

Abstract
It is now quite well accepted that charge separation in PS2 reaction centers starts predominantly from the accessory chlorophyll BA and not from the special pair P680. To identify spectral signatures of BA, and to further clarify the process of primary charge separation, we compared the femtosecond-infrared pump-probe spectra of the wild-type (WT) PS2 core complex from the cyanobacterium Synechocystis sp. PCC 6803 with those of two mutants in which the histidine residue axially coordinated to PB (D2-His197) has been changed to Ala or Gln. By analogy with the structure of purple bacterial reaction centers, the mutated histidine is proposed to be indirectly H-bonded to the C9=O carbonyl of the putative primary donor BA through a water molecule. The constructed mutations are thus expected to perturb the vibrational properties of BA by modifying the hydrogen bond strength, possibly by displacing the H-bonded water molecule, and to modify the electronic properties and the charge localization of the oxidized donor 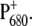 Analysis of steady-state light-induced Fourier transform infrared difference spectra of the WT and the D2-His197Ala mutant indeed shows that a modification of the axially coordinating ligand to PB induces a charge redistribution of
Analysis of steady-state light-induced Fourier transform infrared difference spectra of the WT and the D2-His197Ala mutant indeed shows that a modification of the axially coordinating ligand to PB induces a charge redistribution of 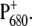 In addition, a comparison of the time-resolved visible/midinfrared spectra of the WT and mutants has allowed us to investigate the changes in the kinetics of primary charge separation induced by the mutations and to propose a band assignment identifying the characteristic vibrations of BA.
In addition, a comparison of the time-resolved visible/midinfrared spectra of the WT and mutants has allowed us to investigate the changes in the kinetics of primary charge separation induced by the mutations and to propose a band assignment identifying the characteristic vibrations of BA.
INTRODUCTION
Light absorption and energy transduction in plants, algae and cyanobacteria occurs in two large transmembrane pigment-protein complexes, photosystem I and photosystem II (PS2), the latter being able to do a one- and two-electron reduction of plastoquinone and to oxidize water to molecular O2. The resolution of the crystal structure of the PS2 core complex, which contains the two antenna proteins, CP43 and CP47, and the D1D2-cytb559 reaction center (RC), has been recently improved, and two crystal structures, at 3.4 and 3.0 Å resolution (1–3), respectively, are now available. The structural data indicates that CP43 binds 14 chlorophylls a (Chla) and two or three β-carotenes. CP47 binds 17 Chla and at least two carotenoids. The D1D2-cytb559 RC binds six Chla, two pheophytins (Phe), two β-carotenes, the primary and secondary plastoquinone acceptors QA and QB, and the water oxidizing Mn-complex.
Light absorption by the antenna proteins is followed by excitation energy transfer and trapping of energy in the RC (4,5). When the excitation energy reaches the D1D2-RC, electron transfer takes place in a few picoseconds, leading to the formation of the radical pair 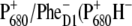 ), where P680 is the so-called Chla special pair and PheD1 is the active branch pheophytin (6–9). The electron is then transferred from the pheophytin to the primary quinone electron acceptor, QA, in a few hundred picoseconds and from there to the secondary quinone, QB, on the hundreds of microseconds timescale (10–12). The special pair
), where P680 is the so-called Chla special pair and PheD1 is the active branch pheophytin (6–9). The electron is then transferred from the pheophytin to the primary quinone electron acceptor, QA, in a few hundred picoseconds and from there to the secondary quinone, QB, on the hundreds of microseconds timescale (10–12). The special pair 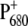 is reduced by a redox active tyrosine (YZ, D1-Tyr161), and the oxidizing equivalent is then transferred to the Mn cluster. For every two turnovers of electron transfer, accompanied by the uptake of two protons, the secondary quinone, QB, is fully reduced to the quinol, QBH2. For every four turnovers, accompanied by the release of four protons, two water molecules are oxidized to generate a molecule of oxygen in the Mn cluster.
is reduced by a redox active tyrosine (YZ, D1-Tyr161), and the oxidizing equivalent is then transferred to the Mn cluster. For every two turnovers of electron transfer, accompanied by the uptake of two protons, the secondary quinone, QB, is fully reduced to the quinol, QBH2. For every four turnovers, accompanied by the release of four protons, two water molecules are oxidized to generate a molecule of oxygen in the Mn cluster.
The dynamics of energy transfer and charge separation in PS2 has been extensively studied by several groups using different spectroscopic techniques such as time-resolved pump-probe absorption (9,13–18), time-resolved fluorescence (19), hole burning (20,21), Stark (22), and Fourier transform infrared (FTIR) (23,24) spectroscopies. However, there is still an open discussion in the literature regarding the exact mechanisms and dynamics of energy and electron transfer occurring in response to light absorption in PS2.
Recent transient absorption and time-resolved fluorescence measurements were interpreted on the basis of an exciton-radical pair equilibrium model, which implies ultrafast equilibration between the CP43 and CP47 pigment-protein complexes with the D1D2-RC followed by primary charge separation on a 9-ps timescale (9,25). However, recent visible (vis)/mid-IR pump-probe measurements performed on intact PS2 core complexes from spinach were analyzed and interpreted on the basis of a kinetic model according to which energy transfer from the core antennas toward the D1D2-RC occurs on a much slower timescale of ∼30–40 ps, whereas direct excitation of the reaction center leads to fast charge separation, on a timescale of 1–2 ps (26). The latter interpretation is more in line with the available structural information on the PS2 core, showing that the minimum distance between the pigments contained in the antenna complexes and those contained in the D1D2-RC is ∼20 Å. Furthermore, slow energy transfer components are also observed in the isolated antenna complexes: both vis/vis and vis/mid-IR pump-probe measurements on isolated CP43 and CP47 complexes demonstrate that the intra-antenna energy transfer dynamics are indeed highly multiphasic, showing rapid equilibration components, which are complete on a 2-ps timescale, and a slow energy transfer phase toward a rather isolated red-shifted Chla occurring in ∼10–20 ps for both complexes (27–29).
As concerns the mechanism of primary charge separation, it was initially proposed, by analogy to the nonoxygenic purple bacterial RCs, that in PS2, as well, a “special pair” of chlorophylls exists and acts as a primary donor. However, in contrast to what is observed in RCs from purple bacteria, there is no spectroscopic evidence in PS2 to support this idea, as all the Chla and pheophytin absorb at nearly the same wavelength. The absence of a strong excitonically-coupled special pair led to the proposition of the “multimer model” (30), according to which the electronic interactions between the different chlorins contained in the D1D2-RC are all similar, allowing charge separation to start from any pigment, depending on the particular realization of the disorder. Support for this idea was first obtained in bacterial reaction centers where it was shown that after selective excitation of the monomer chlorophyll located on the active branch, BA, ultrafast charge separation occurred without the involvement of the excited special pair (31,32).
Recently, it was inferred, using time-resolved visible-pump/mid-IR spectroscopy (8), and later confirmed, also using vis/vis experiments (9), that in isolated D1D2-RC, the electron hole in the primary radical pair is localized on a site other than P680, most probably on the monomer chlorophyll BA. The electron hole subsequently migrates to P680, giving rise to the formation of the radical pair 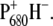 The justification for this kinetic mechanism in the infrared work was based on the observation that absorption signatures characteristic of the formation of H− appeared within 0.8 ps, whereas the signals belonging to
The justification for this kinetic mechanism in the infrared work was based on the observation that absorption signatures characteristic of the formation of H− appeared within 0.8 ps, whereas the signals belonging to 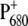 only appeared after 6 ps (8).
only appeared after 6 ps (8).
Although the formation of the pheophytin anion, H−, on a subpicosecond timescale was clearly demonstrated by this experiment, a definite assignment of the 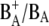 absorption bands could not be made. One of the most straightforward ways to identify spectral signatures of a particular pigment is to construct a site-specific mutant that modifies its immediate protein environment. This is particularly useful where infrared spectroscopy is concerned, as absorption in this spectral region is highly sensitive to the environment in which the pigment is located. In the case of photosynthetic proteins, one of the most informative regions is that between 1600 and 1800 cm−1, which mainly probes the 9-keto and 10a-ester chlorophyll groups. For these features, spectral shifts of several tens of cm−1 are expected due to H-bonds, pigment-pigment interactions, or changes in polarity of the environment relative to the frequency observed for Chla dissolved in a nonpolar solvent (33–35). Thus, in principle, the comparison of time-resolved absorbance changes in the infrared of wild-type (WT) and specific mutants that modify the environment of BA, and, in particular, the strength of the hydrogen bond to its 9-keto carbonyl group, should allow us to identify the characteristic spectral features of this pigment. Furthermore, the analysis of the changes induced by the specified mutations on the kinetics of primary energy transfer and charge separation should give additional information on the role played from BA in these processes. It is preferable to use PS2 core complexes, which are the basis for the x-ray crystallographic structures and are more stable and structurally intact than the D1D2-RC preparations. This of course complicates the spectral analysis because of additional dynamic processes associated with the CP43 and CP47 antenna complexes. Time-resolved vis-pump/mid-IR-probe spectroscopy measurements, recently performed on intact PS2 core complexes from spinach, have demonstrated that this technique has the sensitivity necessary to study the complex dynamics of these systems. The application of a kinetic scheme to the time-resolved data by means of target analysis has furthermore demonstrated that the core complex can be spectrally described as the sum of its components, namely CP43, CP47, and the D1D2-RC (26). Based on these results, we have employed time-resolved vis-pump/mid-IR spectroscopy to compare the initial dynamics of energy transfer and charge separation in core complexes isolated from the WT and two PS2 reaction center mutants of Synechocystis sp. PCC 6803. Our goal was to identify spectral signatures of BA, and to further clarify the process of primary charge separation. In the mutants analyzed, the histidine residue axially coordinated to PB (D2-His197) has been changed to Ala or Gln. Analysis of the available crystal structure of PS2 cores (2,3) shows the possibility of an indirect H-bond between the mutated D2-His197 and the 9-keto carbonyl of the putative primary donor BA, which, by analogy to purple bacterial reaction centers (36), would occur through a water molecule. The mutation of the axially coordinating histidine of PB with a smaller, noncoordinating (Ala) or differently coordinating (Gln) residue is expected to perturb the H-bond to the 9-keto carbonyl of BA. The structural arrangement of P680 is shown in Fig. 1, together with the mutated histidine.
absorption bands could not be made. One of the most straightforward ways to identify spectral signatures of a particular pigment is to construct a site-specific mutant that modifies its immediate protein environment. This is particularly useful where infrared spectroscopy is concerned, as absorption in this spectral region is highly sensitive to the environment in which the pigment is located. In the case of photosynthetic proteins, one of the most informative regions is that between 1600 and 1800 cm−1, which mainly probes the 9-keto and 10a-ester chlorophyll groups. For these features, spectral shifts of several tens of cm−1 are expected due to H-bonds, pigment-pigment interactions, or changes in polarity of the environment relative to the frequency observed for Chla dissolved in a nonpolar solvent (33–35). Thus, in principle, the comparison of time-resolved absorbance changes in the infrared of wild-type (WT) and specific mutants that modify the environment of BA, and, in particular, the strength of the hydrogen bond to its 9-keto carbonyl group, should allow us to identify the characteristic spectral features of this pigment. Furthermore, the analysis of the changes induced by the specified mutations on the kinetics of primary energy transfer and charge separation should give additional information on the role played from BA in these processes. It is preferable to use PS2 core complexes, which are the basis for the x-ray crystallographic structures and are more stable and structurally intact than the D1D2-RC preparations. This of course complicates the spectral analysis because of additional dynamic processes associated with the CP43 and CP47 antenna complexes. Time-resolved vis-pump/mid-IR-probe spectroscopy measurements, recently performed on intact PS2 core complexes from spinach, have demonstrated that this technique has the sensitivity necessary to study the complex dynamics of these systems. The application of a kinetic scheme to the time-resolved data by means of target analysis has furthermore demonstrated that the core complex can be spectrally described as the sum of its components, namely CP43, CP47, and the D1D2-RC (26). Based on these results, we have employed time-resolved vis-pump/mid-IR spectroscopy to compare the initial dynamics of energy transfer and charge separation in core complexes isolated from the WT and two PS2 reaction center mutants of Synechocystis sp. PCC 6803. Our goal was to identify spectral signatures of BA, and to further clarify the process of primary charge separation. In the mutants analyzed, the histidine residue axially coordinated to PB (D2-His197) has been changed to Ala or Gln. Analysis of the available crystal structure of PS2 cores (2,3) shows the possibility of an indirect H-bond between the mutated D2-His197 and the 9-keto carbonyl of the putative primary donor BA, which, by analogy to purple bacterial reaction centers (36), would occur through a water molecule. The mutation of the axially coordinating histidine of PB with a smaller, noncoordinating (Ala) or differently coordinating (Gln) residue is expected to perturb the H-bond to the 9-keto carbonyl of BA. The structural arrangement of P680 is shown in Fig. 1, together with the mutated histidine.
FIGURE 1.

Relative positions of PA, PB, and BA, and of the mutated histidine D2-H197.
We report here the comparison of time-resolved vis-pump/midIR-probe spectra of WT PS2 cores with those of the D2-His197Ala and D2-His197Gln mutants. Furthermore, we have employed light-induced FTIR difference spectroscopy to characterize the modification induced by the mutation on the electronic structure of P680 and on the charge distribution of 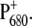 The observed spectral differences are discussed in terms of the charge redistribution of
The observed spectral differences are discussed in terms of the charge redistribution of 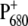 and possible spectral assignments for BA.
and possible spectral assignments for BA.
MATERIALS AND METHODS
Sample preparation and mutant construction
PS2 core samples from Synechocystis sp. PCC 6803 were prepared as described previously (37).
FTIR measurements
Samples for FTIR measurements were suspended in 50 mM MES buffer (20 mM CaCl2, 5 mM MgCl2, and 0.03% n-dodecyl-β-D-maltoside, pH 6.1) in D2O, in the presence of 50 mM silicomolybdate, to promote oxidation of the pheophytin anion.
The samples were sandwiched between two BaF2 plates and the FTIR difference spectra were recorded at 250 K on a Nicolet Magna 860 spectrophotometer equipped with an MCT detector and a gas-flow cryostat. Difference spectra were calculated from data sets consisting of ∼1000 cycles of 32 scans recorded before and after continuous illumination. The spectral resolution was 4 cm−1.
Vis-pump/mid-IR-probe spectroscopy
For these experiments, the sample was concentrated to an optical density (OD) of 0.5 (WT), 0.68 (D2-His197Ala), and 0.4 (D2-His197Gln) at 670 nm for a 20-μm optical path length and suspended in 50 mM MES buffer (20 mM CaCl2, 5 mM MgCl2, and 0.03% n-dodecyl-β-D-maltoside, pH 6.1) in D2O. All measurements were performed on closed reaction centers, and no mediators were added to oxidize QA. The experimental setup consisted of an integrated Ti:sapphire oscillator-regenerative amplifier laser system (Hurricane, SpectraPhysics, Mountain View, CA) operating at 1 kHz and 800 nm, producing 85-fs pulses of 0.6 mJ. A portion of the 800-nm light was used to pump a noncollinear optical parametric amplifier to produce the excitation pulses at 680 nm, which were focused on the sample with a 20-cm lens to a spot size of ∼150 μm in diameter. A second portion of the 800-nm light was used to pump an optical parametric generator and amplifier with a difference frequency generator (TOPAS, Light Conversion, Vilnius, Lithuania) to produce the mid-IR probe pulses, which were focused on the sample with a 6-cm lens. The probe and pump pulses were spatially overlapped in the sample. After passing through the sample, the probe pulses were dispersed by a spectrograph, imaged onto a 32-element MCT detector, and fed into 32 home-built integrate-and-hold devices that were read out at every shot with a National Instruments (Austin, TX) acquisition card. To ensure a fresh spot for each laser shot, the sample was displaced by a home-built Lissajous scanner. The polarization of the excitation pulse was set to the magic angle (54.7°) with respect to the IR probe pulses. A phase-locked chopper operating at 500 Hz was used to ensure that the sample was excited by every other shot and that the change in transmission could be measured. The instrument response function was ∼150 fs. The excitation wavelength was 680 nm for WT and the D2-His197Gln mutant, and 675 nm for the D2-His197Ala mutant. The excitation energy was kept at 100 nJ for all of the experiments. At least 200 scans for each sample were averaged to produce the finally analyzed time-resolved spectra. The data were subjected to global and target analysis (38). The noise level in the row data is ∼380 μOD, which mainly consists of structureless baseline noise due to the fact that no reference probe pulse has been used. Subtraction of the baseline noise by singular vector decomposition of the residual matrix leads to a reduction of the noise to 200 μOD as estimated by the fitting procedure.
RESULTS
Light-induced FTIR difference spectra
Previous characterization of a set of PS2 mutants in which the axially coordinating histidine of both PA (D1-His198) and PB (D2-His197) had been replaced by different residues, showed that the reduction potential of the redox couple 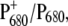 as well as the charge distribution among
as well as the charge distribution among 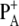 and
and 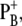 could be modulated by ligand replacement at position D1-198 and, to a lesser extent, at position D2-197 (37). Of all of the mutations constructed at D2-197, the replacement of histidine by alanine showed the largest effect on the spectral and redox properties of P680. In this case, the
could be modulated by ligand replacement at position D1-198 and, to a lesser extent, at position D2-197 (37). Of all of the mutations constructed at D2-197, the replacement of histidine by alanine showed the largest effect on the spectral and redox properties of P680. In this case, the 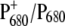 difference absorption spectrum was red-shifted with respect to the WT and the reduction potential of PB decreased by 20–40 mV, enhancing the stabilization of the positive charge on
difference absorption spectrum was red-shifted with respect to the WT and the reduction potential of PB decreased by 20–40 mV, enhancing the stabilization of the positive charge on 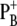 and, consequently, a different amount of charge sharing between the two chlorophylls forming the dimer. It is likely that in the D2-His197Ala mutant a water molecule has replaced His as the axial ligand. The modification of the electronic and redox properties of P680 is expected to influence the dynamics of the primary charge separation and possibly also the spectral properties of the radical pair
and, consequently, a different amount of charge sharing between the two chlorophylls forming the dimer. It is likely that in the D2-His197Ala mutant a water molecule has replaced His as the axial ligand. The modification of the electronic and redox properties of P680 is expected to influence the dynamics of the primary charge separation and possibly also the spectral properties of the radical pair 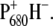 To characterize these effects, we compared the
To characterize these effects, we compared the 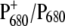 light-induced FTIR difference spectrum of the WT PS2 core complex with that of the D2-His197Ala mutant. Fig. 2 shows the light-induced FTIR difference spectra of PS2 core complexes from Synechocystis sp. PCC 6803 isolated from the WT and from the D2-His197Ala mutant measured at 250 K, in the presence of silicomolybdate as external redox mediator, which is able to oxidize the pheophytin, thus allowing photoaccumulation of
light-induced FTIR difference spectrum of the WT PS2 core complex with that of the D2-His197Ala mutant. Fig. 2 shows the light-induced FTIR difference spectra of PS2 core complexes from Synechocystis sp. PCC 6803 isolated from the WT and from the D2-His197Ala mutant measured at 250 K, in the presence of silicomolybdate as external redox mediator, which is able to oxidize the pheophytin, thus allowing photoaccumulation of 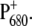
FIGURE 2.

Light-induced 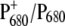 FTIR difference spectra from Synechocystis PS2 core complexes from the WT (a) and D2-His197Ala (b) (T = 250 K; 4 cm−1 resolution).
FTIR difference spectra from Synechocystis PS2 core complexes from the WT (a) and D2-His197Ala (b) (T = 250 K; 4 cm−1 resolution).
The WT and mutant spectra show several differences. Signals from the 9-keto and 10-ester carbonyl vibrations of P680 are expected to be located in the region between 1750 and 1650 cm−1. In particular, the two positive signals at 1709 and 1725 cm−1 have been assigned to the 9-keto carbonyls of 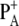 and
and 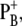 which are upshifted upon cation formation (24,39). The corresponding bleaching signals are observed at 1699 and 1681 cm−1, but the assignment of these bands is not without controversy. Recently, the
which are upshifted upon cation formation (24,39). The corresponding bleaching signals are observed at 1699 and 1681 cm−1, but the assignment of these bands is not without controversy. Recently, the 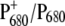 FTIR spectra obtained from BBY membranes, PS2 core, and D1D2-RC preparations have been compared and characterized (40). On the basis of the measured spectra, the authors concluded that both the PA and PB 9-keto carbonyls have similar frequencies at ∼1700 cm−1, and that neither are implicated in hydrogen bonds. A close look at the crystal structure of the PS2 core complex, however, shows that D2-Ser282 can directly hydrogen-bond to the 10a-ester carbonyl, and possibly—through an intervening water molecule—to the 9-keto carbonyl, of PB, thus suggesting that its absorption might be downshifted with respect to the corresponding group of PA, located in a less polar environment. Assuming this to be the case, the bleaching at 1681 cm−1 could be attributed to PB. A definitive assignment of these bands can be obtained only by analyzing more site-specific mutants. Work in this direction is in progress. The idea that the 9-keto groups of PA and PB absorb at different frequencies is, however, also supported by the comparison of the spectra shown in Fig. 2. Here, we observe that the relative intensity of the two positive signals at 1709 and 1724 cm−1 and of the two bleaching signals at 1699 and 1680 cm−1 is different between WT and mutant. We expect, based on previous measurements (37), that the mutation at D2-His197 influences the charge redistribution on
FTIR spectra obtained from BBY membranes, PS2 core, and D1D2-RC preparations have been compared and characterized (40). On the basis of the measured spectra, the authors concluded that both the PA and PB 9-keto carbonyls have similar frequencies at ∼1700 cm−1, and that neither are implicated in hydrogen bonds. A close look at the crystal structure of the PS2 core complex, however, shows that D2-Ser282 can directly hydrogen-bond to the 10a-ester carbonyl, and possibly—through an intervening water molecule—to the 9-keto carbonyl, of PB, thus suggesting that its absorption might be downshifted with respect to the corresponding group of PA, located in a less polar environment. Assuming this to be the case, the bleaching at 1681 cm−1 could be attributed to PB. A definitive assignment of these bands can be obtained only by analyzing more site-specific mutants. Work in this direction is in progress. The idea that the 9-keto groups of PA and PB absorb at different frequencies is, however, also supported by the comparison of the spectra shown in Fig. 2. Here, we observe that the relative intensity of the two positive signals at 1709 and 1724 cm−1 and of the two bleaching signals at 1699 and 1680 cm−1 is different between WT and mutant. We expect, based on previous measurements (37), that the mutation at D2-His197 influences the charge redistribution on 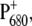 which modifies the intensity of these signals. Changes in the relative intensity and/or position of the carbonyl bands upon ligand mutation could result from a partial change in bond order for the 9-keto carbonyl group, a possible consequence of distortion of the chlorophyll ring, or a redistribution of charge on ring V of the macrocycle.
which modifies the intensity of these signals. Changes in the relative intensity and/or position of the carbonyl bands upon ligand mutation could result from a partial change in bond order for the 9-keto carbonyl group, a possible consequence of distortion of the chlorophyll ring, or a redistribution of charge on ring V of the macrocycle.
Further evidence for charge redistribution on 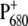 can be observed in the 1550–1500 cm−1 region, which is characteristic of amide II vibrations, but where vibrational modes of the Chla ring are also expected. In particular, the signal observed at 1554 cm−1 for the WT is downshifted by 2 cm−1 and is much more intense for the mutant, which could indicate a ring distortion due to the mutation of the axially coordinating residue. Two small negative bands observed at 1536 and 1521 cm−1 in the case of WT coalesce in a broader band centered at 1527 cm−1 in case of the mutant. Positive bands in the region 1500–1430 cm−1 are also slightly enhanced for the mutant. Probably the most direct evidence for charge redistribution on
can be observed in the 1550–1500 cm−1 region, which is characteristic of amide II vibrations, but where vibrational modes of the Chla ring are also expected. In particular, the signal observed at 1554 cm−1 for the WT is downshifted by 2 cm−1 and is much more intense for the mutant, which could indicate a ring distortion due to the mutation of the axially coordinating residue. Two small negative bands observed at 1536 and 1521 cm−1 in the case of WT coalesce in a broader band centered at 1527 cm−1 in case of the mutant. Positive bands in the region 1500–1430 cm−1 are also slightly enhanced for the mutant. Probably the most direct evidence for charge redistribution on 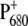 comes from the downshift of the peak at 1308 cm−1, which is observed at 1302 cm−1 for the mutant. This band should correspond to the one observed at 1290 cm−1 in the P+/P FTIR difference spectra from Rhodobacter sphaeroides RCs, which, together with modes at 1480 and 1560 cm−1, have been attributed to a “phonon mode”, forbidden by symmetry in monomer chlorophyll, but allowed when charge sharing between two Chla molecules is established (41–44). The observed shifts are thus indicative of a modification in the electronic properties of P680 and charge distribution on
comes from the downshift of the peak at 1308 cm−1, which is observed at 1302 cm−1 for the mutant. This band should correspond to the one observed at 1290 cm−1 in the P+/P FTIR difference spectra from Rhodobacter sphaeroides RCs, which, together with modes at 1480 and 1560 cm−1, have been attributed to a “phonon mode”, forbidden by symmetry in monomer chlorophyll, but allowed when charge sharing between two Chla molecules is established (41–44). The observed shifts are thus indicative of a modification in the electronic properties of P680 and charge distribution on 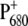 determined by the mutation of D2-His197.
determined by the mutation of D2-His197.
Visible-pump/mid-IR probe measurements
Wild-type
WT PS2 core particles were excited with 100-nJ, 680-nm laser pulses and absorption changes were recorded in the region between 1560 and 1780 cm−1. Time-resolved data were globally analyzed using a sequential model with increasing lifetimes (38). Four components were necessary to properly fit the data, which resulted in lifetimes of 2.0 ps, 17 ps, 114 ps, and 1.5 ns. The corresponding evolution-associated difference spectra (EADS) are shown in Fig. 3.
FIGURE 3.

EADS resulting from global analysis of time-resolved data from Synechocystis sp. PCC 6803 PS2 WT core complexes using a sequential kinetic scheme with increasing lifetimes. Pump-probe data in the mid-IR region between 1560 and 1780 cm−1 were obtained upon excitation at 680 nm, 100 nJ excitation power.
In the case of PS2 from spinach, a comparison with the previously measured spectra of CP43 (27), CP47 (28), and isolated D1D2-RC (8) has shown that the spectra obtained using core complexes contain spectral features representative of the isolated pigment-protein complexes (26). The first spectral component, with a lifetime of 2 ps, shows two bleaching signals in the region 1705–1680 cm−1, which are representative of the 9-keto stretch absorption bands of the different chlorins contained both in the D1D2-RC and in the antenna proteins. As already mentioned, the 9-keto bands are very sensitive to their surroundings and downshifts >20 cm−1 can be expected with respect to the frequency observed in nonpolar solvents (e.g., 1695 cm−1 in tetrahydrofuran) (33–35) due to the presence of hydrogen bonds or differences in polarity of the surrounding medium. The less intense bleaching observed at higher wavenumbers (∼1740 cm−1) can be assigned to 10a-ester absorption. In the excited state, both 9-keto carbonyl and 10a-ester absorption bands downshift by several wavenumbers, whereas upshifts are observed when a positive charge is created on a chlorophyll molecule (8,28,45). The intense and broad band observed in the initial spectral component centered around 1650 cm−1 can thus be assigned to excited-state chlorophylls located both in the antennas and in the reaction center, whereas the two positive bands at 1728 and 1709 cm−1 are probably due to a mixture of 10a-ester downshift in the excited state, charge transfer between strongly interactive molecules (27,40), and initial charge separation upon direct excitation of the RC.
In the following component, we observe a partial recovery of the two bleaching carbonyl bands and a decay of the induced absorption band, due to annihilation and excited-state decay as a result of the excitation density being >1 photon/PS2 core complex. After 17 ps, the spectrum represented by the green line appears. Here, we observe a further decay of excited-state absorption and we clearly see the appearance of a bleaching at ∼1656 cm−1, which has been attributed to a protein response after charge separation (8). The bleaching at ∼1702 cm−1 downshifts by several wavenumbers, both in this and in the last spectral component.
The last spectrum should represent the final charge-separated state 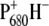 since, although core preparations contain the primary acceptor quinone, QA, our measurements were executed on “closed centers”, with QA reduced. Indeed, there is good agreement between the last spectral component measured by vis-pump/mid-IR-probe spectroscopy with the FTIR spectrum representative of the radical pair
since, although core preparations contain the primary acceptor quinone, QA, our measurements were executed on “closed centers”, with QA reduced. Indeed, there is good agreement between the last spectral component measured by vis-pump/mid-IR-probe spectroscopy with the FTIR spectrum representative of the radical pair 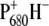 (Fig. 4).
(Fig. 4).
FIGURE 4.

Comparison between the long-lived component obtained from the global analysis of the vis-pump/mid-IR-probe data and the 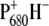 spectrum measured by light-induced FTIR measurements for WT PS2 core complexes from Synechocystis sp. PCC 6803.
spectrum measured by light-induced FTIR measurements for WT PS2 core complexes from Synechocystis sp. PCC 6803.
Mutants
The D2-His197Gln mutant was excited with 100-nJ, 680-nm laser pulses, whereas in the case of D2-His197Ala, 100-nJ, 675-nm pulses were used. In both cases, the kinetic traces were globally analyzed using a sequential model with increasing lifetimes, and again four components were necessary to properly fit the data. A comparison of the EADS obtained by global analysis of WT PS2 and the two mutants is shown in Fig. 5.
FIGURE 5.

Comparison between the EADS obtained from global analysis of the time-resolved data from Synechocystis sp. PCC 6803 PS2 core WT and D2-His197Gln, and D2-His197Ala mutants. Pump-probe data in the mid-IR region between 1560 and 1780 cm−1 were obtained using 100-nJ excitation pulses at 680 nm for the WT and D2-His197Gln samples and at 675 nm for the D2-His197Ala mutant.
A comparison of the WT EADS with those of the two mutants shows that the overall aspect of the spectral evolution remains unchanged, although significant differences are apparent, particularly in the carbonyl bleaching region. In the case of WT, two bleaching signals are evident in the early spectral component, an intense one at 1702 cm−1 and a less pronounced one at 1680 cm−1, whereas for the D2-His197Gln, an additional small bleaching at ∼1690 cm−1 is visible in the first spectral component, which increases in the 16-ps component and then partially recovers in the following spectral evolution. In the case of the D2-His197Ala mutant, again two bleaching signals are visible, the positions of which are similar to those observed for the WT. However, in the early spectral components, the intensity ratio between them is reversed, with the bleaching at 1680 cm−1 being the most intense.
The dynamics of the charge separation process is somewhat affected by the mutations, to a greater extent for the D2-His197Ala mutant, where the first two components appear faster than the WT, whereas the third one is significantly slower. The spectral differences between the three sets of measurements are evident already in the EADS of the fastest time component, which probably implies that the mutation also affects the distribution of the initial excitation between the D1D2-RC and the antenna complexes CP43 and CP47 because of a modification of the electronic levels of P680/BA.
Target analysis
Global analysis, based on a simple sequential scheme with increasing lifetimes, shows that there exist notable spectral differences between the time-resolved data sets for the WT and mutants. However, the EADS obtained for such global analyses are not representative of a particular state of the system, but, particularly in the case of the fastest components, can represent a mixture of excited and charge-separated states. To extract the spectra corresponding to the different states of the system as it evolves over time, it is necessary to introduce a specific kinetic model and to analyze the data by means of a target or compartmental analysis. We describe here the kinetic model used to perform such target analysis and the spectral assignments derived from a comparison of the resulting spectral components of the WT and mutant samples.
Given the number of pigments contained within the system, and on which the excitation can be initially localized, it is necessary to find a simplified scheme with a limited number of compartments, which is able to give a satisfactory description of the processes occurring within the system. We apply here a model used recently to analyze the dynamics of energy transfer and primary charge separation in PS2 cores from spinach (26). The kinetic scheme used in this and in the previous work is represented in Fig. 6, where the species-associated difference spectra (SADS) obtained for the WT are also shown.
FIGURE 6.

(A) Five-compartment model employed for the target analysis of the time-resolved data; the rates given in the scheme are those of the WT. (B) Species-associated difference spectra derived from target analysis for PS2 core complexes from WT Synechocystis sp. PCC 6803: (a) CP43; (b) CP47; (c) RP*/RP1; and (d) RP2.
The kinetic scheme includes five compartments: two antenna compartments representing the excited states of CP43 and CP47, respectively; an RC*/RP1 compartment representing a mixture of the excited state of the reaction center and the first radical pair 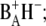 and two ensuing radical pair compartments, RP2 and RP3, both representing the secondary radical pair
and two ensuing radical pair compartments, RP2 and RP3, both representing the secondary radical pair 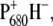 which is the final state reached in closed reaction centers. As observed in previous measurements on spinach PS2 core complexes, the population of the primary radical pair
which is the final state reached in closed reaction centers. As observed in previous measurements on spinach PS2 core complexes, the population of the primary radical pair 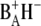 is very low, making it difficult to extract from our data a pure spectrum for this state. This is because there are a number of excited states whose energy is only slightly higher than that of the first charge-separated state, causing the transient population of
is very low, making it difficult to extract from our data a pure spectrum for this state. This is because there are a number of excited states whose energy is only slightly higher than that of the first charge-separated state, causing the transient population of 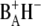 to be very low at all times during the experiment. However, differences between the RC*/RP1 spectrum obtained for the WT and those obtained for the two mutants are indicative of effects of the introduced mutation on the dynamics of energy transfer and primary charge separation, and lead to a plausible assignment for the 9-keto absorption of BA. Since the 680-nm excitation wavelength used in the experiment excites both the antenna and the reaction center, the initial input population is distributed according to the number of pigments contained in each subunit. The scheme depicted in Fig. 6 also takes into account the utrafast annihilation processes occurring in the excited state both for the antennas and for the RC, by means of an additional intracompartment component describing a fast annihilation phase that is complete in ∼2 ps. The rates connecting the different compartments, and hence the lifetimes, of the five states of the system for the WT are, as expected, very similar to what was obtained in the analogous analysis of vis/mid-IR pump-probe data collected for spinach PS2 cores (26). The application of the target model depicted in Fig. 6 required the excited-state spectra of CP43 and CP47 measured by vis/mid-IR pump-probe spectroscopy as an additional input (27,28); furthermore, time-resolved fluorescence data (46), measured on closed PS2 core complexes from spinach, have been fit simultaneously with femto-IR data to better describe the loss of excited states on the longer timescale. The comparison of the antenna spectra obtained by target analysis with those measured independently by vis/mid-IR pump-probe spectroscopy on the isolated pigment-protein complexes is shown in Fig. 7.
to be very low at all times during the experiment. However, differences between the RC*/RP1 spectrum obtained for the WT and those obtained for the two mutants are indicative of effects of the introduced mutation on the dynamics of energy transfer and primary charge separation, and lead to a plausible assignment for the 9-keto absorption of BA. Since the 680-nm excitation wavelength used in the experiment excites both the antenna and the reaction center, the initial input population is distributed according to the number of pigments contained in each subunit. The scheme depicted in Fig. 6 also takes into account the utrafast annihilation processes occurring in the excited state both for the antennas and for the RC, by means of an additional intracompartment component describing a fast annihilation phase that is complete in ∼2 ps. The rates connecting the different compartments, and hence the lifetimes, of the five states of the system for the WT are, as expected, very similar to what was obtained in the analogous analysis of vis/mid-IR pump-probe data collected for spinach PS2 cores (26). The application of the target model depicted in Fig. 6 required the excited-state spectra of CP43 and CP47 measured by vis/mid-IR pump-probe spectroscopy as an additional input (27,28); furthermore, time-resolved fluorescence data (46), measured on closed PS2 core complexes from spinach, have been fit simultaneously with femto-IR data to better describe the loss of excited states on the longer timescale. The comparison of the antenna spectra obtained by target analysis with those measured independently by vis/mid-IR pump-probe spectroscopy on the isolated pigment-protein complexes is shown in Fig. 7.
FIGURE 7.

Comparison between the excited states of CP43 and CP47 obtained by target analysis on WT samples (solid lines) with those measured for the isolated antenna complexes from spinach (dash-dotted lines).
The spectral comparison reported in Fig. 7 shows that the agreement between the measured spectrum of CP43 with the corresponding SADS obtained by target analysis is very good, whereas differences are observed in the case of CP47. There can be two main reasons for this discrepancy. A first possibility is that there exist some structural differences between the CP47 and CP43 pigment-protein complexes contained in spinach cores as compared to those from Synechocystis, as the spectra of the isolated antennas used as input in the target analysis have been measured on samples extracted from spinach. A second reason for the observed discrepancy may be that the spectra of the isolated antenna complexes were obtained upon excitation at 585 nm, whereas in this case an excitation wavelength of 680 nm was used. It is thus possible that the use of a different excitation wavelength determines the bleaching of different chlorophylls in this case compared to previous experiments, giving rise to the differences observed between the SADS extracted by target analysis and the spectra previously measured for CP43 and CP47. It is worth noting that when a similar target analysis was applied to femto-IR data measured for spinach core complexes, agreement was best between measured spectra and SADS for CP47, whereas some differences were noticed for CP43 (26).
The forward and backward connecting rates between the five compartments are reported in Fig. 6, and they result in the following lifetimes for WT Synechocystis sp. PCC 6803: 2.7 ps, 29 ps, 39 ps, 360 ps, and 35 ns. In agreement with previous data obtained from spinach, the dynamics of energy transfer from the antennas to the RC is relatively slow: this process mainly occurs in the 29-ps component, whereas the faster 2.7-ps component principally represents the decay of the RC*/RP1 compartment as a result both of the formation of RP2 (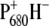 ) and of annihilation. The 39-ps lifetime reflects residual energy flow between CP43 and CP47, which have a small free-energy difference because of the different numbers of chlorophylls contained in the two antennas. The 360-ps component constitutes most of the RP3 compartment, and corresponds to the relaxation of the
) and of annihilation. The 39-ps lifetime reflects residual energy flow between CP43 and CP47, which have a small free-energy difference because of the different numbers of chlorophylls contained in the two antennas. The 360-ps component constitutes most of the RP3 compartment, and corresponds to the relaxation of the 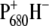 radical pair. The spectrum of the RC*/RP1 component is similar to the one previously obtained in the case of spinach core complexes, and it also shows a good agreement with the sum of the excited state and first radical pair state obtained by performing a target analysis on isolated RCs from spinach (8), as shown in Fig. 8, further confirming to a first approximation that the PS2 core can be described as the sum of its antennas and RC components.
radical pair. The spectrum of the RC*/RP1 component is similar to the one previously obtained in the case of spinach core complexes, and it also shows a good agreement with the sum of the excited state and first radical pair state obtained by performing a target analysis on isolated RCs from spinach (8), as shown in Fig. 8, further confirming to a first approximation that the PS2 core can be described as the sum of its antennas and RC components.
FIGURE 8.

Comparison between the RC*/RP1 spectrum obtained by target analysis on PS2 core complex samples (solid line) with the sum of the excited-state spectra (PC and BC) and first radical pair spectra obtained for isolated reaction centers from spinach (dash-dotted line). The PC and BC spectra have been obtained from a target analysis of time-resolved data measured for isolated RCs. In the target model, two excited-state compartments were introduced, one from which charge separation can readily occur (PC), and the other of which transfers on a slow timescale to the first (BC) (for discussion, see Groot et al. (8)).
The kinetic model of Fig. 6 has also been applied to the time-resolved data collected for the two mutants, D2-His197Ala and D2-His197Gln. The lifetimes obtained in these cases are reported in Table 1, where lifetimes of the WT are also reported for comparison. The individual rate for all the energy and electron transfer processes specified by the target model are reported in Table 2 for the WT and both mutants.
TABLE 1.
Lifetimes resulting from target analysis of PS2 core complexes from WT Synechocystis sp. PCC 6803 and D2-His197Gln and D2-His197Ala mutants
| Lifetime 1 | Lifetime 2 | Lifetime 3 | Lifetime 4 | Lifetime 5 | |
|---|---|---|---|---|---|
| WT | 2.7 ps | 29 ps | 39 ps | 360 ps | 35 ns |
| D2-His197Gln | 4.0 ps | 24 ps | 31 ps | 415 ps | 36 ns |
| D2-His197Ala | 2.3 ps | 22 ps | 27 ps | 520 ps | 40 ns |
The analysis is based on the five-compartment kinetic scheme of Fig. 6.
TABLE 2.
Individual rates (in ps−1) for the energy and electron transfer processes occurring in PS2 core complexes
| WT | D2-His197Gln | D2-His197Ala | |
|---|---|---|---|
| CP43→RC*/RP1 | 0.025 | 0.026 | 0.04 |
| CP43←RC*/RP1 | 0.045 | 0.053 | 0.083 |
| CP47→RC*/RP1 | 0.033 | 0.036 | 0.05 |
| CP47←RC*/RP1 | 0.066 | 0.053 | 0.09 |
| RP1→RP2 | 0.25 | 0.31 | 0.22 |
| RP1←RP2 | 0.0625 | 0.078 | 0.064 |
| RP2→RP3 | 0.0037 | 0.0035 | 0.0026 |
| RP2←RP3 | 7.1E-04 | 7.2E-04 | 5.26E-04 |
Transfer processes were modeled using the target scheme of Fig. 6. The annihilation rates, included in the target model by means of an additional intradecay component for the antenna and the RC*/RP1 compartments, are not reported in the table. In all cases, annihilation is completed in ∼2–3 ps.
Although the lifetimes obtained in the case of the two mutants are not dramatically different from those obtained in the case of WT, it appears that in both cases the introduced mutation has had an effect on the dynamics of energy equilibration between the antenna and the RC compartments, most likely due to a variation of the electronic levels of P680 and/or BA. Energy transfer from the antenna complexes to the RC is in fact faster for both mutants. This finding is in agreement with the previous characterization of a set of mutants in which the histidine ligand of both moieties of P680 was changed to different residues, which influenced both the redox potential of P680 and the sharing of the positive charge on 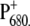 In particular, it was shown that for both mutants analyzed in this article, the reduction potential of the couple
In particular, it was shown that for both mutants analyzed in this article, the reduction potential of the couple 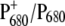 was lowered by 20–40 mV, thus providing a stabilization of oxidizing equivalents on
was lowered by 20–40 mV, thus providing a stabilization of oxidizing equivalents on 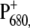 confirmed by the red shift in its absorption. The variation of the redox potential was found to have only limited influence on the rate of forward electron transfer (37), as also reported here. The lifetime that appears to be most affected by the mutation is the decay of RP2 in RP3, which occurs in 360 ps for the WT and in 415 and 520 ps for the two mutants. The back rate constant for the RP2-to-RP3 transition is also influenced by the fluorescence data, which have been fit together with the femto-IR data for the mutants also, although much less weight has been given to them in the analysis. The RP2→RP3 transition represents the relaxation of the radical pair
confirmed by the red shift in its absorption. The variation of the redox potential was found to have only limited influence on the rate of forward electron transfer (37), as also reported here. The lifetime that appears to be most affected by the mutation is the decay of RP2 in RP3, which occurs in 360 ps for the WT and in 415 and 520 ps for the two mutants. The back rate constant for the RP2-to-RP3 transition is also influenced by the fluorescence data, which have been fit together with the femto-IR data for the mutants also, although much less weight has been given to them in the analysis. The RP2→RP3 transition represents the relaxation of the radical pair 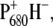 the rate of which can be affected by the ligand substitution on PB due to electronic (different charge localization and energy levels on
the rate of which can be affected by the ligand substitution on PB due to electronic (different charge localization and energy levels on 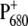 ) and environmental factors (possible chlorophyll distortion and different interaction with the surrounding amino acids).
) and environmental factors (possible chlorophyll distortion and different interaction with the surrounding amino acids).
The comparison between the SADS representing CP43 and CP47 obtained for the two mutants and the spectra measured for the isolated antennas show a less satisfactory agreement with respect to the WT, in particular for the D2-His197Ala sample and for the CP47 SADS (spectra shown in the Supplementary Material). Again, the different excitation energies used in this experiment, compared with what was used for isolated antennas, should be taken into account, but in the case of the mutants the discrepancy could also be due to a different distribution of the initial excitation energy among the antennas and the RC. This implies that the excited-state spectrum of the antenna complexes can change because different Chla are now bleached compared to what is observed for the isolated CP43 and CP47 and for the WT. As mentioned above, the major differences are observed for CP47 probably because the chlorophylls connected to this antenna subunit are closer to the mutation site on PB than the pigments linked to CP43. The discrepancy is again more pronounced for the D2-His197Ala mutant, which, according to the previous characterization, is the one that has the greater effect on the spectral properties of P680 (37). In this case, the best fit is obtained if only the CP43 spectrum is introduced as input. Since the deviations of the Synechocystis PS2 antenna spectra from the spectra of the isolated spinach PS2 CP43 and CP47 complexes may have some influence on the spectrum of the RC*/RP1 compartment, we have tested to what extent this region is affected by a variation of the spectral shape of the CP43 and CP47 SADS. In particular, in the case of the D2-His197Ala mutant, we have compared the RC*/RP1 SADS obtained when only the CP43 spectrum is introduced as input with that obtained when the spectra of both antenna complexes are used. The results of this comparison show that, though differences and compensations in the C=O region of the two antenna SADS are significant, the carbonyl region of the RC*/RP1 SADS is not substantially affected. This is because the lifetimes of the CP43 and CP47 compartments, which mainly decay on the 30–40 ps timescale, is substantially different from that of the RC*/RP1 compartment, which mainly decays on a faster, 3- to 4-ps timescale.
DISCUSSION
Charge redistribution on P680+ in the mutants
In this work, we have studied the dynamics of energy transfer and primary charge separation in intact PS2 core complexes from Synechocystis sp. PCC 6803 by using vis-pump/mid-IR-probe spectroscopy. By comparing the time-resolved IR spectra of WT PS2 cores with those of specific mutants in which the environment of the primary donor has been modified by ligand substitution on PB, we expect to be able to gain information on the dynamics of charge separation and to spectrally identify the putative primary donor, BA. To extract information on charge redistribution of 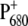 induced by the introduced mutation, we compared the light-induced FTIR spectra of WT and D2-His197Ala mutant. Analogous mutants have been previously produced also for RCs from nonoxygenic purple bacteria. In that case, ligand substitution with leucine of HisM200 and HisL173, coordinated, respectively, to the PM and PL bacteriochlorophylls forming the special pair, caused the loss of the central magnesium from the bacteriochlorophyll with the mutated ligand and, consequently, the formation of a heterodimer consisting of a bacteriochlorophyll and a bacteriopheophytin (47). It is worth noting that, in bacterial systems, ligand substitution by Gly retained both PM and PL chlorophylls, inducing only minor perturbations on the structural and functional properties of the special pair, as shown by the comparison of Raman spectra of WT RCs with those containing the Gly substituted ligand (48). For Rb. sphaeroides, comparison of FTIR difference spectra of the WT and the leucine mutants at the positions L173 and M200, allowed a spectral assignment of the vibrations characteristic of PM and PL. In particular, the intensity of the bands at ∼1290, 1500–1430, and 1580–1530 cm−1 was strongly reduced in the mutants, thus indicating that those vibrations are characteristic for an interacting dimer of Chls. Although to a minor extent, we also observe perturbation of bands located at similar positions in the FTIR spectrum of D2-His197Ala mutant when compared with the WT spectrum, thus indicating charge redistribution on
induced by the introduced mutation, we compared the light-induced FTIR spectra of WT and D2-His197Ala mutant. Analogous mutants have been previously produced also for RCs from nonoxygenic purple bacteria. In that case, ligand substitution with leucine of HisM200 and HisL173, coordinated, respectively, to the PM and PL bacteriochlorophylls forming the special pair, caused the loss of the central magnesium from the bacteriochlorophyll with the mutated ligand and, consequently, the formation of a heterodimer consisting of a bacteriochlorophyll and a bacteriopheophytin (47). It is worth noting that, in bacterial systems, ligand substitution by Gly retained both PM and PL chlorophylls, inducing only minor perturbations on the structural and functional properties of the special pair, as shown by the comparison of Raman spectra of WT RCs with those containing the Gly substituted ligand (48). For Rb. sphaeroides, comparison of FTIR difference spectra of the WT and the leucine mutants at the positions L173 and M200, allowed a spectral assignment of the vibrations characteristic of PM and PL. In particular, the intensity of the bands at ∼1290, 1500–1430, and 1580–1530 cm−1 was strongly reduced in the mutants, thus indicating that those vibrations are characteristic for an interacting dimer of Chls. Although to a minor extent, we also observe perturbation of bands located at similar positions in the FTIR spectrum of D2-His197Ala mutant when compared with the WT spectrum, thus indicating charge redistribution on 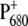 due to the introduced mutation. Information on the charge distribution on
due to the introduced mutation. Information on the charge distribution on 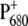 can also be obtained from the amount of up-shift observed for the 9-keto absorption upon charge separation, which is 25–32 cm−1 for Chl in tetrahydrofuran, and is 26 or 24 cm−1, respectively, for PS2 WT and the D2-His197Ala mutant, if measured between the most intense bands in the carbonyl region (1699(−)/1725(+) for WT and 1702(−)/1726(+) for the mutant.) By comparing the shift observed for the WT and the mutant, it would appear that the charge redistribution on
can also be obtained from the amount of up-shift observed for the 9-keto absorption upon charge separation, which is 25–32 cm−1 for Chl in tetrahydrofuran, and is 26 or 24 cm−1, respectively, for PS2 WT and the D2-His197Ala mutant, if measured between the most intense bands in the carbonyl region (1699(−)/1725(+) for WT and 1702(−)/1726(+) for the mutant.) By comparing the shift observed for the WT and the mutant, it would appear that the charge redistribution on 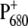 due to the mutation is not very significant, since the variation of only 2 cm−1 is minor. It is difficult to exactly quantify the amount of charge redistribution in the dimer based on this measurement, but a qualitative idea can be obtained by comparison with the up-shift measured for WT Rb. sphaeroides RC. In that case, an up-shift of 21 cm−1 was observed, corresponding to a charge ratio of 2:1 between the two halves of the special pair, as measured by ENDOR experiments (49). If in WT PS2 core the positive charge is for the 86% on PA, as also estimated from ENDOR experiments (50), an up-shift difference of 2 cm−1 between WT and mutant would imply a charge localization of ∼76% on PA for the mutant. This estimation has to be taken with extreme care, since it is based on the comparison between data collected from purple bacteria and PS2 core, which are completely different organisms. A more quantitative estimation of charge redistribution on
due to the mutation is not very significant, since the variation of only 2 cm−1 is minor. It is difficult to exactly quantify the amount of charge redistribution in the dimer based on this measurement, but a qualitative idea can be obtained by comparison with the up-shift measured for WT Rb. sphaeroides RC. In that case, an up-shift of 21 cm−1 was observed, corresponding to a charge ratio of 2:1 between the two halves of the special pair, as measured by ENDOR experiments (49). If in WT PS2 core the positive charge is for the 86% on PA, as also estimated from ENDOR experiments (50), an up-shift difference of 2 cm−1 between WT and mutant would imply a charge localization of ∼76% on PA for the mutant. This estimation has to be taken with extreme care, since it is based on the comparison between data collected from purple bacteria and PS2 core, which are completely different organisms. A more quantitative estimation of charge redistribution on 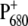 due to the modification of PA or PB ligand would require the application of experimental methods better suited to measuring the spin distribution on the dimer, such as EPR or ENDOR. It is worth noting that changes are also observed in the structure of a broad band centered at ∼2000 cm−1, which has been assigned to an electronic transition of
due to the modification of PA or PB ligand would require the application of experimental methods better suited to measuring the spin distribution on the dimer, such as EPR or ENDOR. It is worth noting that changes are also observed in the structure of a broad band centered at ∼2000 cm−1, which has been assigned to an electronic transition of 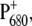 and is taken as an indication of partial charge delocalization in the dimer. The intensity of this transition is higher in the mutant, as expected if the charge is more delocalized between PA and PB (51). The band also looks more structured, showing peaks at 1880, 2090, and 2200 cm−1, whereas in the case of the WT only a broad maximum centered around 2000 cm−1 is visible. The comparison of spectral features in this region is reported in the Supplementary Material (see Fig. S3 in Data S1).
and is taken as an indication of partial charge delocalization in the dimer. The intensity of this transition is higher in the mutant, as expected if the charge is more delocalized between PA and PB (51). The band also looks more structured, showing peaks at 1880, 2090, and 2200 cm−1, whereas in the case of the WT only a broad maximum centered around 2000 cm−1 is visible. The comparison of spectral features in this region is reported in the Supplementary Material (see Fig. S3 in Data S1).
Mutation-induced changes on the dynamics of primary energy transfer and charge separation and spectral assignment of the 9-keto band of BA
The comparison of the time-resolved infrared spectra of the WT and the two P680 mutants analyzed in this work can give new insights into the mechanism of primary charge separation in PS2 and, in particular, on the role of the accessory chlorophyll BA in this process. Both global and target analysis of the time-resolved data showed differences in the dynamics of primary energy and charge-separation processes induced by the mutations of the P680 ligand, which may therefore be due to a modification of the electronic levels of P680 and BA and of the reduction potential of P680. The dynamical components mostly affected by the mutations are those related to energy transfer and equilibration between the antenna complexes and the reaction center and to the stabilization of the radical pair 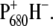 These effects are consistently captured both by the global analysis, which for both mutants, but in particular for the D2-His197Ala, shows a faster kinetics for the two earliest time components and a slower rate for the following spectral evolution, and by the target analysis of the data, which shows that the most affected lifetimes are those related to the equilibration between antennas and reaction center (∼30 ps) and relaxation of the secondary radical pair (360–500 ps).
These effects are consistently captured both by the global analysis, which for both mutants, but in particular for the D2-His197Ala, shows a faster kinetics for the two earliest time components and a slower rate for the following spectral evolution, and by the target analysis of the data, which shows that the most affected lifetimes are those related to the equilibration between antennas and reaction center (∼30 ps) and relaxation of the secondary radical pair (360–500 ps).
The faster energy transfer is likely due to the changed absorption properties of P680/BA due to ligand substitution (37) causing better spectral overlap with the CP43 and CP47 transferring states. The slight difference in the faster lifetime (2.3–4.0 ps) we interpret with caution, since it may be influenced by the relative amount of annihilation occurring in the first picosecods. This lifetime is representative for the decay of the RC*/RP1 compartment as a result both of the formation of RP2 (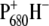 ) and of annihilation. Inspection of Table 2 indeed shows that the charge separation rate is not significantly affected by the introduced mutation, in agreement with the idea that the primary donor resides on a pigment other than P680, whose electronic properties are, as discussed before, influenced by ligand substitution. It appears that in the case of the D2-His197Gln mutant, the amount of annihilation is less than for the other two samples, determining the 4.0-ps lifetime to be longer compared to the corresponding 2.7- and 2.3-ps lifetimes, respectively, measured for the WT and D2-His197Ala samples. This is also evident from the EADS obtained in the sequential analysis (Fig. 5), showing that the excited-state decay occurring in the first picoseconds, and evidenced by the decay of the induced absorption signal in the 1650 cm−1 region between the first two EADS, is less in the case of this mutant than for the WT and the D2-His197Ala mutant.
) and of annihilation. Inspection of Table 2 indeed shows that the charge separation rate is not significantly affected by the introduced mutation, in agreement with the idea that the primary donor resides on a pigment other than P680, whose electronic properties are, as discussed before, influenced by ligand substitution. It appears that in the case of the D2-His197Gln mutant, the amount of annihilation is less than for the other two samples, determining the 4.0-ps lifetime to be longer compared to the corresponding 2.7- and 2.3-ps lifetimes, respectively, measured for the WT and D2-His197Ala samples. This is also evident from the EADS obtained in the sequential analysis (Fig. 5), showing that the excited-state decay occurring in the first picoseconds, and evidenced by the decay of the induced absorption signal in the 1650 cm−1 region between the first two EADS, is less in the case of this mutant than for the WT and the D2-His197Ala mutant.
We also find that the dynamics of RP2 relaxation, i.e., a drop in the free energy of the 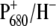 radical pair, is slower in both mutants, possibly because of a variation in the reorganization energy associated with electron transfer and different interactions of the radical pair with the protein environment, due, for instance, to the PB macrocycle distortions or to the direct effect of the altered nearest coordinating amino acid. This finding is in agreement with the idea that protein dynamics has a strong influence on electron transfer kinetics, as recently pointed out in a study considering the kinetics of primary charge separation in bacterial reaction center mutants with altered driving force for electron transfer (52). According to the results of that investigation, it appears that protein response to the initial excitation events is fast and independent on the driving force for electron transfer, whereas charge separation readily occurs only when the protein has achieved a suitable nuclear configuration.
radical pair, is slower in both mutants, possibly because of a variation in the reorganization energy associated with electron transfer and different interactions of the radical pair with the protein environment, due, for instance, to the PB macrocycle distortions or to the direct effect of the altered nearest coordinating amino acid. This finding is in agreement with the idea that protein dynamics has a strong influence on electron transfer kinetics, as recently pointed out in a study considering the kinetics of primary charge separation in bacterial reaction center mutants with altered driving force for electron transfer (52). According to the results of that investigation, it appears that protein response to the initial excitation events is fast and independent on the driving force for electron transfer, whereas charge separation readily occurs only when the protein has achieved a suitable nuclear configuration.
Finally, information on the spectral properties of BA can be obtained by comparing the WT and mutant spectra representing the RC*/RP1 state of the system obtained by target analysis. The SADS of the RC*/RP1 compartment obtained for both mutants are compared in Fig. 9 with that obtained for the WT.
FIGURE 9.

Comparison between the spectra of the RC*/RP1 compartment (a) on WT PS2 core complexes with those of the D2-His197Gln (b) and D2-His197Ala (c) mutants, obtained by target analysis.
The three SADS differ significantly in the carbonyl bleaching region. The WT spectrum shows two bleaching signals, at 1702 and 1680 cm−1, and positive signals at 1728 and 1716 cm−1, in addition to a broad induced absorption band peaking at 1660 cm−1. The D2-His197Gln SADS shows an additional small bleaching at 1691 cm−1 whereas for the D2-His197Ala mutant, bleaching signals at similar positions to those observed for the WT are evident. Signals in the RC*/RP1 spectrum for the WT can be assigned by comparison with steady-state FTIR measurements (23,40). Since this spectral component represents a mixture of RC excited state and the first radical pair, we expect that signals both from 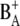 and H−, forming the primary radical pair, and from P680*, which is involved in the excited state of the reaction center, will contribute to this spectral component. According to the literature, the signal at 1702 cm−1 can be assigned to the 9-keto carbonyl of PA, although, as mentioned before, less agreement has been reached on the position of the 9-keto bleaching of PB. Absorption from the primary electron acceptor H− is expected to contribute to the bleaching at ∼1680 cm−1, in agreement with the assignment from steady-state FTIR measurements (23). No bleaching signals directly attributable to BA are visible.
and H−, forming the primary radical pair, and from P680*, which is involved in the excited state of the reaction center, will contribute to this spectral component. According to the literature, the signal at 1702 cm−1 can be assigned to the 9-keto carbonyl of PA, although, as mentioned before, less agreement has been reached on the position of the 9-keto bleaching of PB. Absorption from the primary electron acceptor H− is expected to contribute to the bleaching at ∼1680 cm−1, in agreement with the assignment from steady-state FTIR measurements (23). No bleaching signals directly attributable to BA are visible.
There have been a few suggestions in the literature concerning possible spectral assignment of the 9-keto carbonyl of BA. One possibility is that the 9-keto carbonyl of BA absorbs at 1687 cm−1, as suggested by target analysis on femto-IR data collected from isolated RC samples from spinach (8), whereas another possibility comes from the measurement of light-induced FTIR difference spectra of the triplet state in PS2 RCs from spinach, where it has been shown that the Chla on which the triplet is principally localized at low temperature, which has been recently identified as BA (53), absorbs at ∼1670 cm−1 (45,54). We expect that by introducing the mutation, which should displace the H-bonding water molecule on BA, the absorption band of the latter would upshift. By comparing the SADS reported in Fig. 9, we favor the assignment based on the triplet FTIR measurements, also supported by the analysis of the vibronic structure observed in the 5K line narrowed emission spectrum of PSII (55). According to this interpretation, the small bleaching appearing at 1691 cm−1 in the RC*/RP1 spectral component in the case of the D2-His197Gln mutant could be assigned to the upshifted 9-keto carbonyl of BA. We note that the spectra from the global analysis of the D2-His197Gln mutant apparently indicate an incomplete recovery of this bleaching on the long timescales, which would be expected to occur for a BA signal. This finding could possibly be related to an electrochromic signal arising from BA that is sensitive to the charge on PA. Furthermore, in WT, and, to a minor extent, in the Ala mutant (see Fig.5), we observe at later times a downshift of the bleaching that is initially located at ∼1700 cm−1. This spectral evolution is principally due to the pheophytin reduction leading to compensations between 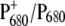 and H−/H absorptions as we could establish from the good agreement between our
and H−/H absorptions as we could establish from the good agreement between our 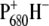 spectra and the P+/P and H−/H spectra as obtained by FTIR (Fig. 4). In the case of the Ala mutant, the 9-keto carbonyl of BA is also expected to upshift, in this case possibly to 1678 cm−1, where, however, contributions from H and possibly PB are also present. The relative intensity of the two bleaching signals observed at 1702 and 1678 cm−1 is different between WT and D2-His197Ala mutant, with a relatively more intense signal at 1678 cm−1 in the case of the mutant. This observation is in line with the hypothesis of a contribution from BA to this signal in the D2-His197Ala sample, but not in the WT, where the 1670 cm−1 signal is hidden by the induced absorption band. Our assignment is thus based on the comparison between the SADS obtained by target analysis of time-resolved data, whose interpretation is in line with the expected effect of hydrogen-bond strength variation on the carbonyl group absorption and the agreement with data previously reported in the literature.
spectra and the P+/P and H−/H spectra as obtained by FTIR (Fig. 4). In the case of the Ala mutant, the 9-keto carbonyl of BA is also expected to upshift, in this case possibly to 1678 cm−1, where, however, contributions from H and possibly PB are also present. The relative intensity of the two bleaching signals observed at 1702 and 1678 cm−1 is different between WT and D2-His197Ala mutant, with a relatively more intense signal at 1678 cm−1 in the case of the mutant. This observation is in line with the hypothesis of a contribution from BA to this signal in the D2-His197Ala sample, but not in the WT, where the 1670 cm−1 signal is hidden by the induced absorption band. Our assignment is thus based on the comparison between the SADS obtained by target analysis of time-resolved data, whose interpretation is in line with the expected effect of hydrogen-bond strength variation on the carbonyl group absorption and the agreement with data previously reported in the literature.
CONCLUSIONS
The results presented in this work confirm that the dynamics of energy transfer and primary charge separation in PS2 core can be explained very well by using a kinetic model containing two slowly transferring antenna compartments connected to the reaction center, which, once excited, induces fast primary charge separation.
The comparison of vis-pump/mid-IR-probe spectra of WT PS2 core with those recorded for mutants where the histidine residue coordinated to PB has been changed to alanine or glutamine allowed us to examine the changes in the dynamics of primary energy transfer and charge separation in PS2 induced by ligand substitution on P680. All of the observations reported are in line with participation of the monomer chlorophyll BA in the process of primary charge separation, and give substantial support to the hypothesis that the primary charge separation in PS2 predominantly starts from BA and not from P680. Finally, the comparison between the SADS obtained by target analysis of our time-resolved data and information previously reported in the literature allowed us to suggest a plausible assignment for the absorption band BA.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
The authors thank Wim Vermaas and Dexter Chisholm for the D2-197 site-directed mutants.
This work was supported by the Netherlands Organization for Scientific Research and by the National Research Initiative of the United States Department of Agriculture Cooperative State Research, Education and Extension Service, grant 2003-35318-13589 (to B.A.D.).
Editor: Jose Onuchic.
References
- 1.Biesiadka, J., B. Loll, J. Kern, K.-D. Irrgang, and A. Zouni. 2004. Crystal structure of cyanobacterial photosystem II at 3.2 Å resolution: a closer look at the Mn-cluster. Phys. Chem. Chem. Phys. 6:4733–4736. [Google Scholar]
- 2.Ferreira, K. N., T. M. Iverson, K. Maghlaoui, J. Barber, and S. Iwata. 2004. Architecture of the photosynthetic oxygen-evolving center. Science. 303:1831–1838. [DOI] [PubMed] [Google Scholar]
- 3.Loll, B., J. Kern, W. Saenger, A. Zouni, and J. Biesiadka. 2005. Towards complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II. Nature. 438:1040–1044. [DOI] [PubMed] [Google Scholar]
- 4.van Grondelle, R., J. P. Dekker, T. Gillbro, and V. Sundstrom. 1994. Energy transfer and trapping in photosynthesis. Biochim. Biophys. Acta. 1187:1–65. [Google Scholar]
- 5.de Weerd, F. L., I. H. M. van Stokkum, H. van Amerongen, J. P. Dekker, and R. van Grondelle. 2002. Pathways for energy transfer in the core light-harvesting complexes CP43 and CP47 of photosystem II. Biophys. J. 82:1586–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vasil'ev, S., P. Orth, A. Zouni, T. G. Owens, and D. Bruce. 2001. Excited-state dynamics in photosystem II: Insights from the x-ray crystal structure. Proc. Natl. Acad. Sci. USA. 98:8602–8607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barter, L. M. C., M. Bianchietti, C. Jeans, M. J. Schilstra, B. Hankamer, B. A. Diner, J. Barber, J. R. Durrant, and D. R. Klug. 2001. Relationship between excitation energy transfer, trapping, and antenna size in photosystem II. Biochemistry. 40:4026–4034. [DOI] [PubMed] [Google Scholar]
- 8.Groot, M. L., N. P. Pawlowicz, L. J. G. W. van Wilderen, J. Breton, I. H. M. van Stokkum, and R. van Grondelle. 2005. Initial electron donor and acceptor in isolated photosystem II reaction centers identified with femtosecond mid-IR spectroscopy. Proc. Natl. Acad. Sci. USA. 102:13087–13092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holzwarth, A. R., M. G. Muller, M. Reus, M. Nowaczyk, J. Sander, and M. Rogner. 2006. Kinetics and mechanism of electron transfer in intact photosystem II and in the isolated reaction center: pheophytin is the primary electron acceptor. Proc. Natl. Acad. Sci. USA. 103:6895–6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barter, L. M. C., D. R. Klug, and R. van Grondelle. 2005. Energy trapping and equilibration: a balance of regulation and efficiency. In Photosystem II: the Light-Driven Water Plastoquinone Oxidoreductase. T. J. Wydrzynsky, K. Satho, and J. A. Freeman, editors. Springer, Dordrecht, The Netherlands. 491–514.
- 11.Dekker, J. P., and R. van Grondelle. 2000. Primary charge separation in photosystem II. Photosynth. Res. 63:195–208. [DOI] [PubMed] [Google Scholar]
- 12.Renger, G., and A. R. Holzwarth. 2005. Primary electron transfer. In Photosystem II: The Light-Driven Water Plastoquinone Oxidoreductase. T. J. Wydrzynsky, K. Satho, and J. A. Freeman, editors. Springer, Dordrecht, The Netherlands. 139–175.
- 13.Durrant, J. R., G. Hastings, D. M. Joseph, J. Barber, G. Porter, and D. R. Klug. 1992. Subpicosecond equilibration of excitation energy in isolated photosystem II reaction centers. Proc. Natl. Acad. Sci. USA. 89:11632–11636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hastings, G., J. R. Durrant, J. Barber, G. Porter, and D. R. Klug. 1992. Observation of pheophytin reduction in photosystem two reaction centers using femtosecond transient absorption spectroscopy. Biochemistry. 31:7638–7647. [DOI] [PubMed] [Google Scholar]
- 15.Muller, M. G., M. Hucke, M. Reus, and A. R. Holzwarth. 1996. Primary processes and structure of the photosystem II reaction center. 4. Low-intensity femtosecond transient absorption spectra of D1–D2-cyt-b559 reaction centers. J. Phys. Chem. 100:9527–9536. [Google Scholar]
- 16.Groot, M.-L., F. van Mourik, C. Eijckelhoff, I. H. M. van Stokkum, J. P. Dekker, and R. van Grondelle. 1997. Charge separation in the reaction center of photosystem II studied as a function of temperature. Proc. Natl. Acad. Sci. USA. 94:4389–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenfield, S. R., M. Seibert, Govindjee, and M. R. Wasielewski. 1997. Direct measurement of the effective rate constant for primary charge separation in isolated photosystem II reaction centers. J. Phys. Chem. B. 101:2251–2255. [Google Scholar]
- 18.Greenfield, S. R., M. Seibert, and M. R. Wasielewski. 1999. Time-resolved absorption changes of the pheophytin Qx band in isolated photosystem II reaction centers at 7 K. Energy transfer and charge separation. J. Phys. Chem. B. 103:8364–8374. [Google Scholar]
- 19.Andrizhiyevskaya, E. G., D. Frolov, R. van Grondelle, and J. P. Dekker. 2004. On the role of the CP47 core antenna in the energy transfer and trapping dynamics of Photosystem II. Phys. Chem. Chem. Phys. 6:4810–4819. [Google Scholar]
- 20.Groot, M.-L., J. P. Dekker, R. van Grondelle, F. T. H. den Hartog, and S. Volker. 1996. Energy transfer and trapping in isolated photosystem II reaction centers of green plants at low temperature. A study by spectral hole burning. J. Phys. Chem. 100:11488–11495. [Google Scholar]
- 21.Zazubovich, V., R. Jankowiak, K. Riley, R. Picorel, M. Seibert, and G. J. Small. 2003. How fast is excitation energy transfer in the photosystem II reaction center in the low temperature limit? Hole burning vs photon echo. J. Phys. Chem. B. 107:2862–2866. [Google Scholar]
- 22.Frese, R. N., M. Germano, F. L. de Weerd, I. H. M. van Stokkum, A. Y. Shkuropatov, V. A. Shuvalov, H. J. van Gorkom, R. van Grondelle, and J. P. Dekker. 2003. Electric field effects on the chlorophylls, pheophytins, and β-carotenes in the reaction center of photosystem II. Biochemistry. 42:9205–9213. [DOI] [PubMed] [Google Scholar]
- 23.Nabedryk, E., S. Andrianambinintsoa, G. Berger, M. Leonhard, W. Mantele, and J. Breton. 1990. Characterization of bonding interactions of the intermediary electron acceptor in the reaction center of photosystem II by FTIR spectroscopy. Biochim. Biophys. Acta. 1016:49–54. [Google Scholar]
- 24.Noguchi, T., T. Tomo, and Y. Inoue. 1998. Fourier transform infrared study of the cation radical of P680 in the photosystem II reaction center: evidence for charge delocalization on the chlorophyll dimer. Biochemistry. 37:13614–13625. [DOI] [PubMed] [Google Scholar]
- 25.Miloslavina, Y., M. Szczepaniak, M. G. Muller, J. Sander, M. Nowaczyk, M. Rogner, and A. R. Holzwarth. 2006. Charge separation kinetics in intact photosystem II core particles is trap-limited. A picosecond fluorescence study. Biochemistry. 45:2436–2442. [DOI] [PubMed] [Google Scholar]
- 26.Pawlowicz, N. P., M. L. Groot, I. H. M. van Stokkum, J. Breton, and R. van Grondelle. 2007. Charge separation and energy transfer in the photosystem II core complex studied by femtosecond midinfrared spectroscopy. Biophys. J. 93:2732–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Donato, M., R. van Grondelle, I. H. M. van Stokkum, and M. L. Groot. 2007. Excitation energy transfer in the photosystem II core antenna complex CP43 studied by femtosecond visible/visible and visible/mid-infrared pump probe spectroscopy. J. Phys. Chem. B. 111:7353–7359. [DOI] [PubMed] [Google Scholar]
- 28.Groot, M.-L., J. Breton, L. J. G. W. van Wilderen, J. P. Dekker, and R. van Grondelle. 2004. Femtosecond visible/visible and visible/mid-IR pump-probe study of the photosystem II core antenna complex CP47. J. Phys. Chem. B. 108:8001–8006. [Google Scholar]
- 29.Groot, M.-L., L. J. G. W. van Wilderen, and M. Di Donato. 2007. Time-resolved methods in biophysics. 5. Femtosecond time-resolved and dispersed infrared spectroscopy on proteins. Photochem. Photobiol. Sci. 6:501–507. [DOI] [PubMed] [Google Scholar]
- 30.Durrant, J. R., D. R. Klug, S. L. S. Kwa, R. van Grondelle, G. Porter, and J. P. Dekker. 1995. A Multimer model for P680, the primary electron donor of photosystem II. Proc. Natl. Acad. Sci. USA. 92:4798–4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Brederode, M. E., F. van Mourik, I. H. M. van Stokkum, M. R. Jones, and R. van Grondelle. 1999. Multiple pathways for ultrafast transduction of light energy in the photosynthetic reaction center of Rhodobacter sphaeroides. Proc. Natl. Acad. Sci. USA. 96:2054–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Brederode, M. E., M. R. Jones, F. Van Mourik, I. H. M. Van Stokkum, and R. Van Grondelle. 1997. A new pathway for transmembrane electron transfer in photosynthetic reaction centers of Rhodobacter sphaeroides not involving the excited special pair. Biochemistry. 36:6855–6861. [DOI] [PubMed] [Google Scholar]
- 33.Feiler, U., T. A. Mattioli, I. Katheder, H. Scheer, M. Lutz, and B. Robert. 1994. Effect of vinil substitutions on resonance Raman spectra of (bacterio)chlorophylls. J. Raman Spectrosc. 25:365–370. [Google Scholar]
- 34.Katz, J. J., G. L. Closs, F. C. Pennington, M. R. Thomas, and H. H. Strain. 1963. Infrared spectra, molecular weights, and molecular association of chlorophylls a and b, methyl chlorophyllides, and pheophytins in various solvents. J. Am. Chem. Soc. 85:3801–3809. [Google Scholar]
- 35.Pascal, A. A., L. Caron, B. Rousseau, K. Lapouge, J.-C. Duval, and B. Robert. 1998. Resonance Raman spectroscopy of a light-harvesting protein from the Brown alga Laminaria saccharina. Biochemistry. 37:2450–2457. [DOI] [PubMed] [Google Scholar]
- 36.Stowell, M. H. B., T. M. McPhillips, D. C. Rees, S. M. Soltis, E. Abresch, and G. Feher. 1997. Light-induced structural changes in photosynthetic reaction center: implications for mechanism of electron-proton transfer. Science. 276:812–816. [DOI] [PubMed] [Google Scholar]
- 37.Diner, B. A., E. Schlodder, P. J. Nixon, W. J. Coleman, F. Rappaport, J. Lavergne, W. F. J. Vermaas, and D. A. Chisholm. 2001. Site-directed mutations at D1-His198 and D2-His197 of photosystem II in Synechocystis PCC 6803: sites of primary charge separation and cation and triplet stabilization. Biochemistry. 40:9265–9281. [DOI] [PubMed] [Google Scholar]
- 38.van Stokkum, I. H. M., D. S. Larsen, and R. van Grondelle. 2004. Global and target analysis of time-resolved spectra. Biochim. Biophys. Acta. 1657:82–104. [DOI] [PubMed] [Google Scholar]
- 39.Breton, J., R. Hienerwadel, and E. Nabedryk. 1997. Spectroscopy of Biological Molecules. P. Carmona, R. Navarro, and A. Hernanz, editors. Kluwer Academic, Dordrecht, The Netherlands. 101–102.
- 40.Okubo, T., T. Tomo, M. Sugiura, and T. Noguchi. 2007. Perturbation of the structure of P680 and the charge distribution on its radical cation in isolated reaction center complexes of photosystem II as revealed by Fourier transform infrared spectroscopy. Biochemistry. 46:4390–4397. [DOI] [PubMed] [Google Scholar]
- 41.Johnson, E. T., F. Muh, E. Nabedryk, J. C. Williams, J. P. Allen, W. Lubitz, J. Breton, and W. W. Parson. 2002. Electronic and vibronic coupling of the special pair of Bacteriochlorophylls in photosynthetic reaction centers from wild-type and mutant strains of Rhodobacter sphaeroides. J. Phys. Chem. B. 106:11859–11869. [Google Scholar]
- 42.Gasyna, Z., and P. N. Schatz. 1996. Analysis of the intervalence band in the oxidized photosynthetic bacterial reaction center. J. Phys. Chem. 100:1445–1448. [Google Scholar]
- 43.Reimers, J. R., and N. S. Hush. 1996. The effects of couplings to symmetric and antisymmetric modes and minor asymmetry on the spectral properties of mixed-valence and related charge-transfer systems. Chem. Phys. 208:177–193. [Google Scholar]
- 44.Reimers, J. R., and N. S. Hush. 1995. Nature of the ground and first excited states of the radical cations of photosynthetic bacterial reaction centres. Chem. Phys. 197:323–332. [Google Scholar]
- 45.Noguchi, T., Y. Inoue, and K. Satoh. 1993. FT-IR studies on the triplet state of P680 in the photosystem II reaction center: Triplet equilibrium within a chlorophyll dimer. Biochemistry. 32:7186–7195. [DOI] [PubMed] [Google Scholar]
- 46.Andrizhiyevskaya, E. G., J. A. Bautista, B. A. Diner, R. van Grondelle, and J. P. Dekker. 2005. Energy transfer and charge separation in the Photosystem II core complex studied by time-resolved fluorescence. PhD thesis. Vrije Universiteit Amsterdam, Amsterdam, The Netherlands.
- 47.Nabedryk, E., S. J. Robles, E. Goldman, D. C. Youvan, and J. Breton. 1992. Probing the primary donor environment in the histidineM200. fwdarw. leucine and histidineL173. fwdarw. leucine heterodimer mutants of Rhodobacter capsulatus by light-induced Fourier transform infrared difference spectroscopy. Biochemistry. 31:10852–10858. [DOI] [PubMed] [Google Scholar]
- 48.Goldsmith, J. O., B. King, and S. G. Boxer. 1996. Mg Coordination by amino acid side chain is not required for assembly and function of the special pair in bacterial photosynthetic reaction centers. Biochemistry. 35:2421–2428. [DOI] [PubMed] [Google Scholar]
- 49.Rautter, J., F. Lendzian, W. Lubitz, S. Wang, and J. P. Allen. 1994. Comparative study of reaction centers from photosynthetic purple bacteria: electron paramagnetic resonance and electron nuclear double resonance spectroscopy. Biochemistry. 33:12077–12084. [DOI] [PubMed] [Google Scholar]
- 50.Rigby, S. E. J., J. H. A. Nugent, and P. J. O′Malley. 1994. ENDOR and special triple resonance studies of chlorophyll cation radicals in photosystem 2. Biochemistry. 33:10043–10050. [DOI] [PubMed] [Google Scholar]
- 51.Breton, J., E. Nabedryk, and W. W. Parson. 1992. A new infrared electronic transition of the oxidized primary electron donor in bacterial reaction centers: a way to assess resonance interactions between the bacteriochlorophylls. Biochemistry. 31:7503–7510. [DOI] [PubMed] [Google Scholar]
- 52.Wang, H., S. Lin, J. P. Allen, J. C. Williams, S. Blankert, C. Laser, and N. W. Woodbury. 2007. Protein dynamics control the kinetics of initial electron transfer in photosynthesis. Science. 316:747–750. [DOI] [PubMed] [Google Scholar]
- 53.Schlodder, E., W. J. Coleman, P. J. Nixon, R. O. Cohen, T. Renger, and B. A. Diner. 2007. Site-directed mutations at D1-His198 and D1-Thr179 of photosystem II in Synechocystis sp. PCC 6803: deciphering the spectral properties of the PSII reaction centre. Philos. Trans. R. Soc. Lond. B: Biol. Sci. 363:1197–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Noguchi, T., T. Tomo, and C. Kato. 2001. Triplet formation on a monomeric chlorophyll in the photosystem ii reaction center as studied by time-resolved infrared spectroscopy. Biochemistry. 40:2176–2185. [DOI] [PubMed] [Google Scholar]
- 55.Peterman, E. J. G., H. van Amerongen, R. van Grondelle, and J. P. Dekker. 1998. The nature of the excited state of the reaction center of photosystem II of green plants: A high-resolution fluorescence spectroscopy study. Proc. Natl. Acad. Sci. USA. 95:6128–6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.