

Abstract
We present a new method for computing interaction potentials of solvated proteins directly from small-angle x-ray scattering data. An ensemble of proteins is modeled by Monte Carlo or molecular dynamics simulation. The global x-ray scattering of the whole model ensemble is then computed at each snapshot of the simulation, and averaged to obtain the x-ray scattering intensity. Finally, the interaction potential parameters are adjusted by an optimization algorithm, and the procedure is iterated until the best agreement between simulation and experiment is obtained. This new approach obviates the need for approximations that must be made in simplified analytical models. We apply the method to lambda repressor fragment 6-85 and fyn-SH3. With the increased availability of fast computer clusters, Monte Carlo and molecular dynamics analysis using residue-level or even atomistic potentials may soon become feasible.
INTRODUCTION
Small angle x-ray scattering (SAXS) is a convenient tool for determining protein-protein interaction potentials in solution. A major driving force of this work has been the need for determining ideal conditions for protein crystallization. Thus, the focus has been on the effect of the concentration of precipitation agents and cosolvents (1,2).
Two additional areas could benefit greatly from the effective potentials provided by SAXS studies. One is the study of solvation shells around proteins. Neutron scattering, NMR spectroscopy, simulation, and terahertz spectroscopy have shown that solvent shells of substantial thickness exist around proteins (3–6). Dynamical solvation effects studied by terahertz spectroscopy extend to >10 Å from the protein surface (7). Protein-protein interactions are mediated by such solvent shells, and thus contain information about the solvent shells when measured at sufficiently high concentrations. The other area is the study of transient protein aggregation. Very rapidly folding proteins have folding timescales comparable to the lifetime of transient aggregates (8,9). Such transient aggregates can nucleate irreversible aggregation (10,11), a process linked with numerous diseases. Protein-protein interaction potentials play a key role in defining how easily such nuclei form.
Effective interaction potentials are currently extracted from SAXS data with the aid of analytical approximations to speed up the calculation (1). The random phase approximation treats each protein molecule as an independent scatterer characterized by a form factor. The form factor can be obtained approximately by extrapolating SAXS measurements to infinite dilution (12). The observed scattering intensity is then assumed to be a product of the form factor and a scattering factor, an approximation strictly valid over the full range of scattering angles only for dilute particles. From the scattering factor, a radial pair distribution function and corresponding radial effective potential are obtained. Square-well and Yukawa potentials are used because they have simple Fourier transforms (2). The commonly used DLVO form consists of a hard-sphere cutoff, and two Yukawa potentials (∼exp[−(r − r0)/δ]/r) for long-range repulsion and short-range attraction between proteins.
Increases in computing power enable a more direct approach, which we introduce here. Simulation of multiprotein ensemble dynamics is followed by evaluation of the x-ray scattering of the whole ensemble. Iteration can then be used to refine force fields “on the fly” without any low-concentration approximations or scattering analysis approximations.
Fig. 1 outlines our approach. We first simulate the dynamics of an ensemble of dozens to hundreds of model proteins that interact via an adjustable interaction potential. Either Monte Carlo or molecular dynamics simulations are used to sample configurations of the ensemble. We then calculate the global x-ray scattering intensity of the entire model ensemble at each configuration, eliminating the need for low-concentration or random phase approximations. The resulting series of scattering intensities is averaged to obtain the steady-state SAXS intensity as a function of scattering angle. An optimization algorithm compares the computed signal with the experimental signal, and modifies the adjustable interaction potential for the next round of simulation. The process repeats iteratively until the experimental data is matched with the smallest least-squares deviation. Any form of potential can thus be fitted exactly for polydisperse model particles at any concentration.
FIGURE 1.

Method for extracting the protein-protein potential from SAXS data. In step 1, a protein ensemble of up to 100 molecules is simulated by Monte Carlo or molecular dynamics. In step 2, the exact x-ray scattering for the model ensemble is evaluated at each simulation snapshot. In step 3, the average x-ray scattering curve is obtained and compared with experimental data. In step 4, the interaction potential is adjusted by steepest descent for the next round of simulation.
In this first application, we determine isotropic interaction potentials, and hence assume spherical model protein monomers. Aggregates can have any shape made from these monomer building blocks, up to the size of the box used for simulation, typically 20 monomer diameters or more. Thus the analysis must be truncated at large scattering angles, but it does not assume spherical aggregates or low monomer concentration. We illustrate the method by fitting experimental data for the two proteins λ6-85 and fyn-SH3 to several potential models. The ethylene glycol-water solvent we use is similar to the one used in recent SAXS studies of folding kinetics (13). With the advent of interaction potentials based on sums of amino-acid-pair interactions, the simulation direct fitting approach could yield anisotropic interaction potentials in the near future, revealing potential aggregation sites or local changes in the protein solvation shell.
METHODS
Proteins
λ*6–85 is an 80-residue, five-helix globular protein of molecular mass 9.2 kDa (see Fig. 4, inset). The protein we used in SAXS experiments contained the mutations Tyr22Trp, Glu33Tyr, Gly46Ala, and Gly48Ala, engineered by site-directed mutagenesis (Stratagene Quickchange kit, La Jolla, CA) based on a wild-type plasmid donated by Terry Oas (14). fyn-SH3 is a predominantly β-sheet protein (molecular mass 9.3 kDa) (see Fig. 4, inset) with 78 residues and a tag of six histidine residues. The sequence, donated by Alan Davidson, has mutations Val1Ser, Val5Glu, Ala39Val, and Val55Phe (15).
FIGURE 4.

Experiment (circles with error bars) and molecular dynamics simulation (thick solid line) of the scattering intensity versus magnitude of the scattering vector for  (A) and fyn-SH3 (B), confirming the quality of the parameter set obtained by MC modeling. The estimated 1σ sampling error we achieved in the MD simulations is indicated by the envelopes. (Insets) Native PDB structures for the protein fragments, as visualized with VMD (29).
(A) and fyn-SH3 (B), confirming the quality of the parameter set obtained by MC modeling. The estimated 1σ sampling error we achieved in the MD simulations is indicated by the envelopes. (Insets) Native PDB structures for the protein fragments, as visualized with VMD (29).
Genes for the two proteins were inserted into the pET-15b vector, expressed in Rosetta TM (DE3) pLysS cells (Novagen, San Diego, CA), and grown in LB broth at 37°C for 8 h. After induction with isopropyl-ß-D-thiogalactopyranoside at 25°C for 12 h, cells were lysed with a French press, and the supernatant was collected after centrifugation. Proteins were selectively bound to a nickel-agarose his-tag binding column (Pharmacia, Peapack, NJ) and eluted with a 250 mM imidazole buffer. The six-histidine tag of λ6–85 was cleaved by thrombin (VWR, West Chester, PA), and additional purification was performed with Amicon 3 kDa and 30 kDa membranes (Fisher Scientific, Hampton, NH). fyn-SH3 was used with the his tag. The identity of λ6–85 and fyn-SH3 was confirmed by electrospray ionization mass spectroscopy and their purity by sodium dodecyl sulfate polyacrylamide gel electrophoresis.
Final protein concentrations in buffers used for experiments were determined by near-UV absorption spectroscopy at 280 nm of the tryptophan and tyrosine residues as described by Edelhoch (16). We have found this procedure to yield similar results in aqueous and aqueous-osmolyte buffers. We estimate a relative accuracy of ±1% for dilution series from the same sample, and an absolute accuracy of ∼±5%. Results are rounded to the nearest 10 μM.
SAXS measurements
SAXS measurements were performed at the Biophysics Collaborative Access Team Beamline of the Advanced Photon Source at Argonne National Laboratory (Argonne, IL) (17). An Aviex CCD camera with an active area of ∼160 × 80 mm2 (2084 × 1042 pixels, 78 μm gap between pixels), located 1.9 m from sample, was used to collect data in the scattering angle range of Q = 4πsinθ/λ = 0.03–0.12 Å−1, at a nominal wavelength of 1 Å. Low concentration data for fyn-SH3 were also collected with a Pilatus CCD camera. The x-ray beam was collimated to a spot size of 300 × 130 μm2 at the sample cell.
To reduce radiation damage, and to enable a direct comparison with our previous SAXS folding study of λ6–85, we performed our experiments in a 45:55 vol % ethylene glycol/water buffer. The ionic strength was 50 mM phosphate at pH 7.0. The temperature in all experiments was −28 ± 1°C, cooled by a Neslab ULT-80DD recirculator. Steady-state SAXS data were collected in a UNISOKU sample cell with 80 μl volume and 50 μm thick sapphire windows. The exposure time was 500 ms for λ6-85, and 300 ms for fyn-SH3 (four frames of 200 ms on the Pilatus detector), based on extensive exposure/concentration tests for protein damage. We measured steady-state SAXS data of λ6-85 up to 2.92 mM, and of fyn-SH3 up to 1.68 mM, without any visible aggregation at room temperature or at −28°C. Each sample was filtered with a 0.2 μM pore syringe filter (Corning, Toledo, OH) before use. The raw data were angle-averaged with logarithmic weighting in Q, and a reference buffer curve was subtracted.
Interaction potentials
To enable Monte Carlo or molecular dynamics simulations, a protein-protein interaction potential has to be chosen. We tested several pairwise-additive isotropic interaction potentials not easily fitted by analytical methods. At short distance, an r12 repulsive term was used instead of the commonplace hard sphere wall:
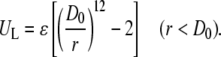 |
(1) |
Past the potential minimum at D0, exponential, Gaussian and Yukawa forms were used in various combinations to model both attractive and repulsive-attractive potentials:
 |
(2) |
where ɛ is the potential depth, 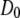 is the center-of-mass distance between proteins where the repulsive potential wall begins, and δ is the attractive potential range.
is the center-of-mass distance between proteins where the repulsive potential wall begins, and δ is the attractive potential range.
The softer than hard-sphere potential wall, not easily amenable to the analytical treatment, highlights the fact that no reference potential assumptions need to be made. In our first application, we assumed isotropically interacting particles and pairwise additive potentials, although nonspherical particles and n-body potentials could be implemented in the future because our approach requires only that the total potential energy for the multiprotein system can be evaluated.
Configurational sampling
To avoid the need for low-concentration approximations, we sample a whole protein ensemble much larger than the typical aggregate size. Protein configurations were sampled by two methods: Metropolis Monte Carlo sampling (MMC), which illustrates computation of the scattering curve from a thermal simulation, and Langevin molecular dynamics (LMD), to illustrate computation of scattering curves from real-time dynamics simulations. In both approaches, we distributed n = 25–100 spherically symmetric model protein particles in a spherical or cube-shaped volume, the latter with periodic boundary conditions. The diameter of the simulation volume was determined by the experimental protein concentration. To reduce oscillatory boundary artifacts in the SAXS calculation, the diameter of the volume was varied randomly about the average. Test runs with up to 20,000 protein particles confirmed that full convergence over the desired range of Q could be achieved rapidly with 25 particles for fyn-SH3 and with 100 particles for 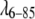 over the full experimental concentration range.
over the full experimental concentration range.
For MMC sampling, we started out with a random distribution of particles. Single particles were then chosen at random, and moved by random displacements inside the spherical volume. Each move was accepted or rejected based on the Metropolis criterion by computing the change in total energy, ΔE (18). When the net energy change was negative, the move was accepted, whereas a positive energy change was accepted with a probability of exp(−ΔE/kBT). Equilibration of the total energy to within the statistical noise typically required 50n moves for λ6−85. This sampling was repeated until the scattering intensity (see below) was a smooth function of Q. The longest runs provide estimated error bounds for the computed scattering curve.
For molecular dynamics sampling in real time, we used an LMD approach in a cubic volume with periodic boundary conditions. Each protein particle was subject to a vectorial force resulting from the other protein particles, and to a random Brownian force simulating the implicit solvent dynamics. In addition, the Brownian motion was countered by a vectorial damping term. Inertial forces were neglected, resulting in 3n equations of motion
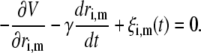 |
(3) |
For nonspherical particles subject to anisotropic interaction potentials, an additional set of 3n equations for rotational diffusion would have to be solved, but no additional complications are introduced by our approach. In Eq. 3, V is the interaction potential summed over all protein pairs (Eqs. 1 and 2). Protein particle m is at position 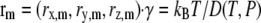 denotes the velocity relaxation rate, which depends on the diffusion coefficient D, assumed independent of coordinate. ξi(t) is Gaussian white noise with zero mean, and a variance set to satisfy the Onsager fluctuation-dissipation theorem that relates ξ and γ (19). The equations of motion were integrated by a standard integrator using finite-difference derivatives (thus, Brownian noise or discontinuities in the potential derivative are not a problem). Derivatives with respect to a single particle, like the energy change ΔE, could be evaluated efficiently. The protein distribution was allowed to evolve to a mean particle deviation of at least 3.4 Rg before sampling the next configuration, to ensure that the scattering calculation did not needlessly sample very similar configurations.
denotes the velocity relaxation rate, which depends on the diffusion coefficient D, assumed independent of coordinate. ξi(t) is Gaussian white noise with zero mean, and a variance set to satisfy the Onsager fluctuation-dissipation theorem that relates ξ and γ (19). The equations of motion were integrated by a standard integrator using finite-difference derivatives (thus, Brownian noise or discontinuities in the potential derivative are not a problem). Derivatives with respect to a single particle, like the energy change ΔE, could be evaluated efficiently. The protein distribution was allowed to evolve to a mean particle deviation of at least 3.4 Rg before sampling the next configuration, to ensure that the scattering calculation did not needlessly sample very similar configurations.
Scattering signal
For each multiprotein configuration from the MMC or LMD simulations, we calculated the exact x-ray scattering by evaluating
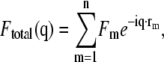 |
(4) |
where Fm is the scattering amplitude for particle m. Because we are determining isotropic interaction potentials here, we approximated each protein particle by a sphere and used the corresponding Fm (20,21). The assumption of individual spherical particles sets an upper limit on the Q values that can be fitted. A more realistic electron distribution based on diffraction data would have to be used if anisotropic potentials and large Q values are to be used in fitting. Equation 4 treats the scattering of the model protein ensemble exactly at any concentration and for any aggregation state that is small compared to the size of the simulation box. Thus, no extrapolations to dilute samples or analytical approximations usually needed for polydisperse systems need to be made. The total scattering intensity is obtained from
 |
(5) |
and averaged over all configurations sampled by the simulations to yield the average SAXS scattering intensity, I(Q), for direct comparison with experiment.
Approximately 100,000 configurations were averaged for each concentration to obtain a smooth I(Q) for comparison with experiment. To minimize boundary effects and oscillations of the intensity at low Q below the experimental noise level, either a spherical volume was chosen, and its volume was changed randomly about the average value required by each protein concentration (22), or a spherical volume from the center of a periodic boundary condition box was chosen for the x-ray scattering calculation.
Data fitting
We fitted three potential parameters: potential depth, ɛ; potential range, δ; and potential wall, 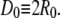 An efficient Levenberg-Marquardt optimization algorithm (23,24) was applied to fit the potential parameters to the experimental concentration-dependent scattering data. Minimal evaluation of I(Q) is desirable because each concentration point requires a large number of MMC/LMD simulations to yield a smooth curve.
An efficient Levenberg-Marquardt optimization algorithm (23,24) was applied to fit the potential parameters to the experimental concentration-dependent scattering data. Minimal evaluation of I(Q) is desirable because each concentration point requires a large number of MMC/LMD simulations to yield a smooth curve.
We also fitted a fourth parameter, the radius of gyration, Rg, of the model proteins, to account for the direct effect of particle size on the scattering data. R0 measures monomer size from the point of view of the interaction potential, whereas Rg measures monomer size from the point of view of the scattering intensity. Rg is not entirely independent of R0. For an ideal hard-sphere monomer, 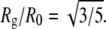 Deviations from spherical shape, and a tapering of the electron density distribution due to solvation or a soft potential wall (as in Eq. 1), are both effectively accounted for by allowing deviations from this ratio. A large deviation would indicate that a better model for the monomeric proteins is needed.
Deviations from spherical shape, and a tapering of the electron density distribution due to solvation or a soft potential wall (as in Eq. 1), are both effectively accounted for by allowing deviations from this ratio. A large deviation would indicate that a better model for the monomeric proteins is needed.
RESULTS
Concentration-dependent SAXS of λ6–85
Fig. 2 shows the concentration dependence of the scattering intensity as a function of Q for the 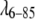 Q33Y G46A G48A mutant. A Guinier plot (ln(I) vs. Q2, not shown) deviates from linearity below Q2 = 0.006 Å−2, indicating some aggregation. Dilution of samples shows that this aggregation is reversible over the concentration range we studied. No deviations were observed at concentrations <100 μM or for Q up to 0.11 Å−1, indicating that the spherical approximation for protein monomers is good for
Q33Y G46A G48A mutant. A Guinier plot (ln(I) vs. Q2, not shown) deviates from linearity below Q2 = 0.006 Å−2, indicating some aggregation. Dilution of samples shows that this aggregation is reversible over the concentration range we studied. No deviations were observed at concentrations <100 μM or for Q up to 0.11 Å−1, indicating that the spherical approximation for protein monomers is good for 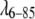 over the range of scattering angles considered here.
over the range of scattering angles considered here.
FIGURE 2.

Scattering intensity versus magnitude of the scattering vector for  (A) and fyn-SH3 (B). The lines going through the experimental data points are fits from Monte Carlo simulation.
(A) and fyn-SH3 (B). The lines going through the experimental data points are fits from Monte Carlo simulation.
MMC fitting results for λ6–85
Simulations were performed by the MMC method. The best fit to experimental data (Fig. 2 A) was obtained with a UL + UE potential (Lennard-Jones wall, attractive exponential). The calculated radius of gyration is 13.52 Å, the potential depth is 1.5 kT0, with 3.6 Å of potential range, and the potential wall beginning at D0 ≈ 31.8 Å (Table 1 and Fig. 3 A; T0 = 245 K). A total of 100 proteins was used in 5000 Metropolis iterations to obtain equilibrated results for each configuration, and 100,000 configurations were sampled. As one might expect, two parameters of this fit are somewhat correlated, the potential range and depth.
TABLE 1.
Best fit of the 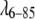 SAXS data to a UL + UE (r12 repulsive, exponential attractive) potential
SAXS data to a UL + UE (r12 repulsive, exponential attractive) potential
| Potential type | Rg (Å) | Potential depth ɛ (kT0) | Potential range δ (Å) | Potential wall D0 (Å) | RMSE |
|---|---|---|---|---|---|
| UL + UE | 13.5 ± 0.2 | 1.5 ± 0.5 | 3.6 ± 0.5 | 31.8 ± 3.0 | 0.0073 |
| Best fit | 2920 μM | 2300 μM | 1470 μM | 750 μM | 520 μM | 210 μM | Weighted average |
| RMSE | 0.0050 | 0.0049 | 0.0040 | 0.0048 | 0.0072 | 0.013 | 0.0073 |
Also shown in the table are the root mean-square errors (RMSE) for the best fit at individual concentrations. All RMSEs of the fit lie within the experimental error. kT0 corresponds to 245 K.
FIGURE 3.

(A) Best-fit interaction potential for  and fyn-SH3 (in 45% ethylene glycol buffer at −28°C). (B) Comparison of the MMC and analytical best-fit hard sphere plus Yukawa potentials for
and fyn-SH3 (in 45% ethylene glycol buffer at −28°C). (B) Comparison of the MMC and analytical best-fit hard sphere plus Yukawa potentials for  The greatest variation between the three
The greatest variation between the three  shown is in D0. (MMC parameters: D0 = 35.5 Å, δ = 4.14 Å, ɛ = 1.65 kT0, and Rg = 13.8 Å; analytical: D0 = 37.8 Å, δ = 4.14 Å (fixed), ɛ = 1.71 kT0, and Rg = 13.6 Å).
shown is in D0. (MMC parameters: D0 = 35.5 Å, δ = 4.14 Å, ɛ = 1.65 kT0, and Rg = 13.8 Å; analytical: D0 = 37.8 Å, δ = 4.14 Å (fixed), ɛ = 1.71 kT0, and Rg = 13.6 Å).
LMD simulation for λ6–85
We also performed an LMD simulation with the same potential as MMC at 2920 μM concentration, to confirm consistency of the MMC fitting results with molecular dynamics. We tested a range of different timescales (500 ns, 50 ns, 5 μs, 20 μs) for 25 proteins in a cube having periodic boundary condition. The resulting I(Q) is shown in Fig. 4 A, and agrees with the experimental data within sampling uncertainty. The sampling uncertainty of the molecular dynamics simulations is shown by the error bars. The timescale between successive configurations chosen for scattering calculations was estimated form the diffusion equation 〈r2〉 = 6Dt in 3-D, allowing the protein ensemble to move enough so that successive configurations were independent of one another.
Concentration-dependent SAXS of fyn-SH3
Fig. 2 B shows the concentration dependence I(Q) for fyn-SH3. The slope of a Guinier plot (not shown) deviates more strongly from linearity at low Q than for  indicating more extensive aggregation and a stronger interaction potential. As in the case of
indicating more extensive aggregation and a stronger interaction potential. As in the case of  the spherical monomer approximation works to the largest Q values for which data were collected.
the spherical monomer approximation works to the largest Q values for which data were collected.
Fitting results for fyn-SH3
Ensemble configurations were generated by MMC simulation. The best fit was obtained with a UL + UY + UE potential (Lennard Jones repulsive wall, attractive Yukawa potential well and repulsive exponential potential). Potentials without a repulsive long-range interaction produced significantly worse fits ( ). For the three-term potential, the calculated radius of gyration is 14.85 Å and the potential wall size is 42.0 Å. The attractive Yukawa potential depth is 11.2 kT0 with a 1 Å range, The repulsive exponential potential depth is 7.5 kT0, with a range of 2.0 Å, which results in a net potential depth of 3.65 kT (Table 2 and Fig. 3 A). We used 625,000 Metropolis iterations to obtain converged results, and 50,000 final configurations were sampled. Compared to
). For the three-term potential, the calculated radius of gyration is 14.85 Å and the potential wall size is 42.0 Å. The attractive Yukawa potential depth is 11.2 kT0 with a 1 Å range, The repulsive exponential potential depth is 7.5 kT0, with a range of 2.0 Å, which results in a net potential depth of 3.65 kT (Table 2 and Fig. 3 A). We used 625,000 Metropolis iterations to obtain converged results, and 50,000 final configurations were sampled. Compared to 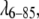 SH3 consistently produced fits with shorter range but deeper potential wells.
SH3 consistently produced fits with shorter range but deeper potential wells.
TABLE 2.
Best fit of the fyn-SH3 SAXS data to a UL + UY + UE (r12 repulsive, exponential attractive, exponential repulsive) potential
| Attractive
|
Repulsive
|
Net well depth ɛ (kT0) | ||||||
|---|---|---|---|---|---|---|---|---|
| Potential type | Rg (Å) | ɛ1 (kT0) | δ1 (Å) | ɛ2 (kT0) | δ2 (Å) | D0 (Å) | RMSE | |
| UL + UY + UE | 14.85 ± 0.2 | 11.2 ± 0.5 | 1.0 | 7.5 ± 0.2 | 2.0 | 42.0 ± 4.0 | 3.7 | 0.035 |
| Best fit | 1680 μM | 1020 μM | 690 μM | 470 μM | 190 μM | 60 μM | Weighted average |
| RMSE | 0.016 | 0.0074 | 0.010 | 0.034 | 0.028 | 0.060 | 0.035 |
Also shown in the table are the root mean-square errors (RMSE) for the best overall fit at individual concentrations. kT0 corresponds to 245 K.
LMD simulation for fyn-SH3
We also performed an LMD simulation with the converged MMC potential at 1690 μM concentration, to confirm consistency of the MMC fitting results and the molecular dynamics simulations. Again, we tested a range of different timescales (from 50 ns to 5 μs) for 25 proteins in a cube having periodic boundary condition. The resulting I(Q) is shown in Fig. 4 B, and also agrees with the experimental data within sampling uncertainty. The timescale between successive configurations was chosen by the same criterion as for 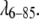
DISCUSSION
We have obtained interaction potentials for two proteins under identical buffer conditions by using the four-step procedure in Fig. 1. First, Monte Carlo or molecular dynamics simulations of a model protein ensemble compute thermally averaged or time-averaged particle distributions for up to 100 protein particles. Next, x-ray scattering functions,  are computed directly for the whole ensemble. These are essentially exact for scattering angles corresponding to the size range from monomer particle to simulation box. In the third step, the resulting scattering intensity is computed without further approximations and then compared to SAXS data. In the last step, a least-squares algorithm refines the potential parameters, so that a new simulation can be started to iterate until the best fit is obtained. The best fits are summarized in Tables 1 and 2.
are computed directly for the whole ensemble. These are essentially exact for scattering angles corresponding to the size range from monomer particle to simulation box. In the third step, the resulting scattering intensity is computed without further approximations and then compared to SAXS data. In the last step, a least-squares algorithm refines the potential parameters, so that a new simulation can be started to iterate until the best fit is obtained. The best fits are summarized in Tables 1 and 2.
Although MMC sampling and MD simulations are computationally much more expensive than the analytical approximations commonly used, direct simulation provides a correct description of the scattering amplitude at any concentration, for any monomer size, and for any aggregate shape consistent with the model monomers and up to the size of the simulation box. Any functional form of the potential, rather than a perturbing potential added to a hard-sphere repulsion, can be fitted without additional effort simply by replacing the two-body interaction potential in the simulation.
The simplifying assumptions we retained in the present application are an isotropic interaction potential and hence an isotropic monomer shape, limiting the maximum Q values that could be fitted. The ratio 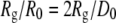 provides a connection between the interaction potential (characterized by D0) and how the protein scatters (characterized by Rg). Both proteins had a ratio within 9% of the
provides a connection between the interaction potential (characterized by D0) and how the protein scatters (characterized by Rg). Both proteins had a ratio within 9% of the 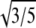 ratio expected for spherical monomers (Tables 1 and 2). Over the Q-range we examined, neither deviations of protein shapes from a sphere nor electron-density variations are likely to fully account for the difference from the ideal
ratio expected for spherical monomers (Tables 1 and 2). Over the Q-range we examined, neither deviations of protein shapes from a sphere nor electron-density variations are likely to fully account for the difference from the ideal 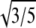 ratio. More likely, hydration water that interacts strongly with the protein surface could explain the discrepancy between the fitted values of Rg and D0, because the effective size of the hydrated protein could simply be different for the two different physical processes of x-ray scattering and protein-protein interaction.
ratio. More likely, hydration water that interacts strongly with the protein surface could explain the discrepancy between the fitted values of Rg and D0, because the effective size of the hydrated protein could simply be different for the two different physical processes of x-ray scattering and protein-protein interaction.
Indeed, our fitted radii of gyration in Tables 1 and 2 are larger than the values obtained by taking the bare protein structures from the Protein Data Bank. For example, one would expect Rg = 11.85 Å for bare 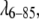 not the 13.1–13.8 Å range obtained from our fits, the best of which has Rg = 13.5 Å (Table1). It has been shown previously that the hydration layer around proteins perturbs SAXS such as to increase the effective radius by 1–2 Å. The program CRYSOL takes this effect into account (25,26). Its predicted hydrated radius of gyration is 13.5–13.8 Å, depending on the method used, in excellent agreement with the value we derived from fitting interaction potentials to the SAXS experiment. A similar result is obtained for fyn-SH3, although our experimentally fitted radius of gyration is yet another 0.5 Å larger than the one obtained from CRYSOL. This could be due to the histidine tag on our fyn-SH3 protein, which was not included in the CRYSOL calculation (no structure is available for the tag).
not the 13.1–13.8 Å range obtained from our fits, the best of which has Rg = 13.5 Å (Table1). It has been shown previously that the hydration layer around proteins perturbs SAXS such as to increase the effective radius by 1–2 Å. The program CRYSOL takes this effect into account (25,26). Its predicted hydrated radius of gyration is 13.5–13.8 Å, depending on the method used, in excellent agreement with the value we derived from fitting interaction potentials to the SAXS experiment. A similar result is obtained for fyn-SH3, although our experimentally fitted radius of gyration is yet another 0.5 Å larger than the one obtained from CRYSOL. This could be due to the histidine tag on our fyn-SH3 protein, which was not included in the CRYSOL calculation (no structure is available for the tag).
Extrapolations of the scattering data in Fig. 1 to zero concentration are fitted well by CRYSOL with Protein Data Bank structural data as input, showing that the folded monomer shapes remain consistent throughout the concentration range. Our fitting approach clearly does not require a low concentration extrapolation to yield reliable results.
This leads to the question: What range of concentrations is needed to reliably fit the potential parameters, and which parameters remain least reliably determined? The fitting uncertainties are largest for D0. We confirm in two ways that D0 is the least well constrained parameter in our fits of 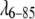 and fyn-SH3. First, we fixed it at the hard-sphere value
and fyn-SH3. First, we fixed it at the hard-sphere value 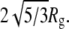 This yielded radii of gyration, Rg, well depths, ɛ, and potential ranges, δ, that agreed with Tables 1 and 2 within the indicated uncertainties. D0, on the other hand, shifted by up to 11%, showing that Rg is much more strongly constrained by the SAXS data than is D0. Still, the χ2 of the fits did increase by up to 70% when the constraint relating D0 and Rg was introduced. Thus the differences between D0 and Rg cannot be explained just by parameter uncertainties.
This yielded radii of gyration, Rg, well depths, ɛ, and potential ranges, δ, that agreed with Tables 1 and 2 within the indicated uncertainties. D0, on the other hand, shifted by up to 11%, showing that Rg is much more strongly constrained by the SAXS data than is D0. Still, the χ2 of the fits did increase by up to 70% when the constraint relating D0 and Rg was introduced. Thus the differences between D0 and Rg cannot be explained just by parameter uncertainties.
To investigate how many concentrations are needed to determine parameters, we performed fits with as few as two of the concentration series. For example, 2920 and 520 μM for 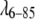 yielded a very similar potential shape (ɛ = −1.6 kT0, δ = 3.8 Å for comparison with Table 1), but the parameter D0 varies greatly (as low as D0 = 25 Å). When more concentrations are added, D0 approaches values more consistent with Rg. We conclude, at least for
yielded a very similar potential shape (ɛ = −1.6 kT0, δ = 3.8 Å for comparison with Table 1), but the parameter D0 varies greatly (as low as D0 = 25 Å). When more concentrations are added, D0 approaches values more consistent with Rg. We conclude, at least for 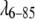 and fyn-SH3, that two concentrations are sufficient to define the shape of the potential, but that D0 must either be constrained by Rg, or requires at least 5 or 6 concentrations, including high concentrations, to be adequately constrained.
and fyn-SH3, that two concentrations are sufficient to define the shape of the potential, but that D0 must either be constrained by Rg, or requires at least 5 or 6 concentrations, including high concentrations, to be adequately constrained.
It is worth noting that analytical fitting methods also have problems determining D0 accurately. For example, two studies of the lysozyme interaction potential had to fix D0 at values ranging from 28 to 36 Å to fit the other potential parameters (2,27). The value for an ideal sphere is ∼37 Å in that case. Our numerical scattering method can be used to validate the analytical approximations usually used to obtain isotropic interaction potentials. To do so, we compared an analytical potential for 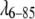 to a simulation-derived potential. To make the comparison feasible within the limitations of the analytical approach, we used a hard-sphere reference potential, coupled with an attractive Yukawa term, to yield a potential similar in shape to our best fit in Table 1. We employed the analytical method described by Winter and co-workers (2), after verifying that our analytical code reproduced their experimental SAXS intensities from their potential parameters. Fig. 3 B compares the numerical
to a simulation-derived potential. To make the comparison feasible within the limitations of the analytical approach, we used a hard-sphere reference potential, coupled with an attractive Yukawa term, to yield a potential similar in shape to our best fit in Table 1. We employed the analytical method described by Winter and co-workers (2), after verifying that our analytical code reproduced their experimental SAXS intensities from their potential parameters. Fig. 3 B compares the numerical  potential with the analytical potential. Either D0 or the potential range δ was highly correlated with potential depth in the analytical fit, so we had to fix one at the MMC value (δ in Fig. 3 B; the result looks even closer with D0 fixed). With that restriction, reasonable agreement is obtained between the analytical and simulation result. However, as already discussed above, the simulation yields a much more robust fit than the analytical model when more than two concentrations are used; it does not treat the potential as a small perturbation to a hard-sphere wall. In particular, D0 can be floated as a free parameter and yields results consistent with Rg (<9% discrepancy) when enough concentrations are fitted. To the best of our knowledge, we did not find any analytical treatments in the literature where adjusting D0 and Rg independently was possible, let alone yielded consistent results.
potential with the analytical potential. Either D0 or the potential range δ was highly correlated with potential depth in the analytical fit, so we had to fix one at the MMC value (δ in Fig. 3 B; the result looks even closer with D0 fixed). With that restriction, reasonable agreement is obtained between the analytical and simulation result. However, as already discussed above, the simulation yields a much more robust fit than the analytical model when more than two concentrations are used; it does not treat the potential as a small perturbation to a hard-sphere wall. In particular, D0 can be floated as a free parameter and yields results consistent with Rg (<9% discrepancy) when enough concentrations are fitted. To the best of our knowledge, we did not find any analytical treatments in the literature where adjusting D0 and Rg independently was possible, let alone yielded consistent results.
We examined a number of isotropic interaction potentials in addition to the best-fit and hard-wall shapes, and found that Gaussian attractive potentials generally performed more poorly than the exponential or Yukawa forms used in the DLVO model. In all fits, the 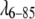 potential was longer-range than the fyn-SH3, which resembles a “sticky sphere”. A long-range but weak attractive potential for
potential was longer-range than the fyn-SH3, which resembles a “sticky sphere”. A long-range but weak attractive potential for 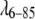 is compatible with recent terahertz measurements of hydration shells around the same mutant (7). These measurements indicated that the dynamics of water molecules are affected by the protein to >10 Å from the protein surface. Such hydration water may significantly mediate protein-protein interactions. It is even possible that the protein-protein interaction potential depends on protein concentration because of concentration-induced changes in the hydration shell. However, our current SAXS data was adequately modeled by a concentration-independent interaction potential.
is compatible with recent terahertz measurements of hydration shells around the same mutant (7). These measurements indicated that the dynamics of water molecules are affected by the protein to >10 Å from the protein surface. Such hydration water may significantly mediate protein-protein interactions. It is even possible that the protein-protein interaction potential depends on protein concentration because of concentration-induced changes in the hydration shell. However, our current SAXS data was adequately modeled by a concentration-independent interaction potential.
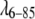 has a significantly lower propensity for aggregation than fyn-SH3, but only the latter requires a repulsive potential in the fit to match the data within experimental uncertainty (Fig. 3 A). Both proteins were examined in identical buffer solutions of 45:55 vol % ethylene glycol/water, 50 mM phosphate at pH 7.0 and −28°C. As discussed by Winter and co-workers (2), the size of the repulsive potential is very sensitive to the ionic strength and ionic composition of the buffer. Given the isoelectric points of pI = 8.25 (
has a significantly lower propensity for aggregation than fyn-SH3, but only the latter requires a repulsive potential in the fit to match the data within experimental uncertainty (Fig. 3 A). Both proteins were examined in identical buffer solutions of 45:55 vol % ethylene glycol/water, 50 mM phosphate at pH 7.0 and −28°C. As discussed by Winter and co-workers (2), the size of the repulsive potential is very sensitive to the ionic strength and ionic composition of the buffer. Given the isoelectric points of pI = 8.25 ( ) and pI = 4.84 (fyn-SH3), it is not surprising that there are differences between
) and pI = 4.84 (fyn-SH3), it is not surprising that there are differences between 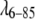 and fyn-SH3 in the screening of the long-range electrostatic repulsion.
and fyn-SH3 in the screening of the long-range electrostatic repulsion.
As measurements over wide Q-ranges become available with new high brightness synchrotron sources, the direct fitting approach will also be useful for determining anisotropic interaction potentials. This requires two additions to our treatment: the potential itself must treat anisotropic interactions, and the scattering calculation can no longer assume spherical monomers. Regarding the potential, Ha-Duong and co-workers have developed residue-residue pair potentials that can be applied to surface residues of interacting proteins (28). To treat arbitrary protein shapes, one adds a rotational diffusion term to Eq. 3 and replaces Fm in Eq. 4 with the orientation-dependent structure factor of the monomeric protein computed using a program such as CRYSOL (25). It remains to be seen how much information might be extracted from scattering data at larger angles using this approach.
In conclusion, direct fitting of SAXS data to interaction potentials via Monte Carlo or molecular dynamics simulation of a model protein ensemble provides a useful alternative to analytical approximations. The form of the potential is unrestricted and no approximations regarding the scattering amplitude of the model protein ensemble need to be made. A range of concentrations still provides the best sampling of protein-protein distances to determine the potential (the potential wall location D0 in particular), but extrapolations to zero concentration are not necessary. When the potential is restricted to have a hard-sphere wall, our method validates the analytical methods used to date, but actually fits D0 more consistently with the protein size determined by the scattering amplitude (Rg). With the advent of higher-power computing, the numerical approach demonstrated here can be extended straightforwardly to include coarse-grained anisotropic interaction potentials, and randomly reorienting nonspherical protein shapes.
Acknowledgments
We thank Dr. Liang Guo at Argonne National Laboratory for helpful assistance in setting up the beam line for experiments, and Mr. Y. Matsumura for assistance during data collection.
This work was supported by National Science Foundation grant MCB 0613643. Time on BioCAT-18, managed by Prof. Thomas Irving, was supported by proposal GUP-6360 of the Advanced Photon Source at the Argonne National Laboratory. M.G. was supported by a Lycan Professorship.
Editor: Jill Trewhella.
References
- 1.Tardieu, A., A. Le Verge, M. Malfois, F. Bonneté, S. Finet, M. Riés-Kautt, and L. Belloni. 1999. Proteins in solution: from x-ray scattering intensities to interaction potentials. J. Cryst. Growth. 196:193–203. [Google Scholar]
- 2.Javid, N., K. Vogtt, C. Krywka, M. Tolan, and R. Winter. 2007. Protein-protein interactions in complex cosolvent solutions. Phys. Chem. Chem. Phys. 8:679–689. [DOI] [PubMed] [Google Scholar]
- 3.Otting, G., and K. Wüthrich. 1989. Studies of protein hydration in aqueous solution by direct NMR observation of individual protein-bound water molecules. Biochemistry. 111:1871–1875. [Google Scholar]
- 4.Zanotti, J. M., M. C. Bellissent-Funel, and J. Parello. 1999. Hydration-coupled dynamics in proteins studied by neutron scattering and NMR: the case of the typical EF-hand calcium-binding parvalbumin. Biophys. J. 76:2390–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rösgen, J., B. M. Pettitt, and D. W. Bolen. 2005. Protein folding, stability, and solvation structure in osmolyte solutions. Biophys. J. 89:2988–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leitner, D. M., M. Havenith, and M. Gruebele. 2006. Biomolecule large-amplitude motion and solvation dynamics: modeling and probes from THz to X-rays. Int. Rev. Phys. Chem. 25:553–582. [Google Scholar]
- 7.Ebbinghaus, S., S. J. Kim, M. Heyden, X. Yu, U. Heugen, M. Gruebele, D. Leitner, and M. Havenith. 2007. An extended dynamical solvation shell around proteins. Proc. Natl. Acad. Sci. USA. In press. [DOI] [PMC free article] [PubMed]
- 8.Yang, W. Y., and M. Gruebele. 2003. Folding at the speed limit. Nature. 423:193–197. [DOI] [PubMed] [Google Scholar]
- 9.Silow, M., Y. Tan, A. R. Fersht, and M. Oliveberg. 1999. Formation of short-lived protein aggregates directly from the coil in two-state folding. Biochemistry. 38:13006–13012. [DOI] [PubMed] [Google Scholar]
- 10.Yang, W., and M. Gruebele. 2006. Binary and ternary aggregation with tethered protein constructs. Biophys. J. 90:2930–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Otzen, D. E., S. Miron, M. Akke, and M. Oliveberg. 2004. Transient aggregation and stable dimerization induced by introducing an Alzheimer sequence into a water-soluble protein. Biochemistry. 43:12964–12978. [DOI] [PubMed] [Google Scholar]
- 12.Niehbur, M., and M. H. J. Koch. 2005. Effects of urea and trimethylamine-N-oxide (TMAO) on the interactions of lysozyme in solution. Biophys. J. 89:1978–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dumont, C., Y. Matsumura, S. J. Kim, J. Li, E. Kondrashkina, H. Kihara, and M. Gruebele. 2006. Solvent-tuning collapse and helix formation time scales of λ6-85. Protein Sci. 15:2596–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghaemmaghami, S., J. M. Word, R. E. Burton, J. S. Richardson, and T. G. Oas. 1998. Folding kinetics of a fluorescent variant of monomeric lambda repressor. Biochemistry. 37:9179–9185. [DOI] [PubMed] [Google Scholar]
- 15.Larson, S. M., A. A. D. Nardo, and A. R. Davidson. 2000. Analysis of covariation in an SH3 domain sequence alignment: applications in tertiary contact prediction and the design of compensating hydrophobic core substitutions. J. Mol. Biol. 303:433–446. [DOI] [PubMed] [Google Scholar]
- 16.Edelhoch, H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 6:1948–1954. [DOI] [PubMed] [Google Scholar]
- 17.Fischetti, R., S. Stepanov, G. Rosenbaum, R. Barrea, E. Black, D. Gore, R. Heurich, E. Kondrashkina, A. J. Kropf, S. Wang, K. Zhang, T. C. Irving, and G. B. Bunke. 2004. The BioCAT undulator beamline 18ID: a facility for biological non-crystalline diffraction and X-ray absorption spectroscopy at the Advanced Photon Source. J. Synchrotron Radiat. 11:399–405. [DOI] [PubMed] [Google Scholar]
- 18.Metropolis, N., A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller, and E. Teller. 1953. Equation of state calculations by fast computing machines. J. Chem. Phys. 21:1087–1092. [Google Scholar]
- 19.Chandler, D. 1989. Modern Statistical Mechanics. Oxford University Press, Oxford, UK.
- 20.Rayleigh, L. 1914. On the diffraction of light by spheres of small relative index. Proc. Roy. Soc. Lond. A-90:219–225. [Google Scholar]
- 21.Rayleigh, L. 1910. The incidence of light upon a transparent sphere of dimension comparable to the wavelength. Proc. Roy. Soc. Lond. A-84:25–46. [Google Scholar]
- 22.Suhonen, H. 2005. Simulation of Small-Angle X-Ray Scattering from Collagen Fibrils. Master's thesis. University of Helsinki, Helsinki, Finland.
- 23.Moré, J. J., B. S. Garbow, and K. E. Hillstrom. 1980. User Guide for MINPACK-1. In Argonne National Laboratory Report ANL-80-74. Argonne National Laboratory, Argonne, IL. 1–48.
- 24.Moré, J. J., D. C. Sorensen, and K. E. Hillstrom. 1984. The MINPACK Project. In Sources and Development of Mathematical Software. W. J. Cowell, editor. Prentice-Hall, New York. 88–111.
- 25.Svergun, D., C. Barberato, and M. H. J. Koch. 1995. CRYSOL: a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 28:768–773. [Google Scholar]
- 26.Svergun, D., S. Richard, M. H. J. Koch, Z. Sayers, S. Kuprin, and G. Zaccai. 1998. Hydration shell in solution: validation by x-ray and neutron scattering. Proc. Natl. Acad. Sci. USA. 95:2267–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narayanan, J., and X. Y. Liu. 2003. Protein interactions in undersaturated and supersaturated solutions: a study using light and x-ray scattering. Biophys. J. 84:523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bosdevant, N., D. Borgis, and T. Ha-Duong. 2007. A coarse-grained protein-protein potential derived from an all-atom force field. J. Phys. Chem. B. 111:9390–9399. [DOI] [PubMed] [Google Scholar]
- 29.Humphrey, W. F., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]