroscopy. The amide I′ band of the peptide in the presence of DMPC, DMPG, and DMPA in D2O buffer at a lipid/peptide molar ratio of 15:1 displayed a maximum at 1626 cm−1, 1621 cm−1, and 1618 cm−1, respectively (not shown in the interests of brevity). The maximum of the band did not change significantly upon increasing the temperature (not shown), which suggests a high degree of conformational stability of the peptide in solution. The assignment of the amide I′ component bands to specific structural features has been described previously (45). Bands at ∼1620–1626 cm−1 would correspond to either β-sheet structures or self-aggregated peptides forming a intermolecular network of hydrogen-bonded β-structures or both (56), so that those structures should be the main ones of E2FP in the presence of membranes.
DISCUSSION
The fusion of viral and cellular membranes, the critical early events in viral infection, are mediated by class I and II envelope fusion glycoproteins located on the outer surface of the viral membranes (21,57–60). Whereas class I membrane fusion proteins possess a fusion peptide at or near the amino terminus that is critical for fusion, a pair of extended α-helices and, generally, a cluster of aromatic amino acids proximal to a hydrophobic transmembrane domain, class II fusion proteins possess an internal fusion peptide located at a distal location from the transmembrane anchor, as well as different domains comprised mostly of antiparallel β-sheets (58,61). Although their three-dimensional structure is different, their function is identical, and therefore they must share structural and/or functional characteristics in the specific domains, which interact with and disrupt biological membranes (13,61,62). It is known that both HCV E1 and E2 envelope glycoproteins are essential for receptor binding, host-cell entry, and membrane fusion, but their specific roles in the different processes of the viral life cycle are not known (8,15,44,57,63). The E1/E2 heterodimer is thought to be the functional unit, and low pH would induce its dissociation leading to homooligomerization of the active form of the fusion protein (15,63–65). It is already known that, apart from the fusion peptide and the pre- and transmembrane domains, there exist other membranotropic segments along the sequence of membrane fusion proteins which, under a concerted action, are essential for membrane fusion (20). In this context, several hydrophobic patches have been identified in both E1 and E2 envelope glycoproteins which might be important in the mechanism of membrane fusion, not only for modulating membrane binding and interaction, but also for protein-protein interaction (17–19,44,66). These regions should be decisive for membrane fusion to take place, since destabilization of the lipid bilayer and membrane fusion are the result of the binding and interaction of these segments with biological membranes. Recently, it was shown that different segments from E2 participate in the viral fusion process, one of them a segment comprised by residues 600–620 (15). As we showed in a previous work, one of the most membranotropic regions of E2 belongs to the region comprising residues 603–634 (19). In that work, we analyzed the effect of two peptide libraries from HCV E1 and E2 on leakage, hemifusion, and fusion, using model membranes composed of EPC/SM/Chol and liver extract lipids. In this work, we have made an in-depth study of peptide E2FP, which is the combination of the sequence of the previously studied peptides 603-LTPRCLVDYPYRLWHYPC-620, 610-DYPYRLWHYPCTLNFSIF-627, and 617-HYPCTLNFSIFKVRMYVG-634. In this context, the work described here is the natural extension of the previous study, since we have studied the binding and interaction of a new peptide, peptide E2FP, with membrane model systems composed not only of EPC/SM/Chol and liver extract lipids, but also of phospholipids containing specific hydrocarbon acyl chains and headgroups, as well as phospholipids inducing both negative and positive bilayer curvature. We describe leakage, hemifusion, and fusion induced by peptide E2FP, but also its binding, transverse location, aggregation state, and phospholipid polymorphic phase behavior, as well as its structure in the presence of a number of model membrane systems. It should be noted also that this E2 region includes the sequence 613–618, which is supposed to be involved in CD81 binding, and E1/E2 and E2/E2 dimerization (15). Therefore, we present here the results of the study of the interaction of a peptide derived from this region, the E2FP peptide, with model membrane systems.
Peptide E2FP is one of the most membranotropic sequences of HCV E2; accordingly, the E2FP peptide studied in this work displays a high binding constant to model membranes having different phospholipid compositions, as has been found for other peptides (39,46). After binding, the Trp residue of the peptide resides in an environment with a low dielectric constant, showing a significant motional restriction. E2FP showed a slightly higher affinity for anionic-phospholipid compositions than for those containing zwitterionic phospholipids; this difference should be due to the fact that the peptide has a positive net charge at pH 7.4. The existence of a specific interaction with liposomes containing negatively charged phospholipids was corroborated by the change of the dipole potential at the membrane surface, as well as by hydrophilic and lipophylic probe quenching, suggesting that the E2FP peptide was effectively incorporated in the membranes, located in a shallow position but nearer to the interface in the presence of zwitterionic phospholipids than in the presence of negatively charged ones. Nevertheless, the E2FP peptide is capable of binding with high affinity to model membranes containing both negatively charged and zwitterionic phospholipids.
The E2FP peptide was also capable of disrupting the membrane bilayer, causing the release of fluorescent probes. This effect is dependent on lipid composition and on the lipid/peptide molar ratio. The highest effect was observed for liposomes containing negatively charged phospholipids, but leakage values observed for liposomes composed of zwitterionic phospholipids, although lower, were also significant. Although the specific disrupting effect should be due primarily to hydrophobic interactions within the bilayer, the specific charge of the phospholipid headgroups affect the extent of membrane leakage, albeit slightly. The induction of hemifusion and fusion by E2FP were also studied, and similar results were obtained: specific and large membrane hemifusion and fusion values were found in the presence of liposomes composed of both negatively charged and zwitterionic phospholipids. It is interesting that liposomes containing TPE (phospholipid inducing negative curvature) displayed relatively high hemifusion and fusion values, whereas liposomes containing LPC (phospholipid inducing positive curvature) were the ones that elicited the lowest values. This inhibitory effect produced by inverted-cone-shaped phospholipids has been previously observed in some viral and nonviral fusion systems (67–70). This effect could be due to the fact that E2FP, located at the bilayer interface and interacting with the phospholipid headgroups, could be capable of changing the polymorphic phase behavior of the membrane bilayer, since it was able to inhibit the presence of hexagonal phases but induce the presence of other lamellar phases (isotropic 31P-NMR signal, but lamellar SAXD diffraction pattern). As observed by SAXD, the presence of the peptide increased slightly the membrane thickness and increased the disorder of the membrane. The increase in the membrane thickness, the increase in lateral tension within the membrane, and the packing stress could disorder the lipid molecules in a way that could be detected by NMR as an isotropic signal. These data reveal that E2FP affects the fluidity behavior of the phospholipids in the membrane, suggesting, as commented previously, that the E2FP peptide, although interacting with the membrane, should be located at the lipid-water interface (41).
We have also shown that E2FP peptide is capable of affecting the steady-state fluorescence anisotropy of fluorescent probes located in the palisade structure of the membrane, since the peptide, in general, was able of decreasing the mobility of the phospholipid acyl chains above but not below the Tm when compared to the pure phospholipids. It is significant that the peptide sensed the phospholipid main phase transition, indicating its incorporation into the palisade structure of the membrane. The infrared spectra of the amide I′ region of the fully hydrated peptide did not change with temperature, indicating a high stability of its conformation, where extended β-strands with strong intermolecular interactions predominated. The binding to the surface and the modulation of the phospholipid biophysical properties that take place when E2FP is bound to the membrane, i.e., partitioning into the membrane surface and perturbation of the bilayer architecture, could be related to the conformational changes that might occur during the activity of the HCV E2 glycoprotein. For all assayed phospholipids, and at increasing peptide concentrations, we observed a decrease in cooperativity and mobility. These features would indicate that E2FP would interact with the membrane through both electrostatic and hydrophobic effects, and that it would be adsorbed at the membrane interface; however, it is possible that part of the peptide could be inserted deeper than the membrane interface, in this way increasing the membrane permeability, as we have already shown. These and previous data suggest the notion that the E2 region where E2FP resides might be a fusion determinant and have an essential role in the membrane fusion process. If that is true, it would imply that both HCV E1 and E2 glycoproteins are directly implicated in the mechanism that makes possible the entry of the HCV virus into its cellular host: in this way, fusion peptides would be implicated in the very first steps of membrane fusion, whereas other membranotropic segments would be implicated in membrane destabilization, pore formation, and enlargement. All these sequences should be attractive candidates for antiviral drug development, since they could be targets for antiviral compounds that may lead to new vaccine strategies
Acknowledgments
This work was supported by grant BFU2005-00186-BMC (Ministerio de Ciencia y Tecnologia, Madrid, Spain) to J.V. A.J.P. and J.G. are recipients of predoctoral fellowships from the Autonomous Government of the Comunidad Valenciana, Valencia, Spain.
Editor: Mark Girvin.
References
- 1.Chen, S. L., and T. R. Morgan. 2006. The natural history of hepatitis C virus (HCV) infection. Int. J. Med. Sci. 3:47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Penin, F., J. Dubuisson, F. A. Rey, D. Moradpour, and J. M. Pawlotsky. 2004. Structural biology of hepatitis C virus. Hepatology. 39:5–19. [DOI] [PubMed] [Google Scholar]
- 3.Tan, S. L., A. Pause, Y. Shi, and N. Sonenberg. 2002. Hepatitis C therapeutics: current status and emerging strategies. Nat. Rev. Drug Discov. 1:867–881. [DOI] [PubMed] [Google Scholar]
- 4.Qureshi, S. A. 2007. Hepatitis C virus: biology, host evasion strategies, and promising new therapies on the horizon. Med. Res. Rev. 27:353–373. [DOI] [PubMed] [Google Scholar]
- 5.Reed, K. E., and C. M. Rice. 2000. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr. Top. Microbiol. Immunol. 242:55–84. [DOI] [PubMed] [Google Scholar]
- 6.Vauloup-Fellous, C., V. Pene, J. Garaud-Aunis, F. Harper, S. Bardin, Y. Suire, E. Pichard, A. Schmitt, P. Sogni, G. Pierron, P. Briand, and A. R. Rosenberg. 2006. Signal peptide peptidase-catalyzed cleavage of hepatitis C virus core protein is dispensable for virus budding but destabilizes the viral capsid. J. Biol. Chem. 281:27679–27692. [DOI] [PubMed] [Google Scholar]
- 7.Pozzetto, B., T. Bourlet, F. Grattard, and L. Bonnevial. 1996. Structure, genomic organization, replication and variability of hepatitis C virus. Nephrol. Dial. Transplant. 11(Suppl. 4):2–5. [DOI] [PubMed] [Google Scholar]
- 8.Bartosch, B., J. Dubuisson, and F. L. Cosset. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1–E2 envelope protein complexes. J. Exp. Med. 197:633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cocquerel, L., J. C. Meunier, A. Pillez, C. Wychowski, and J. Dubuisson. 1998. A retention signal necessary and sufficient for endoplasmic reticulum localization maps to the transmembrane domain of hepatitis C virus glycoprotein E2. J. Virol. 72:2183–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cocquerel, L., C. Wychowski, F. Minner, F. Penin, and J. Dubuisson. 2000. Charged residues in the transmembrane domains of hepatitis C virus glycoproteins play a major role in the processing, subcellular localization, and assembly of these envelope proteins. J. Virol. 74:3623–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cocquerel, L., A. Op de Beeck, M. Lambot, J. Roussel, D. Delgrange, A. Pillez, C. Wychowski, F. Penin, and J. Dubuisson. 2002. Topological changes in the transmembrane domains of hepatitis C virus envelope glycoproteins. EMBO J. 21:2893–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Op De Beeck, A., R. Montserret, S. Duvet, L. Cocquerel, R. Cacan, B. Barberot, M. Le Maire, F. Penin, and J. Dubuisson. 2000. The transmembrane domains of hepatitis C virus envelope glycoproteins E1 and E2 play a major role in heterodimerization. J. Biol. Chem. 275:31428–31437. [DOI] [PubMed] [Google Scholar]
- 13.Garry, R. F., and S. Dash. 2003. Proteomics computational analyses suggest that hepatitis C virus E1 and pestivirus E2 envelope glycoproteins are truncated class II fusion proteins. Virology. 307:255–265. [DOI] [PubMed] [Google Scholar]
- 14.Lavillette, D., B. Bartosch, D. Nourrisson, G. Verney, F. L. Cosset, F. Penin, and E. I. Pecheur. 2006. Hepatitis C virus glycoproteins mediate low pH-dependent membrane fusion with liposomes. J. Biol. Chem. 281:3909–3917. [DOI] [PubMed] [Google Scholar]
- 15.Lavillette, D., E. I. Pecheur, P. Donot, J. Fresquet, J. Molle, R. Corbau, M. Dreux, F. Penin, and F. L. Cosset. 2007. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J. Virol. 81:8752–8765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartosch, B., and F. L. Cosset. 2006. Cell entry of hepatitis C virus. Virology. 348:1–12. [DOI] [PubMed] [Google Scholar]
- 17.Yagnik, A. T., A. Lahm, A. Meola, R. M. Roccasecca, B. B. Ercole, A. Nicosia, and A. Tramontano. 2000. A model for the hepatitis C virus envelope glycoprotein E2. Proteins. 40:355–366. [DOI] [PubMed] [Google Scholar]
- 18.Roccasecca, R., H. Ansuini, A. Vitelli, A. Meola, E. Scarselli, S. Acali, M. Pezzanera, B. B. Ercole, J. McKeating, A. Yagnik, A. Lahm, A. Tramontano, R. Cortese, and A. Nicosia. 2003. Binding of the hepatitis C virus E2 glycoprotein to CD81 is strain specific and is modulated by a complex interplay between hypervariable regions 1 and 2. J. Virol. 77:1856–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perez-Berna, A. J., M. R. Moreno, J. Guillen, A. Bernabeu, and J. Villalain. 2006. The membrane-active regions of the hepatitis C virus E1 and E2 envelope glycoproteins. Biochemistry. 45:3755–3768. [DOI] [PubMed] [Google Scholar]
- 20.Peisajovich, S. G., and Y. Shai. 2003. Viral fusion proteins: multiple regions contribute to membrane fusion. Biochim. Biophys. Acta. 1614:122–129. [DOI] [PubMed] [Google Scholar]
- 21.Epand, R. M. 2003. Fusion peptides and the mechanism of viral fusion. Biochim. Biophys. Acta. 1614:116–121. [DOI] [PubMed] [Google Scholar]
- 22.Surewicz, W. K., H. H. Mantsch, and D. Chapman. 1993. Determination of protein secondary structure by Fourier transform infrared spectroscopy: a critical assessment. Biochemistry. 32:389–394. [DOI] [PubMed] [Google Scholar]
- 23.Zhang, Y. P., R. N. Lewis, R. S. Hodges, and R. N. McElhaney. 1992. FTIR spectroscopic studies of the conformation and amide hydrogen exchange of a peptide model of the hydrophobic transmembrane alpha-helices of membrane proteins. Biochemistry. 31:11572–11578. [DOI] [PubMed] [Google Scholar]
- 24.Mayer, L. D., M. J. Hope, and P. R. Cullis. 1986. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta. 858:161–168. [DOI] [PubMed] [Google Scholar]
- 25.Böttcher, C. S. F., C. M. Van Gent, and C. Fries. 1961. A rapid and sensitive sub-micro phosphorus determination. Anal. Chim. Acta. 1061:203–204. [Google Scholar]
- 26.Edelhoch, H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 6:1948–1954. [DOI] [PubMed] [Google Scholar]
- 27.Struck, D. K., D. Hoekstra, and R. E. Pagano. 1981. Use of resonance energy transfer to monitor membrane fusion. Biochemistry. 20:4093–4099. [DOI] [PubMed] [Google Scholar]
- 28.Meers, P., S. Ali, R. Erukulla, and A. S. Janoff. 2000. Novel inner monolayer fusion assays reveal differential monolayer mixing associated with cation-dependent membrane fusion. Biochim. Biophys. Acta. 1467:227–243. [DOI] [PubMed] [Google Scholar]
- 29.Sainz, B., Jr., J. M. Rausch, W. R. Gallaher, R. F. Garry, and W. C. Wimley. 2005. Identification and characterization of the putative fusion peptide of the severe acute respiratory syndrome-associated coronavirus spike protein. J. Virol. 79:7195–7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lakowicz, J. 1999. Principles of Fluorescence Spectroscopy. Kluwer-Plenum Press, New York.
- 31.Eftink, M. R., and C. A. Ghiron. 1977. Exposure of tryptophanyl residues and protein dynamics. Biochemistry. 16:5546–5551. [DOI] [PubMed] [Google Scholar]
- 32.Castanho, M., and M. Prieto. 1995. Filipin fluorescence quenching by spin-labeled probes: studies in aqueous solution and in a membrane model system. Biophys. J. 69:155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wardlaw, J. R., W. H. Sawyer, and K. P. Ghiggino. 1987. Vertical fluctuations of phospholipid acyl chains in bilayers. FEBS Lett. 223:20–24. [DOI] [PubMed] [Google Scholar]
- 34.Santos, N. C., M. Prieto, and M. A. Castanho. 1998. Interaction of the major epitope region of HIV protein gp41 with membrane model systems. A fluorescence spectroscopy study. Biochemistry. 37:8674–8682. [DOI] [PubMed] [Google Scholar]
- 35.Wall, J., F. Ayoub, and P. O'Shea. 1995. Interactions of macromolecules with the mammalian cell surface. J. Cell Sci. 108:2673–2682. [DOI] [PubMed] [Google Scholar]
- 36.Golding, C., S. Senior, M. T. Wilson, and P. O'Shea. 1996. Time resolution of binding and membrane insertion of a mitochondrial signal peptide: correlation with structural changes and evidence for cooperativity. Biochemistry. 35:10931–10937. [DOI] [PubMed] [Google Scholar]
- 37.Cladera, J., and P. O'Shea. 1998. Intramembrane molecular dipoles affect the membrane insertion and folding of a model amphiphilic peptide. Biophys. J. 74:2434–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gross, E., R. S. Bedlack, Jr., and L. M. Loew. 1994. Dual-wavelength ratiometric fluorescence measurement of the membrane dipole potential. Biophys. J. 67:208–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bernabeu, A., J. Guillen, A. J. Perez-Berna, M. R. Moreno, and J. Villalain. 2007. Structure of the C-terminal domain of the pro-apoptotic protein Hrk and its interaction with model membranes. Biochim. Biophys. Acta. 1768:1659–1670. [DOI] [PubMed] [Google Scholar]
- 40.Giudici, M., J. A. Poveda, M. L. Molina, L. de la Canal, J. M. Gonzalez-Ros, K. Pfuller, U. Pfuller, and J. Villalain. 2006. Antifungal effects and mechanism of action of viscotoxin A3. FEBS J. 273:72–83. [DOI] [PubMed] [Google Scholar]
- 41.Contreras, L. M., F. J. Aranda, F. Gavilanes, J. M. Gonzalez-Ros, and J. Villalain. 2001. Structure and interaction with membrane model systems of a peptide derived from the major epitope region of HIV protein gp41: implications on viral fusion mechanism. Biochemistry. 40:3196–3207. [DOI] [PubMed] [Google Scholar]
- 42.Laggner, P. 1994. X-ray diffraction on biomembranes with emphasis on lipid moiety. Subcell. Biochem. 23:451–491. [DOI] [PubMed] [Google Scholar]
- 43.Pabst, G. 2006. Global properties of biomimetic membranes: perspectives on molecular features. Biophys. Rev. Lett. 1:57–84. [Google Scholar]
- 44.Drummer, H. E., and P. Poumbourios. 2004. Hepatitis C virus glycoprotein E2 contains a membrane-proximal heptad repeat sequence that is essential for E1E2 glycoprotein heterodimerization and viral entry. J. Biol. Chem. 279:30066–30072. [DOI] [PubMed] [Google Scholar]
- 45.Pascual, R., M. Contreras, A. Fedorov, M. Prieto, and J. Villalain. 2005. Interaction of a peptide derived from the N-heptad repeat region of gp41 Env ectodomain with model membranes. Modulation of phospholipid phase behavior. Biochemistry. 44:14275–14288. [DOI] [PubMed] [Google Scholar]
- 46.Moreno, M. R., J. Guillen, A. J. Perez-Berna, D. Amoros, A. I. Gomez, A. Bernabeu, and J. Villalain. 2007. Characterization of the interaction of two peptides from the N terminus of the NHR domain of HIV-1 gp41 with phospholipid membranes. Biochemistry. 46:10572–10584. [DOI] [PubMed] [Google Scholar]
- 47.Pascual, R., M. R. Moreno, and J. Villalain. 2005. A peptide pertaining to the loop segment of human immunodeficiency virus gp41 binds and interacts with model biomembranes: implications for the fusion mechanism. J. Virol. 79:5142–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wall, J., C. A. Golding, M. Van Veen, and P. O'Shea. 1995. The use of fluoresceinphosphatidylethanolamine (FPE) as a real-time probe for peptide-membrane interactions. Mol. Membr. Biol. 12:183–192. [DOI] [PubMed] [Google Scholar]
- 49.Cladera, J., I. Martin, and P. O'Shea. 2001. The fusion domain of HIV gp41 interacts specifically with heparan sulfate on the T-lymphocyte cell surface. EMBO J. 20:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O'Shea, P. 2003. Intermolecular interactions with/within cell membranes and the trinity of membrane potentials: kinetics and imaging. Biochem. Soc. Trans. 31:990–996. [DOI] [PubMed] [Google Scholar]
- 51.Davenport, L., R. E. Dale, R. H. Bisby, and R. B. Cundall. 1985. Transverse location of the fluorescent probe 1,6-diphenyl-1,3,5-hexatriene in model lipid bilayer membrane systems by resonance excitation energy transfer. Biochemistry. 24:4097–4108. [DOI] [PubMed] [Google Scholar]
- 52.Lentz, B. R. 1993. Use of fluorescent probes to monitor molecular order and motions within liposome bilayers. Chem. Phys. Lipids. 64:99–116. [DOI] [PubMed] [Google Scholar]
- 53.Pebay-Peyroula, E., E. J. Dufourc, and A. G. Szabo. 1994. Location of diphenyl-hexatriene and trimethylammonium-diphenyl-hexatriene in dipalmitoylphosphatidylcholine bilayers by neutron diffraction. Biophys. Chem. 53:45–56. [DOI] [PubMed] [Google Scholar]
- 54.Epand, R. M. 1998. Lipid polymorphism and protein-lipid interactions. Biochim. Biophys. Acta. 1376:353–368. [DOI] [PubMed] [Google Scholar]
- 55.Killian, J. A., and B. de Kruijff. 1986. The influence of proteins and peptides on the phase properties of lipids. Chem. Phys. Lipids. 40:259–284. [DOI] [PubMed] [Google Scholar]
- 56.Arrondo, J. L., and F. M. Goni. 1999. Structure and dynamics of membrane proteins as studied by infrared spectroscopy. Prog. Biophys. Mol. Biol. 72:367–405. [DOI] [PubMed] [Google Scholar]
- 57.Kielian, M. 2006. Class II virus membrane fusion proteins. Virology. 344:38–47. [DOI] [PubMed] [Google Scholar]
- 58.Kielian, M., and F. A. Rey. 2006. Virus membrane-fusion proteins: more than one way to make a hairpin. Nat. Rev. Microbiol. 4:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eckert, D. M., and P. S. Kim. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70:777–810. [DOI] [PubMed] [Google Scholar]
- 60.Blumenthal, R., M. J. Clague, S. R. Durell, and R. M. Epand. 2003. Membrane fusion. Chem. Rev. 103:53–69. [DOI] [PubMed] [Google Scholar]
- 61.Schibli, D. J., and W. Weissenhorn. 2004. Class I and class II viral fusion protein structures reveal similar principles in membrane fusion. Mol. Membr. Biol. 21:361–371. [DOI] [PubMed] [Google Scholar]
- 62.Gallo, S. A., C. M. Finnegan, M. Viard, Y. Raviv, A. Dimitrov, S. S. Rawat, A. Puri, S. Durell, and R. Blumenthal. 2003. The HIV Env-mediated fusion reaction. Biochim. Biophys. Acta. 1614:36–50. [DOI] [PubMed] [Google Scholar]
- 63.Ciczora, Y., N. Callens, F. Penin, E. I. Pecheur, and J. Dubuisson. 2007. Transmembrane domains of hepatitis C virus envelope glycoproteins: residues involved in E1E2 heterodimerization and involvement of these domains in virus entry. J. Virol. 81:2372–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ciczora, Y., N. Callens, C. Montpellier, B. Bartosch, F. L. Cosset, A. Op de Beeck, and J. Dubuisson. 2005. Contribution of the charged residues of hepatitis C virus glycoprotein E2 transmembrane domain to the functions of the E1E2 heterodimer. J. Gen. Virol. 86:2793–2798. [DOI] [PubMed] [Google Scholar]
- 65.Keck, Z. Y., A. Op De Beeck, K. G. Hadlock, J. Xia, T. K. Li, J. Dubuisson, and S. K. Foung. 2004. Hepatitis C virus E2 has three immunogenic domains containing conformational epitopes with distinct properties and biological functions. J. Virol. 78:9224–9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brazzoli, M., A. Helenius, S. K. Foung, M. Houghton, S. Abrignani, and M. Merola. 2005. Folding and dimerization of hepatitis C virus E1 and E2 glycoproteins in stably transfected CHO cells. Virology. 332:438–453. [DOI] [PubMed] [Google Scholar]
- 67.Stiasny, K., and F. X. Heinz. 2004. Effect of membrane curvature-modifying lipids on membrane fusion by tick-borne encephalitis virus. J. Virol. 78:8536–8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baljinnyam, B., B. Schroth-Diez, T. Korte, and A. Herrmann. 2002. Lysolipids do not inhibit influenza virus fusion by interaction with hemagglutinin. J. Biol. Chem. 277:20461–20467. [DOI] [PubMed] [Google Scholar]
- 69.Chernomordik, L., E. Leikina, M. S. Cho, and J. Zimmerberg. 1995. Control of baculovirus gp64-induced syncytium formation by membrane lipid composition. J. Virol. 69:3049–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gaudin, Y. 2000. Rabies virus-induced membrane fusion pathway. J. Cell Biol. 150:601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guillen, J., A. J. Perez-Berna, M. R. Moreno, and J. Villalain. 2005. Identification of the membrane-active regions of the severe acute respiratory syndrome coronavirus spike membrane glycoprotein using a 16/18-mer peptide scan: implications for the viral fusion mechanism. J. Virol. 79:1743–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]