

Abstract
Intracerebral infection of susceptible strains of mice, e.g. SJL/J, with Theiler’s murine encephalomyelitis virus (TMEV) leads to a persistent CNS infection accompanied by development of a chronic-progressive inflammatory CNS autoimmune demyelinating disease which is clinically and pathologically similar to human multiple sclerosis. In contrast, resistant strains of mice, e.g. C57BL/6 (B6), effectively clear TMEV from the CNS and do not develop demyelinating disease. Although CD8+ T cells are crucial for viral clearance in B6 mice, SJL mice also mount potent CD8+ T cell responses against virus, thus the reason for the viral persistence in the CNS in these mice is unclear. Here, we examined innate anti-viral responses of CNS-resident astrocytes as a potential determinant of viral persistence and disease susceptibility. We demonstrate that B6 astrocytes produce significantly higher levels of cytokines, chemokines and adhesion molecules in response to TMEV infection, or stimulation with IFN-γ and TNF-α or poly I:C than SJL mice. In addition, TMEV more effectively induces MHC I molecules on B6 astrocytes than SJL, corresponding with an increased ability to activate TMEV-specific CD8+ T cells directly ex vivo. These results suggest that enhanced anti-viral responses of B6 astrocytes contribute to the ability of these mice to clear TMEV from the CNS and therefore to their resistance to the development of autoimmune demyelinating disease.
Keywords: multiple sclerosis, Theiler’s murine encephalomyelitis virus, astrocytes, cytotoxic T lymphocytes, disease susceptibility
Introduction
Intracerebral infection of susceptible strains of mice with the BeAn strain of Theiler’s murine encephalomyelitis virus (TMEV) leads to a persistent central nervous system (CNS) infection accompanied by development of a chronic-progressive inflammatory demyelinating disease beginning approximately 30 days post-infection (dpi) (Dal Canto and Lipton, 1975; Lipton, 1975). TMEV-induced demyelinating disease (TMEV-IDD) is a relevant model for human multiple sclerosis (MS) because many of the clinical symptoms and pathologic correlates are similar, including CD4+ T cell and mononuclear phagocyte infiltration into the CNS, primary demyelination and spastic hindlimb paralysis (Dal Canto and Lipton, 1975; Hafler, 2004; Lipton, 1975). In contrast, resistant mice are able to efficiently clear TMEV from the CNS and do not progress to demyelinating disease (Chamorro et al., 1986).
Resistance to persistent CNS infection by TMEV is controlled by multiple genetic loci, with the strongest linkage to the class I H-2D MHC region (Brahic et al., 2005; Lipton and Melvold, 1984). Mice which carry an H-2s, f, p, r, v or q haplotype are susceptible to TMEV-IDD, while those bearing H-2b, k, or d haplotypes are resistant (Rodriguez and David, 1985). The particular importance of the H-2D region was demonstrated using hybrid strains which express H-2Ks and H-2Db, which are resistant to TMEV-IDD, and those expressing H-2Kb and H-2Ds, which are susceptible (Clatch et al., 1985). Additionally, H-2b mice which are genetically deficient in H-2D are susceptible to TMEV-IDD and the insertion of an H-2Db gene into a normally susceptible H-2q strain renders them resistant, further supporting a critical role of H-2D in determining genetic susceptibility to TMEV-IDD (Azoulay et al., 1994; Azoulay-Cayla et al., 2000; Lipton et al., 1995a).
The strong association of H-2D haplotype with development of demyelinating disease indicates a role of CD8+ T cells in disease which could be pathologic or protective. Most evidence points towards a protective role of CD8+ T cells via the clearance of TMEV from the CNS, because mice on a resistant background which are CD8-depleted via antibody or genetically deficient in CD8+ T cells fail to clear the virus and develop CNS demyelination and inflammation, although they develop only mild clinical symptoms (Borrow et al., 1992; Fiette et al., 1993; Ure and Rodriguez, 2002). Additionally, TMEV-infected CD8-deficient mice on a susceptible background exhibit decreased viral clearance, increased CNS inflammation and a more rapid and severe clinical disease course (Begolka et al., 2001).
Despite evidence demonstrating the importance of the CD8+ T cell response in protection from TMEV-IDD, susceptible strains of mice mount robust CD8+ T cell responses against TMEV, similar in frequency to those in resistant strains (Kang et al., 2002; Lindsley et al., 1991; Lyman et al., 2004), which raises the question of why susceptible strains of mice are unable to completely clear the virus from the CNS. It has recently been demonstrated using bone marrow chimeras that some genetic control of susceptibility depends on cells which are not of hematopoetic origin, suggesting these genes might control responses in the target organ (Aubagnac et al., 2002). Astrocytes are neuroectodermally derived cells which are critical for normal functions of the CNS including synaptic transmission, maintenance of the blood-brain barrier, metabolic support of axons, and ion and neurotransmitter buffering. We and other groups have demonstrated that activated astrocytes can also contribute to immune responses in the CNS through the production of cytokines, chemokines, anti-microbial molecules and the ability to activate CD4+ T cells in the presence of IFN-γ (Bailey et al., 2006; Carpentier et al., 2005; Carpentier et al., 2007). Although astrocytes generally express low to undetectable levels of MHC I molecules under resting conditions (Schnitzer and Schachner, 1981; Traugott, 1987), treatment with cytokines or infection with virus can upregulate MHC I and increase the ability of astrocytes to activate or be lysed by CD8+ T cells (Lavi et al., 1988; Liu et al., 1989; Skias et al., 1987; Suzumura et al., 1986; Wong et al., 1984). The upregulation of MHC I is highly variable between strains of mice and is controlled by genes within the MHC locus (Massa et al., 1989), suggesting that the expression of MHC I on astrocytes could account for some of the MHC-dependent genetic control of TMEV-IDD susceptibility. Astrocytes in susceptible mice are activated in response to TMEV in vivo and harbor viral antigen (Zheng et al., 2001). It has been suggested that astrocytes are the major source of replicating virus in vivo, although other groups contend that microglia/macrophages contribute more to viral load (Lipton et al., 1995b; Zheng et al., 2001).
Here, we demonstrate that astrocytes derived from TMEV-IDD resistant B6 mice are less efficiently infected by TMEV in vitro than those from susceptible SJL mice. Additionally, B6 astrocytes express increased levels of cytokines, chemokines and adhesion molecules in response to various other pro-inflammatory stimuli compared to SJL astrocytes. Finally, we demonstrate that B6 astrocytes infected with TMEV more readily upregulate MHC I and more efficiently activate CD8+ T cells than SJL astrocytes. We hypothesize that the augmented antiviral responses of B6 astrocytes contribute to the enhanced ability of these mice to clear TMEV from the CNS and therefore contribute to their resistance to the development of chronic demyelinating disease.
Results
TMEV infection of SJL and B6 Astrocytes
We have previously observed that astrocytes from TMEV-IDD susceptible SJL mice can be directly infected with TMEV, expressing both viral RNA and protein antigens (Carpentier et al., 2007). To ascertain if there are strain differences in the astrocytic response to TMEV infection between resistant B6 and susceptible SJL mice, we first determined if TMEV infection of B6 astrocytes differs from SJL astrocytes. Fewer B6 astrocytes express TMEV antigens at 48 h post infection, indicating that they are less efficiently infected than SJL astrocytes (Figure 1A,B; p=0.05). Consistent with our previous observations, there was a small but statistically significant increase in cell death after 72 h of infection with TMEV in both SJL and B6 cultures. The amount of cytotoxicity in TMEV-infected cultures is quite small in comparison to a freeze-thaw positive control. There was no difference in the amount of cytotoxicity observed between the mouse strains, indicating that TMEV does not produce a strong lytic infection in astrocytes from either strain (Figure 1C).
Figure 1. SJL astrocytes are infected with TMEV more efficiently than B6 astrocytes.
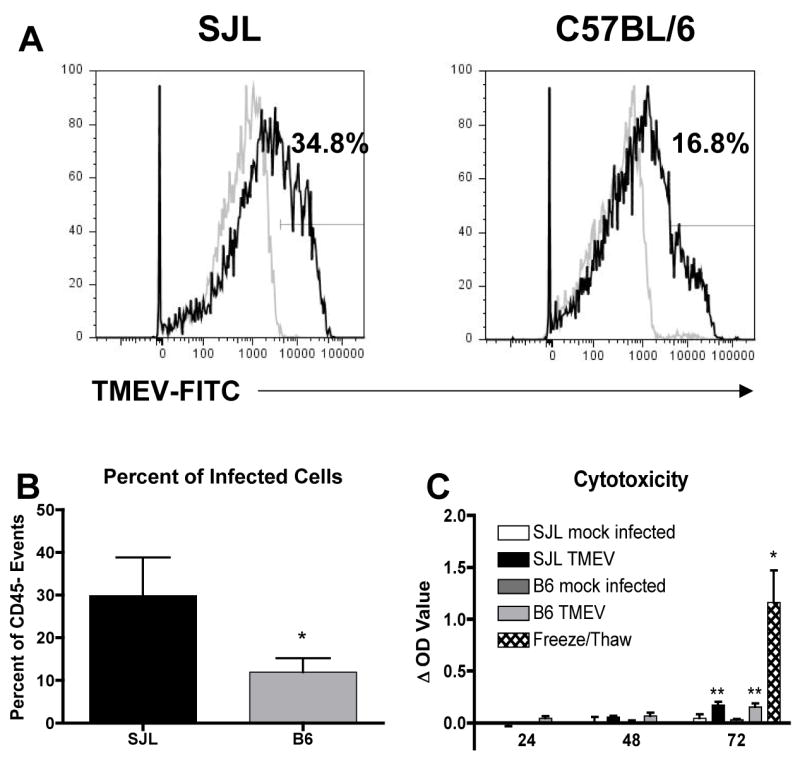
SJL or B6 astrocytes were mock infected or infected with TMEV (MOI=10). (A) Mock infected (grey line) and TMEV infected (black line) cultures were harvested at 48 h and stained with anti-TMEV antibody. The percentage of cells falling within the drawn gate in the TMEV-infected cultures is shown above the gate. Data are representative of 3 separate experiments. (B) The percent of TMEV-infected cells in SJL and B6 cultures was quantified. Data shown are the mean ± SEM of 3 separate experiments. *p=0.05, one-tailed Student’s t test (C) Supernatants were collected at 24, 48 and 72 h post infection and analyzed for lactate dehydrogenase release as a measure of cell death. Data are expressed as the difference between the optical density (OD) of the mock infected samples at 24 h and the experimental samples and are the average ± SEM of 3 separate experiments. Freeze/thawed samples were used as a positive control. A two way ANOVA with repeated measures found significant effects of time (p=0.012) and treatment group (p=0.0014). **OD value significantly increased compared to control, p<0.01 by Bonferroni post test. *OD value significantly increased compared to mock infected cells at 72 h by one-tailed Student’s t test, p<0.05
Innate immune functions of SJL and B6 Astrocytes
We next examined the innate immune functions of astrocytes induced by TMEV infection. We have previously completed time course experiments with astrocytes infected with TMEV and observed that production of type I IFNs, cytokines and chemokines increases through 72 h, and we have therefore chosen this time to examine these innate immune functions (Carpentier et al., 2007). There is a trend towards increased production of type I IFNs in B6 astrocytes after TMEV infection (Figure 2), but it did not reach statistical significance (p=0.19 for IFN-α and p=0.09 for IFN-β). It therefore appears that type I IFN production is likely not the mechanism by which B6 astrocytes better control TMEV infection. B6 astrocytes, however, induce higher levels of a variety of cytokines and chemokines after TMEV infection and poly I:C treatment (Figure 3). In response to treatment with poly I:C, both SJL and B6 astrocytes express similar levels of IL-6, CCL2, CCL5, CXCL1 and CXCL10 (Figure 3A, C, F–H). However, B6 astrocytes treated with poly I:C express significantly higher levels of TNF-α, CCL3, and CCL4 than SJL astrocytes (Figure 3B, D, E; p=0.012 CCL3; p=0.038 CCL4; p=0.032 TNF-α). In response to TMEV infection, as we have previously observed (11), SJL astrocytes express IL-6, CCL2, CCL5 and CXCL10 (Figure 3A, C, F, H). B6 astrocytes infected with TMEV produce significantly increased levels IL-6 and CXCL10 (Figure 3A, H; p=0.026 IL-6; p=0.013 CXCL10) and also show a trend towards increased production of TNF-α, CCL2 and CCL5 (Figure 3B, C, F). B6 astrocytes infected with TMEV also induce CCL3 and CCL4, which is significantly increased compared to SJL (Figure 3D, E; p=0.043 CCL3; p=0.038 CCL4). Combined exposure to IFN-γ and TNF-α induced significant expression of CCL2, CCL5 and CXCL10 in both strains, and CXCL10 production under this condition was slightly enhanced in B6 astrocytes (Figure 3C, F, H; p=0.046). CCL3 and CCL4 were also slightly enhanced in B6 IFN-γ and TNF-α treated astrocytes, but the levels remained quite low compared to the other treatments (Figure 3D, E; p=0.0058 CCL3; p=0.028 CCL4). Importantly, many of these chemokines produced by astrocytes have been demonstrated to be critical for recruitment CD8+ T cells to sites of viral infection (Thomsen et al., 2003).
Figure 2. TMEV infection induces similar levels of type I IFNs in SJL and B6 astrocytes.
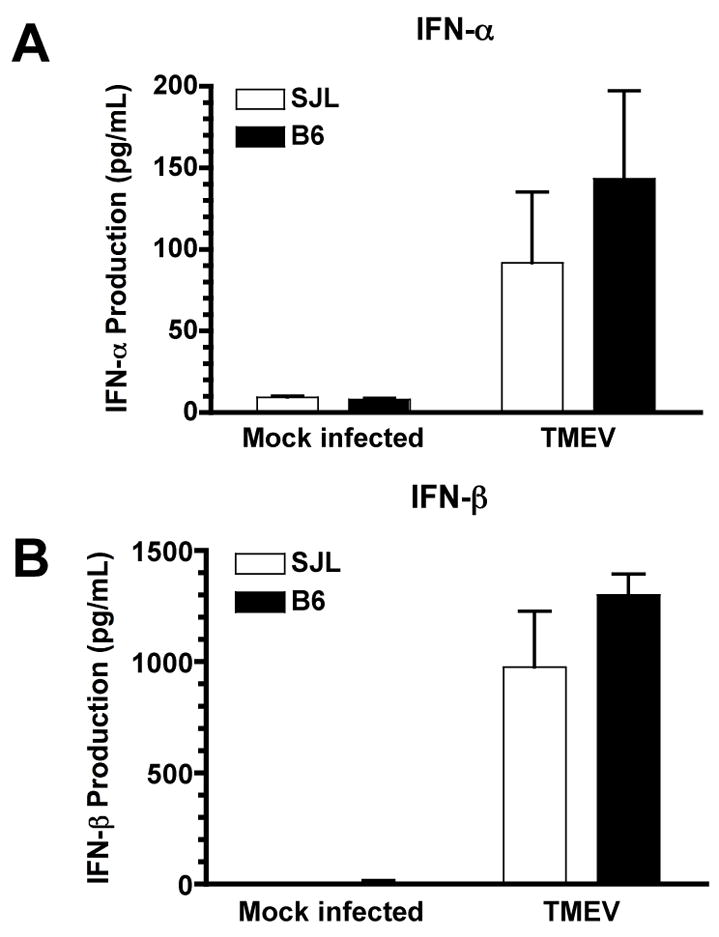
SJL or B6 astrocytes were mock infected or infected with TMEV (MOI=10). Supernatants were harvested at 72 h post-infection and analyzed for the production of IFN-α and IFN-β by ELISA. Data shown are the average ± SEM of 3–4 separate experiments.
Figure 3. B6 astrocytes produce greater amounts of cytokines and chemokines than SJL astrocytes.
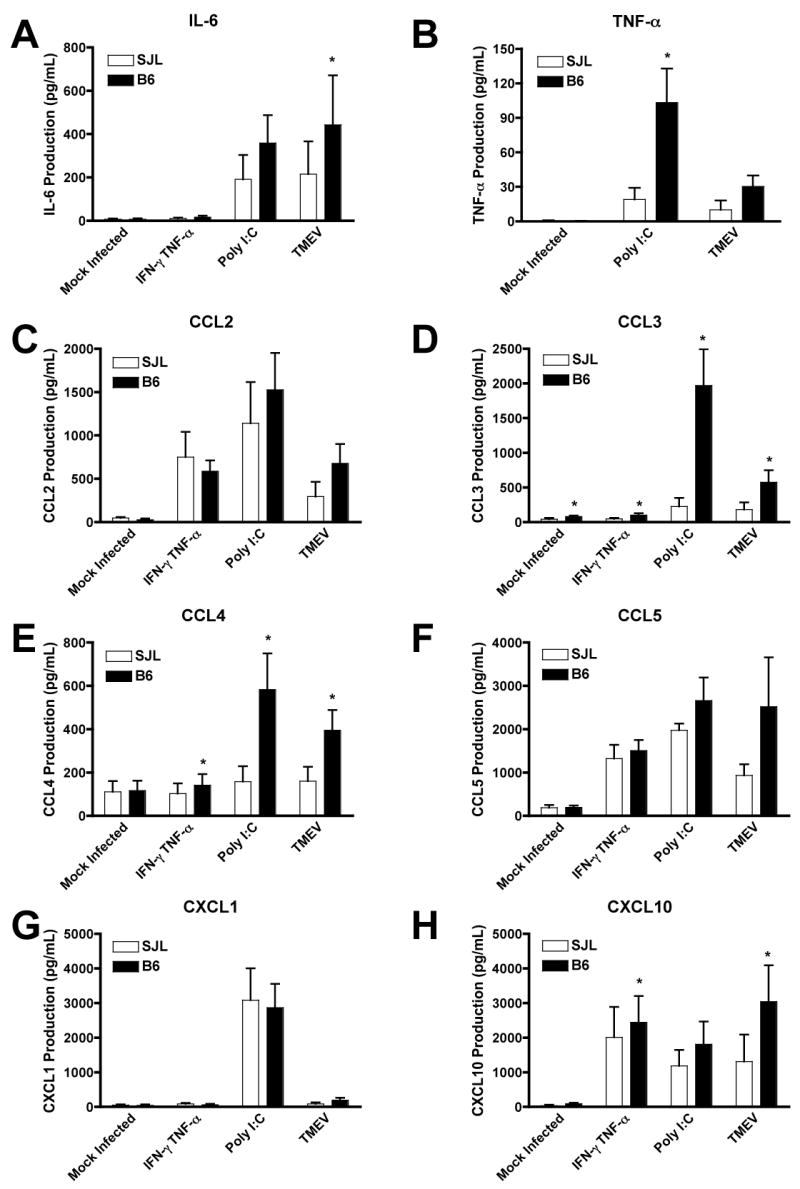
SJL or B6 astrocytes were mock infected, treated with IFN-γ and TNF-α (100 and 500 U/ml), poly I:C (100 mg/ml), or infected with TMEV (MOI=10). Supernatants were harvested at 72 h and analyzed for the production of cytokines and chemokines by Luminex mutiplex bead assay. Data shown are the average ± SEM of 5 separate experiments. *Levels significantly increased compared to SJL astrocytes by paired one-tailed Student’s t test, p<0.05
We have also previously demonstrated that stimulation of astrocytes with TLR ligands or cytokines upregulated their expression of the adhesion molecules ICAM-1 and VCAM-1 (Carpentier et al., 2005). These adhesion molecules are crucial for the infiltration of leukocytes into the CNS (Ransohoff et al., 2003), and it has been proposed that their expression on astrocytes is particularly crucial for the penetration of leukocytes deep into the CNS parenchyma (Gimenez et al., 2004). There is no significant difference in the levels of adhesion molecules expressed in mock infected SJL or B6 astrocytes (Figure 4A,F and data not shown). IFN-γ and TNF-α treatment upregulate ICAM-1 and VCAM-1 similarly in SJL and B6 mice (Figure 4 B,G,E,J). Poly I:C more strongly upregulates ICAM-1 in B6 than SJL (Figure 4 C,E; p=0.04), and there is a trend towards stronger upregulation of VCAM-1 although it does not reach significance (Figure 4 H,J; p=0.07). The increased upregulation of adhesion molecules on B6 astrocytes is most marked and consistent with TMEV infection (Figure 4 D,E,I,J; p=0.03 ICAM-1, p=0.04 VCAM-1), which may contribute towards the increased early infiltration of CD8+ T cells into the CNS of B6 mice. This increased expression of adhesion molecules is not restricted to cells which are actually infected with TMEV, which is a minority of cells in B6 cultures, because there is a whole population shift in ICAM-1 and VCAM-1 expression.
Figure 4. B6 astrocytes express higher levels of adhesion molecules than SJL astrocytes.
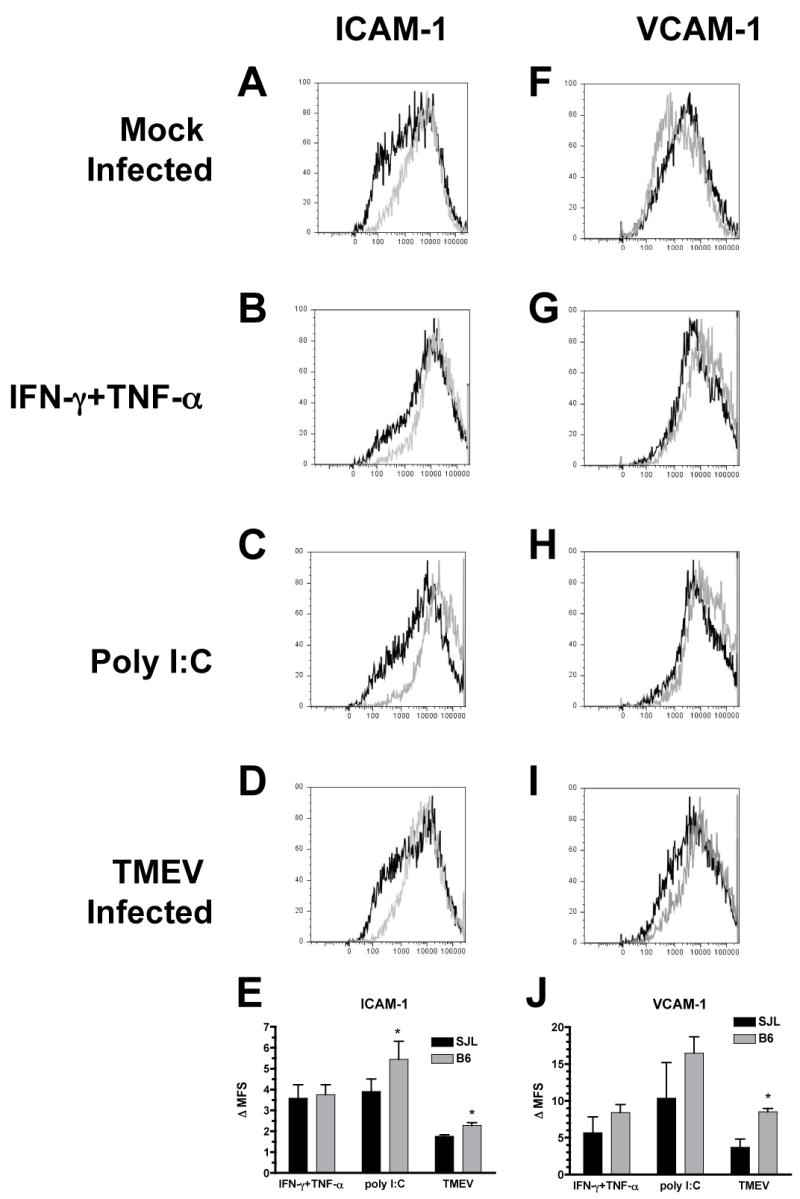
Astrocyte cultures from B6 (gray lines) or SJL (black lines) mice were mock infected, treated with IFN-γ and TNF-α (100 and 500 U/ml) or poly I:C (100 μg/ml), or infected with TMEV (MOI=10). After 48 h, cells were harvested and stained for ICAM-1 (A–D) and VCAM-1 (F–I) and analyzed by flow cytometry. Plots shown are representative of 3–4 separate experiments. (E,J) The fold increase in the mean fluorescence shift (Δ MFS) in the treated samples compared to the control was determined. Data shown are the average ± SEM of 3–4 separate experiments. *Levels significantly increased compared to SJL astrocytes by paired one-tailed student’s T test, p<0.05
CD8+ T cell activation by astrocytes
We next examined the ability of astrocytes to directly activate CD8+ T cells, which are crucial to clearance of TMEV from the CNS (Begolka et al., 2001; Borrow et al., 1992; Fiette et al., 1993). In order to activate CD8+ T cells, a cell must express the appropriate peptide in the context of MHC class I, which astrocytes can express in response to virus infection or cytokine treatment (Fontana et al., 1986; Lavi et al., 1988; Liu et al., 1989; Suzumura et al., 1986; Wong et al., 1984). We thus compared the constitutive expression and regulation of MHC I molecules H-2K and H-2D in SJL and B6 astrocytes, respectively. We chose these molecules because all known TMEV epitopes are restricted to H-2K in SJL and H-2D in B6 mice (Kang et al., 2002; Lyman et al., 2002). SJL astrocytes express little or no H-2Ks in the unstimulated state (Figure 5A), but it is upregulated by treatment with IFN-γ and TNF-α or poly I:C (Figure 5B, C). In contrast, infection with TMEV is a relatively poor inducer of H-2Ks expression by SJL astrocytes (Figure 5D). B6 astrocytes also express low constitutive levels of H-2Db (Figure 5E), but upregulate it in response to IFN-γ and TNF-α, poly I:C, or TMEV infection (Figure 5F-H). Compared to SJL, B6 astrocytes upregulate H-2 expression significantly more in response to IFN-γ and TNF-α (p=0.029) and TMEV (p=0.049), and there is a trend towards higher upregulation in response to poly I:C as well (p=0.09).
Figure 5. TMEV-infected B6 astrocytes upregulate MHC I to higher levels than SJL astrocytes.
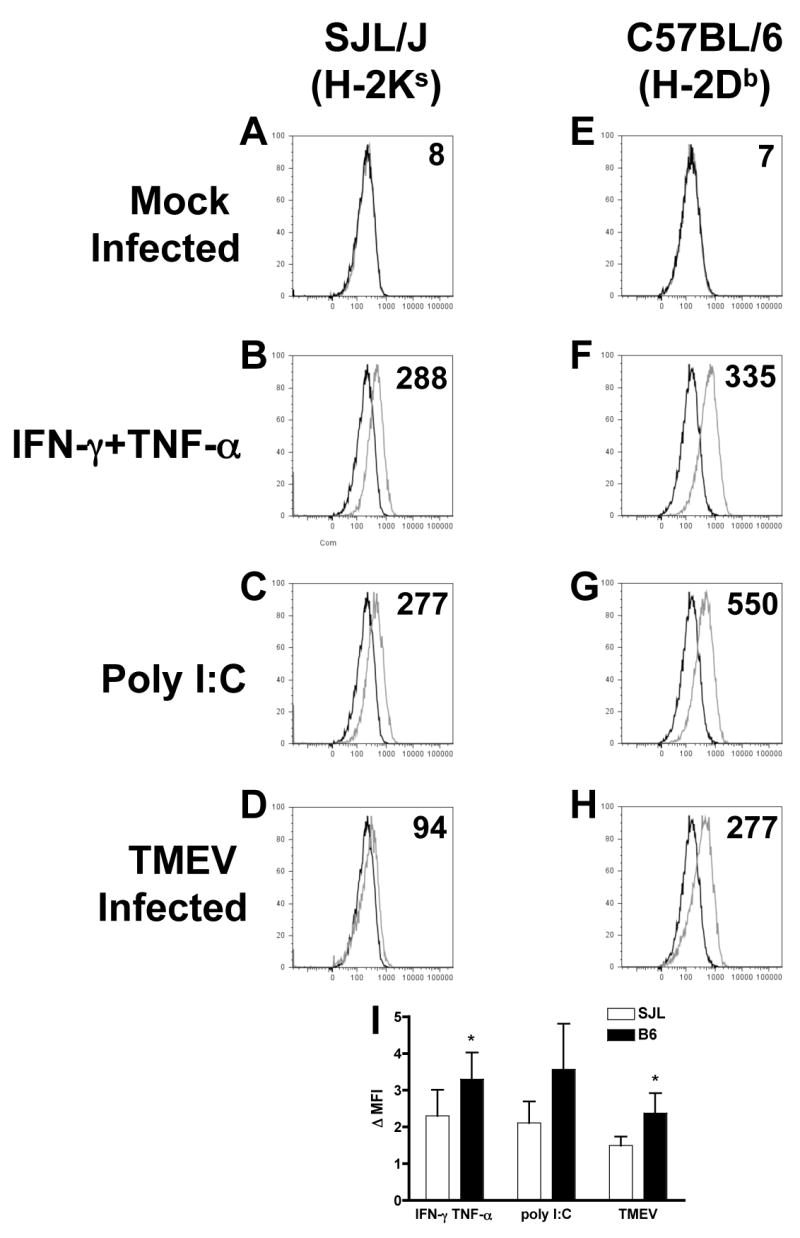
Astrocyte cultures from SJL or B6 mice were mock infected (black line, all graphs), treated with IFN-γ and TNF-α (100 and 500 U/ml) or poly I:C (100 μg/ml) or infected with TMEV (MOI=10; gray lines). After 48 h, cells were harvested and stained for H-2Ks (SJL) or H-2Db (B6) MHC class I molecules and analyzed by flow cytometry. Isotype controls are indicated with gray lines in A, E. Plots shown are representative of 3–4 separate experiments (A–H). Numbers in each graph are the mean fluorescence shift (MFS) of the indicated treatment and strain. (I) The fold increase in the mean fluorescence intensity (Δ MFI) in the treated samples compared to the control was determined. Data shown are the average ± SEM of 3 separate experiments. *Levels significantly increased compared to SJL astrocytes by paired one-tailed student’s T test, p<0.05.
These differences suggested that SJL astrocytes may be less able to activate the antiviral functions of CD8+ T cells due to their relative lack of MHC I expression. However, it is difficult to directly compare the levels of MHC I on B6 and SJL astrocytes because they are distinct molecules and analyzed with different antibodies. Therefore, to further test this hypothesis, we examined the ability of astrocytes to induce IFN-γ production by TMEV-specific CD8+ T cells. CD8+ T cells were isolated from the spleens of naïve or TMEV-infected mice at 8 dpi using magnetic cell sorting with anti-CD8α microbeads. Purified samples averaged approximately 90% CD8α+ cells (data not shown). The majority of CD8α+ cells are also positive for CD90, indicating that they are T cells, although a small subset of CD8α+ cells co-express CD11c (data not shown). This is potentially a concern, because CD8α+ dendritic cells are known for their potent ability to activate CD8+ T cells (Ardavin, 2003). However, we did not observe any IFN-γ production above background in CD8+ T cell cultures from TMEV-infected mice which did not contain astrocytes, even with specific peptide added (Figure 6B). Importantly, virtually none of the sorted cells were CD90+CD8α−, which represents CD4+ T cells and another potential contaminating source of IFN-γ (data not shown). We co-cultured CD8α+ T cells from TMEV-infected SJL and B6 mice with astrocytes that were mock infected, treated with IFN-γ and TNF-α, or infected with TMEV 48 h prior with or without the immunodominant CD8-restricted viral peptides, VP3159–166 for SJL and VP2121–130 for B6 (Kang et al., 2002; Lyman et al., 2002). Culture supernatants were harvested after 96 h and analyzed for IFN-γ levels. Neither SJL nor B6 astrocytes in the absence of CD8+ T cells produce significant levels of IFN-γ under any condition (not shown). CD8+ T cells isolated from naïve mice also failed to produce significant amounts of IFN-γ under any conditions (Figure 6A). SJL and B6 astrocytes were able to induce of IFN-γ from TMEV-specific CD8+ T cells upon the addition of specific peptide (Figure 6B). B6 astrocytes were more efficient at inducing peptide-specific CD8+ T cell activation when TMEV-infected cells were employed (p=0.011 effect of strain by two-way ANOVA; p<0.05, Bonferonni post-test SJL TMEV to B6 TMEV). Mock infected and especially IFN-γ and TNF-α treated astrocytes showed a similar ability to activate CD8+ T cells from TMEV-infected mice, indicating that the difference between these strains is limited to the astrocytic response to TMEV infection. This observation which correlates well with the increased levels of MHC I expression on TMEV-infected astrocytes we have previously noted (Figure 5). Importantly, B6 astrocytes infected with TMEV were also able to induce IFN-γ production from TMEV-specific CD8+ T cells without the addition of exogenous peptide, indicating that they are able to efficiently process and present endogenous viral antigen, whereas SJL astrocytes were not (Figure 6C; p=0.037 effect of strain by two-way ANOVA, p<0.01 by Bonferroni post-test, B6 TMEV to SJL TMEV). This difference is most likely the result of increased MHC I expression in TMEV-infected B6 astrocytes (Figure 5D,H) rather than a difference in viral antigen levels, since we observe somewhat decreased levels of viral antigen in TMEV-infected B6 cultures after 48h (Figure 1A,B). In order to confirm that there are no differences between the frequency or IFN-γ producing ability of SJL and B6 TMEV-specific CD8+ T cells which could potentially account for these results, we performed ELISPOTs from both strains of mice with the immunodominant and subdominant TMEV peptides at 7dpi (Figure 6D,E). Our results demonstrate that similar numbers of CD8+ T cells capable of producing IFN-γ in response to TMEV-specific peptides exist in the spleens of B6 and SJL mice acutely after infection, and further indicate that only the antigen processing and/or presentation ability of astrocytes differ between these two strains. Since astrocytes are infected in vivo with TMEV (Zheng et al., 2001), their ability to process and present viral antigen to CD8+ T cells could be crucial for CNS virus clearance.
Figure 6. B6 astrocytes activate TMEV-specific CD8+ T cells more efficiently than SJL astrocytes.
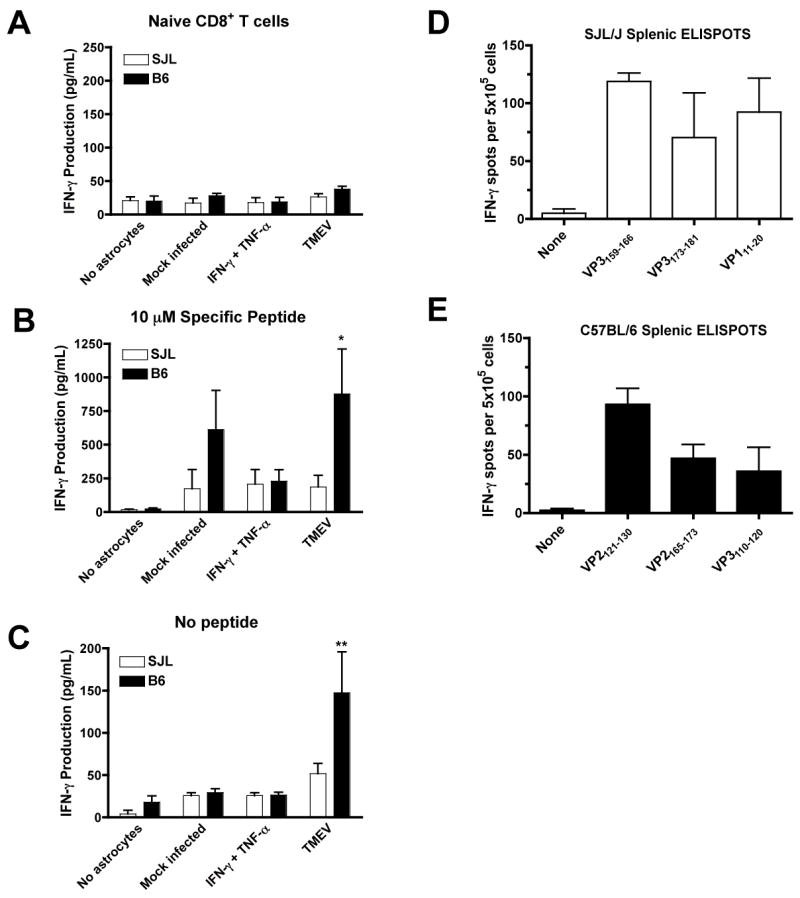
Astrocytes were plated at 105 cells/well and mock infected, treated with IFN-γ and TNF-α (100 and 500 U/ml) or infected with TMEV (MOI=10). At 48 h, CD8+ T cells were isolated by AutoMacs from naive (A) or TMEV-infected mice at day 8 pi (B,C). Astrocytes were washed and incubated with (A,B) or without (C) 10 μM specific peptide (VP3159–166 for SJL and VP2121–130 for B6). Culture supernatants were collected at 96 h and analyzed for IFN-γ production by Luminex multiplex bead assay. Data shown is the average ± SEM of 4 separate experiments. With specific peptide, the effect of strain was significant by two-way ANOVA (p=0.0108). Without specific peptide, both strain (p=0.0371) and treatment (p=0.0022) had significant effects. Levels significantly increased in B6 compared to SJL by Bonferroni post test, *p<0.05; **p<0.01. The frequency of splenic T cells producing IFN-γ in response to the dominant and subdominant viral epitopes from both SJL (D) and B6 (E) mice were analyzed at day 7 PI. Of note the frequency of IFN-γ-producing splenic CD8 T cells specific for the respective immunodominant CTL epitopes did not differ between the two mouse strains. Results are representative of two separate experiments.
Discussion
These findings demonstrate key functional differences between astrocytes from TMEV-IDD resistant B6 and susceptible SJL mice in their ability to mount efficient anti-viral responses. We first demonstrate that B6 astrocytes are more resistant to TMEV infection than SJL astrocytes. Astrocytes are infected in SJL mice in vivo, although whether astrocytes or microglia/macrophages are the major source of replicating virus has been controversial (Lipton et al., 1995b; Zheng et al., 2001). Our data indicates that astrocytes are less efficiently infected in resistant B6 mice, which could account for the somewhat decreased viral loads in B6 compared to SJL brains even early after infection (Lyman et al., 2004), and could significantly contribute to the inability to establish a persistent viral infection and result in demyelinating disease. Our data also demonstrates decreased innate immune functions and decreased ability to process and present antigen to CD8+ T cells in TMEV-infected SJL astrocytes compared to B6. Together, these data suggest the possibility that astrocytes, non-hematopoetic cells resident in the CNS, may significantly contribute to the ability of the immune system to efficiently clear virus in this target organ. We are working to confirm these results in an in vivo system using GFAP-GFP transgenic mice to allow astrocytes to be sorted from TMEV-infected brains and compared for expression of pro-inflammatory molecules and ability to present antigen to CD8+ and CD4+ T cells. We are also pursuing studies in mice with genetic defects in various astrocyte-related inflammatory functions to determine which functions are important for susceptibility to TMEV-IDD in vivo.
The ability to clear TMEV from the CNS is critically dependent on CD8+ T cells (Borrow et al., 1992; Fiette et al., 1993; Ure and Rodriguez, 2002). Additionally, the strongest genetic linkage to TMEV susceptibility lies within the H-2D locus in the MHC region of the genome, further suggesting the critical importance of CD8+ T cells in viral clearance (Brahic et al., 2005). For this reason, most studies to date examining strain-dependent susceptibility to TMEV-IDD have focused on CD8+ T cell functions. Surprisingly, however, susceptible mice mount very strong CD8+ T cell responses to TMEV infection, and the activation, cytokine production and cytolytic functions of SJL CD8+ T cells are similar to those seen in resistant B6 mice (Kang et al., 2002; Lindsley et al., 1991; Lyman et al., 2004).
There is, however, a reported delay in the infiltration of CD8+ T cells into the CNS of TMEV-infected SJL mice compared to B6 (Lyman et al., 2004). Our results correlate well with this study, since we observe here that SJL astrocytes express lower levels of cytokines, chemokines and adhesion molecules, particularly in response to TMEV infection, which may cause less efficient recruitment and infiltration of anti-viral CD8+ T cells into the CNS. B6 astrocytes express higher levels of the cytokines IL-6 and TNF-α after TMEV infection or poly I:C treatment, which may damage the blood-brain barrier (BBB) and contribute to the ability of leukocytes to infiltrate the CNS (de Vries et al., 1996). B6 astrocytes also express significantly higher levels of CXCL10, CCL3 and CCL4 than SJL astrocytes and trend towards higher production of CCL2 and CCL5. These chemokines are of particular interest because they are critical for the recruitment of virus-specific CD8+ T cells to sites of infection throughout the body (Thomsen et al., 2003) and their induction has been described in the CNS in response to many viral infections, including TMEV (Glass et al., 2005; Hoffman et al., 1999; Lane et al., 2000; Nansen et al., 2000; Nuovo and Alfieri, 1996) . Genetic deficiency or blockade of CCL3, CCL5, CXCL10 or CCR2 (the obligate receptor for CCL2) during acute CNS infection generally inhibits the infiltration of T cells and monocytes/macrophages into the CNS and is associated with increased viral loads after infection with MHV or TMEV (Chen et al., 2001; Lane et al., 2000; Liu et al., 2000; Trifilo et al., 2003; Ure et al., 2005). CXCR3, the receptor for CXCL9-11, and CCR5, the receptor for CCL3, CCL4, and CCL5, are also expressed on many CNS-infiltrating CD4+ and CD8+ T cells and likely important for the ability to gain access to the CNS (Giunti et al., 2003; Kivisakk et al., 2002; Nansen et al., 2000; Shacklett et al., 2004; Sorensen et al., 1999). Adhesion molecules expressed at higher levels by B6 astrocytes may also promote more efficient leukocyte entry, and particularly promote their ability to invade deep into the CNS parenchyma (Gimenez et al., 2004).
In addition to being recruited to the CNS, CD8+ T cell cytotoxic functions must be activated in order to clear TMEV from the CNS, which requires the recognition of virus peptides in the context of MHC I. Astrocytes are able to express MHC I in response to cytokines and virus (Fontana et al., 1986; Lavi et al., 1988; Liu et al., 1989; Suzumura et al., 1986; Wong et al., 1984), but a critical role for astrocytes during CD8+ T cell activation in vivo during CNS infection has yet to be demonstrated. We show here that astrocytes from resistant B6 mice are able to upregulate the MHC I molecule H-2Db in response to TMEV infection and to induce the activation of TMEV-specific CD8+ T cells as indicated by the production of IFN-γ. In contrast, upregulation of H-2Ks in TMEV-infected SJL astrocytes is minimal, despite their high level of infection, and SJL astrocytes are inefficient at processing and presenting viral antigen to CD8+ T cells. Importantly, the frequency of splenic TMEV-specific CD8+ T cells able to produce IFN-γ in response to the respective immunodominant TMEV CD8 epitope in SJL and B6 mice is similar. Thus, the decrease in the ability of SJL CNS-resident cells to efficiently activate CD8+ T cells may significantly contribute to the inability of SJL CD8+ T cells to completely clear the virus from the CNS, despite their ability to efficiently carry out anti-viral responses in vitro.
CD8+ T cells are largely protective in TMEV-IDD because of their importance in viral clearance, but there are scenarios in which CD8+ T cells can be pathologic as well. Resistant mice deficient in CD8+ T cells or the CD8+ T cell cytotoxic mediator perforin infected with the DA strain of TMEV develop demyelination, inflammation and persistent virus in the CNS, but display minimal clinical disease (Murray et al., 1998; Ure and Rodriguez, 2002). This has led to the hypothesis that CD8+ T cells may mediate axonal damage in TMEV-IDD and in MS, which is a major pathologic correlate of permanent neurologic disability (McDole et al., 2006; Trapp et al., 1998). Astrocytes reportedly express MHC I in MS lesions, and therefore could contribute to CD8+ T cell-mediated axonal damage by either direct or bystander mechanisms (Hoftberger et al., 2004; Traugott, 1987). Interestingly, SJL astrocytes exposed to the pro-inflammatory cytokines IFN-γ and TNF-α, as would be the case in T cell and macrophage rich MS lesions, are able to upregulate H-2Ks, and in this context MHC I expression by astrocytes may be pathologic. In conclusion, the contribution of the innate and adaptive immune functions of astrocytes therefore appears to be crucial to the balance of protective immunity vs. the development of autoimmune disease after CNS infection.
Materials and methods
Mice
Pregnant (15–17 days) SJL/J (SJL) mice and 5-to 7-week-old SJL mice were purchased from Harlan Labs (Bethesda, MD). Pregnant (11–15 days) and 5-to 7-week old C57BL/6 (B6) mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were housed in the Northwestern University animal facility in accordance with university and National Institutes of Health animal care guidelines and afforded access to food and water ad libitum. Neonatal mice 1 to 3 days old were used for the isolation of astrocytes.
Reagents
Poly I:C was purchased from Sigma (St. Louis, MO) and used at 100 μg/ml. Recombinant murine IFN-γ was purchased from Pharmingen (San Diego, CA) and used at 100 U/ml. Recombinant murine TNF-α was purchased from R&D Systems (Minneapolis, MN) and used at 500 U/ml. These doses selected were experimentally determined to elicit optimal responses (Carpentier et al., 2005). VP3159–166 (FNFTAPFI) and VP2121–130 (FHAGSLLVGM) were purchased from Genemed Synthesis Inc (South San Francisco, CA). The amino acid composition was verified by mass spectrometry and purity (>95%) was assessed by high-performance liquid chromatography.
Media
Mixed glial and astrocyte cultures were maintained in Dulbecco’s modified Eagle medium (DMEM)-F12 (Sigma) supplemented with 10% fetal calf serum (FCS; Sigma), 6 g/l glucose (Sigma), 2.4 g/l sodium bicarbonate (Sigma), 0.37 g/l L-glutamine (Life Technologies, Gaithersburg, MD), 100 U/ml penicillin (Life Technologies) and 100 μg/ml of streptomycin (Life Technologies; complete DMEM-F12 media). T cell assays were performed in DMEM with 5% FCS, 0.37 g/l L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 55 μM 2-Mercaptoethanol (Gibco, Grand Island, NY), and 0.1 mM nonessential amino acids (Sigma; complete D5 media).
Astrocyte Isolation
T75 tissue culture flasks were coated from 3 h to overnight with 10 μg/ml poly-D-lysine (PDL; Sigma). Intact brains were removed from 1-to 3-day-old neonatal mice, hindbrains dissected away, and meninges removed. The cerebral hemispheres were transferred to a nylon mesh bag and gently dissociated. Cells in suspension were passed through #60 and #100 stainless steel screens (Sigma) to remove large pieces of debris and tissue. Cells were seeded at 2–3 brains per flask, and maintained in complete DMEM-F12 media at 37°C and 7.5% CO2. At confluency (generally 14–21 days), microglia and oligodendrocytes were removed from the astroglial bed layer by orbital shaking for 24 h at 300 rpm. Astrocytes that adhered to the flask were removed with 1x trypsin-ethylenediamine-tetracetic acid (EDTA; 0.05 g/l trypsin, 0.02 g/l EDTA; Sigma) and re-plated until experimental use. Cultures prepared in this manner are ≥ 98% pure by staining for glial fibrillary acidic protein (GFAP), an astrocyte specific intermediate filament (Carpentier et al., 2005). We also routinely screen cultures for microglial contamination by flow cytometry for CD45, a marker of immune lineage cells, and typically find ≤ 1% of cells are CD45+ (data not shown).
Treatment/Infection of Primary Astrocytes
At time of infection, all media was removed from astrocyte cultures and replaced with a minimal volume of DMEM. Astrocytes were treated with media alone or stimulated with IFN-γ and TNF-α or poly I:C. TMEV was added at a multiplicity of infection of 10 (MOI; 10 plaque forming units/cell). All cultures were left at room temperature for 1 h to allow the virus to infect. After 1 h, complete F12 media was added to each culture and poly I:C or cytokine was accordingly added such that the total concentration remained constant.
Flow Cytometry
Cells were incubated with Accutase (BD Biosciences, San Jose, CA), removed from flasks, and washed in phosphate buffered saline (PBS) containing 0.5% bovine serum albumin (BSA; Sigma) and 2mM EDTA (Sigma). Intracellular staining with anti-TMEV was performed on cells fixed with Cytofix/Cytoperm solution containing formaldehyde and saponin for 20 minutes at 4°C (Pharmingen). Cells were washed twice in Perm/Wash Buffer containing saponin (Pharmingen) and nonspecific staining was blocked with anti-CD16/32 (eBioscience, San Diego, CA) and 10% normal goat serum (Sigma) for at least 20 minutes at 4°C. Cells were washed once and incubated with anti-TMEV serum generously donated by Dr. Howard Lipton (Northwestern University, Evanston, IL) for 30 minutes at 4°C. Cells were washed twice more and incubated with goat anti-rabbit IgG-FITC (Pharmingen) for 20 minutes at 4°C. Cells were washed at least twice with Perm/Wash buffer and twice with PBS/BSA/EDTA before flow cytometric analysis. For staining of cell surface antigens, cells were blocked with 10% mouse and 10% rat serum (Sigma) and 1:100 purified anti-mouse CD16/32 (eBioscience) in PBS/BSA/EDTA for at least 20 minutes at 4°C. Cells were then incubated with primary antibodies or appropriate isotype controls (eBioscience or Pharmingen) for at least 30 minutes at 4°C. After primary antibody incubation, astrocytes were washed three times with PBS/BSA/EDTA and incubated with secondary antibodies or streptavidin-APC (Pharmingen) as necessary for 20 minutes at 4°C. Cells were washed three more times with PBS/BSA/EDTA prior to analysis. Flow cytometry was performed on a BD Biosciences LSR flow cytometer using FACs Diva acquisition software. Data analysis was performed using Tree Star FlowJo software. Plots shown are gated on forward/side scatter and absence of CD45 staining, to avoid analyzing possible contaminating microglia.
Analysis of Cytotoxicity
Astrocytes were seeded at 7.5×104 cells/ml in PDL coated 48-well plates in complete DMEM-F12 media and allowed to adhere overnight. Cultures were mock infected or infected with TMEV (MOI=10). Supernatants were withdrawn at 24, 48 and 72 h and analyzed for lactate dehydrogenase (LDH) release as a measure of cell death using the Nonradioactive Cytotoxicity Assay kit (Promega) as per manufacturer’s instructions.
Enzyme-Linked Immunoassays (ELISAs)
Astrocytes were seeded at 7.5×104 cells/ml in PDL coated 48-well plates in complete DMEM-F12 media and allowed to adhere overnight. Cultures were mock infected or infected with TMEV (MOI=10) and supernatants were withdrawn at 72 h. IFN-α and IFN-γ ELISAs were performed as per kit instructions (R&D Systems, Minneapolis, MN).
Analysis of Astrocyte Cytokine and Chemokine Protein Expression
Astrocytes were seeded at 7.5×104 cells/ml in PDL coated 48-well plates in complete DMEM-F12 media and allowed to adhere overnight. Cultures were mock infected, treated with IFN-γ (100 U/ml) and TNF-α (500 U/ml), treated with poly I:C (100 μg/ml) or infected with TMEV (MOI=10) and supernatants were withdrawn at 72 h. Supernatants were analyzed for cytokine and chemokine expression by Invitrogen Biosource (Carlsbad, CA) Mouse Individual Flex Kit for IL-6, TNF-α, CCL2, CCL3, CCL4, CCL5, CXCL1, and CXCL10. Analysis was performed on a Luminex IS.100 system (Qiagen, Valencia, CA).
Infection of mice with TMEV
Mice were anesthetized with an inhaled mixture of isoflurane and oxygen and infected intracerebrally with 3×106 plaque forming units (PFU) of the BeAn strain of TMEV. Mice infected for 8 days were used for CD8+ T cell isolation.
CD8+ T Cell Activation Assay
SJL and B6 mice were infected with TMEV at day 0. At day 5, SJL or B6 astrocytes were plated at 1×105/well in a PDL coated 96 well plate. At day 6, astrocyte cultures were mock infected, treated with IFN-γ and TNF-α (100 and 500 U/ml) or infected with TMEV (MOI=10). At day 8, spleens were collected from naïve or TMEV-infected mice and homogenized over a #100 wire mesh screen. Red blood cells were lysed with Tris-ammonium chloride for 5 minutes at room temperature and washed in serum-containing media. Cells were blocked with 10% mouse serum and 10% rat serum in PBS/BSA/EDTA for at least 30 minutes at 4°C and then incubated with anti-CD8α microbeads (Miltenyi Biotec, Auburn, CA) for 15 minutes at 4°C. Positive cells were collected using the positive select double sort (posseld) program on an Automacs system (Miletnyi) and were approximately 90% pure on average as determined by flow cytometry (data not shown). Astrocyte cultures were washed twice with complete D5 media and incubated with 3×105 CD8+ T cells plus 0, 10 or 100 μM of VP3159–166 (SJL) or VP2121–130 (B6) in complete D5 media. At day 12, culture supernatants were harvested and analyzed for cytokine expression using the Upstate Mouse Multi-Cytokine 5plex Kit containing beads specific for IFN-γ, TNF-α, IL-2, IL-4 and IL-10 as per manufacturer’s instructions.
Enzyme-linked Immuno-SPOT (ELISPOT) assays
ELISPOT assays were carried out as previously described (Getts et al., 2007). Briefly, 5×105 bulk splenocytes were isolated at 7dpi and plated at 5×105 cells per well with 0 or 50 μg of the indicated peptide. The number of spots, representing IFN-γ producing cells, was quantified for each strain and peptide. All ELISPOT data are presented as mean number of spots per 5×105 splenocytes ± SEM.
Statistics
A two-way ANOVA with repeated measures and Bonferroni post tests was used to compare cytotoxicity over a 72 h time course and CD8+ T cell activation. Freeze-thaw positive controls were compared to uninfected cultures at 72 h using a one-tailed student’s T test. A paired one-tailed Student’s t test was used to compare means of percentage of infected cells, cytokine and chemokine production and adhesion molecule expression between SJL and B6 astrocytes. In all cases, p<0.05 is considered significant.
Acknowledgments
This work was supported in part by United States Public Health Service, NIH Grant NS-023349. P.A.C. was supported by an NIH Individual Predoctoral Research Fellowship (F31 NS-048807). The authors would like to thank D’Anne Duncan for her technical help.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ardavin C. Origin, precursors and differentiation of mouse dendritic cells. Nat Rev Immunol. 2003;3 (7):582–90. doi: 10.1038/nri1127. [DOI] [PubMed] [Google Scholar]
- Aubagnac S, Brahic M, Bureau JF. Bone marrow chimeras reveal non-H-2 hematopoietic control of susceptibility to Theiler’s virus persistent infection. J Virol. 2002;76 (11):5807–12. doi: 10.1128/JVI.76.11.5807-5812.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azoulay A, Brahic M, Bureau JF. FVB mice transgenic for the H-2Db gene become resistant to persistent infection by Theiler’s virus. J Virol. 1994;68 (6):4049–4052. doi: 10.1128/jvi.68.6.4049-4052.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azoulay-Cayla A, Dethlefs S, Perarnau B, Larsson-Sciard EL, Lemonnier FA, Brahic M, Bureau JF. H-2D(b-/-) mice are susceptible to persistent infection by Theiler’s virus. J Virol. 2000;74 (12):5470–5476. doi: 10.1128/jvi.74.12.5470-5476.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey SL, Carpentier PA, McMahon EJ, Begolka WS, Miller SD. Innate and adaptive immune responses of the central nervous system. Crit Rev Immunol. 2006;26 (2):149–88. doi: 10.1615/critrevimmunol.v26.i2.40. [DOI] [PubMed] [Google Scholar]
- Begolka WS, Haynes LM, Olson JK, Padilla J, Neville KL, Canto MD, Palma J, Kim BS, Miller SD. CD8-deficient SJL mice display enhanced susceptibility to Theiler’s virus infection and increased demyelinating pathology. J Neurovirol. 2001;7 (5):409–420. doi: 10.1080/135502801753170264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow P, Tonks P, Welsh CJR, Nash AA. The role of CD8 + T cells in the acute and chronic phases of Theiler’s murine encephalomyelitis virus-induced disease in mice. J Gen Virol. 1992;73:1861–1865. doi: 10.1099/0022-1317-73-7-1861. [DOI] [PubMed] [Google Scholar]
- Brahic M, Bureau JF, Michiels T. The genetics of the persistent infection and demyelinating disease caused by Theiler’s virus. Annu Rev Microbiol. 2005;59:279–98. doi: 10.1146/annurev.micro.59.030804.121242. [DOI] [PubMed] [Google Scholar]
- Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia. 2005;49 (3):360–374. doi: 10.1002/glia.20117. [DOI] [PubMed] [Google Scholar]
- Carpentier PA, Williams BR, Miller SD. Distinct roles of protein kinase R and toll-like receptor 3 in the activation of astrocytes by viral stimuli. Glia. 2007;55 (3):239–252. doi: 10.1002/glia.20450. [DOI] [PubMed] [Google Scholar]
- Chamorro M, Aubert C, Brahic M. Demyelinating lesions due to Theiler’s virus are associated with ongoing central nervous system infection. J Virol. 1986;57:992–997. doi: 10.1128/jvi.57.3.992-997.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BP, Kuziel WA, Lane TE. Lack of CCR2 results in increased mortality and impaired leukocyte activation and trafficking following infection of the central nervous system with a neurotropic coronavirus. J Immunol. 2001;167 (8):4585–4592. doi: 10.4049/jimmunol.167.8.4585. [DOI] [PubMed] [Google Scholar]
- Clatch RJ, Melvold RW, Miller SD, Lipton HL. Theiler’s murine encephalomyelitis virus (TMEV)-induced demyelinating disease in mice is influenced by the H-2D region: correlation with TMEV-specific delayed-type hypersensitivity. J Immunol. 1985;135:1408–1414. [PubMed] [Google Scholar]
- Dal Canto MC, Lipton HL. Primary demyelination in Theiler’s virus infection. An ultrastructural study Lab Invest. 1975;33:626–637. [PubMed] [Google Scholar]
- de Vries HE, Blom-Roosemalen MC, van Oosten M, de Boer AG, van Berkel TJ, Breimer DD, Kuiper J. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J Neuroimmunol. 1996;64 (1):37–43. doi: 10.1016/0165-5728(95)00148-4. [DOI] [PubMed] [Google Scholar]
- Fiette L, Aubert C, Brahic M, Pena Rossi C. Theiler’s virus infection of beta2-microglobulin-deficient mice. J Virol. 1993;67:589–592. doi: 10.1128/jvi.67.1.589-592.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana A, Erb P, Pircher H, Zinkernagel RM, Weber E, Fierz W. Astrocytes as antigen-presenting cells. Part II : Unlike H-2K-dependent cytotoxic T cells, H-2Ia-restricted T cells are only stimulated in the presence of interferon-gamma. J Neuroimmunol. 1986;12:15–28. doi: 10.1016/0165-5728(86)90093-7. [DOI] [PubMed] [Google Scholar]
- Getts MT, Kim BS, Miller SD. Differential outcome of tolerance induction in naive versus activated Theiler’s virus epitope-specific CD8+ cytotoxic T cells. J Virol. 2007;81 (12):6584–93. doi: 10.1128/JVI.00008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez MA, Sim JE, Russell JH. TNFR1-dependent VCAM-1 expression by astrocytes exposes the CNS to destructive inflammation. J Neuroimmunol. 2004;151 (1–2):116–125. doi: 10.1016/j.jneuroim.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Giunti D, Borsellino G, Benelli R, Marchese M, Capello E, Valle MT, Pedemonte E, Noonan D, Albini A, Bernardi G, Mancardi GL, Battistini L, Uccelli A. Phenotypic and functional analysis of T cells homing into the CSF of subjects with inflammatory diseases of the CNS. J Leukoc Biol. 2003;73 (5):584–90. doi: 10.1189/jlb.1202598. [DOI] [PubMed] [Google Scholar]
- Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J Exp Med. 2005;202 (8):1087–98. doi: 10.1084/jem.20042530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafler DA. Multiple sclerosis. J Clin Invest. 2004;113 (6):788–794. doi: 10.1172/JCI21357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LM, Fige BT, Begolka WS, Miller SD, Karpus WJ. Central nervous system chemokine expression during Theiler’s virus-induced demyelinating disease. J Neurovirol. 1999;5 (6):635–642. doi: 10.3109/13550289909021292. [DOI] [PubMed] [Google Scholar]
- Hoftberger R, Aboul-Enein F, Brueck W, Lucchinetti C, Rodriguez M, Schmidbauer M, Jellinger K, Lassmann H. Expression of major histocompatibility complex class I molecules on the different cell types in multiple sclerosis lesions. Brain Pathol. 2004;14 (1):43–50. doi: 10.1111/j.1750-3639.2004.tb00496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang BS, Lyman MA, Kim BS. The majority of infiltrating CD8+ T cells in the central nervous system of susceptible SJL/J mice infected with Theiler’s virus are virus specific and fully functional. J Virol. 2002;76 (13):6577–6585. doi: 10.1128/JVI.76.13.6577-6585.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivisakk P, Trebst C, Liu Z, Tucky BH, Sorensen TL, Rudick RA, Mack M, Ransohoff RM. T-cells in the cerebrospinal fluid express a similar repertoire of inflammatory chemokine receptors in the absence or presence of CNS inflammation: implications for CNS trafficking. Clin Exp Immunol. 2002;129 (3):510–8. doi: 10.1046/j.1365-2249.2002.01947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane TE, Liu MT, Chen BP, Asensio VC, Samawi RM, Paoletti AD, Campbell IL, Kunkel SL, Fox HS, Buchmeier MJ. A central role for CD4(+) T cells and RANTES in virus-induced central nervous system inflammation and demyelination. J Virol. 2000;74 (3):1415–1424. doi: 10.1128/jvi.74.3.1415-1424.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavi E, Suzumura A, Murasko DM, Murray EM, Silberberg DH, Weiss SR. Tumor necrosis factor induces expression of MHC class I antigens on mouse astrocytes. J Neuroimmunol. 1988;18 (3):245–53. doi: 10.1016/0165-5728(88)90102-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley MD, Thiemann R, Rodriguez M. Cytotoxic T cells isolated from the central nervous systems of mice infected with Theiler’s virus. J Virol. 1991;65:6612–6620. doi: 10.1128/jvi.65.12.6612-6620.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton HL. Theiler’s virus infection in mice: An unusual biphasic disease process leading to demyelination. Infect Immun. 1975;11:1147–1155. doi: 10.1128/iai.11.5.1147-1155.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton HL, Melvold R. Genetic analysis of susceptibility to Theiler’s virus-induced demyelinating disease in mice. J Immunol. 1984;132:1821–1825. [PubMed] [Google Scholar]
- Lipton HL, Melvold R, Miller SD, Dal Canto MC, Jensen K. Mutation of a major histocompatibility class I locus, H-2D, leads to an increased virus burden and disease susceptibility in Theiler’s virus-induced demyelinating disease. J Neurovirol. 1995a;1:138–144. doi: 10.3109/13550289509113960. [DOI] [PubMed] [Google Scholar]
- Lipton HL, Twaddle G, Jelachich ML. The predominant virus antigen burden is present in macrophages in Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J Virol. 1995b;69 (4):2525–2533. doi: 10.1128/jvi.69.4.2525-2533.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MT, Chen BP, Oertel P, Buchmeier MJ, Armstrong D, Hamilton TA, Lane TE. The T cell chemoattractant IFN-inducible protein 10 is essential in host defense against viral-induced neurologic disease. J Immunol. 2000;165 (5):2327–30. doi: 10.4049/jimmunol.165.5.2327. [DOI] [PubMed] [Google Scholar]
- Liu Y, King N, Kesson A, Blanden RV, Mullbacher A. Flavivirus infection up-regulates the expression of class I and class II major histocompatibility antigens on and enhances T cell recognition of astrocytes in vitro. J Neuroimmunol. 1989;21 (2–3):157–68. doi: 10.1016/0165-5728(89)90171-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman MA, Lee HG, Kang BS, Kang HK, Kim BS. Capsid-specific cytotoxic T lymphocytes recognize three distinct H-2D(b)-restricted regions of the BeAn strain of Theiler’s virus and exhibit different cytokine profiles. J Virol. 2002;76 (7):3125–3134. doi: 10.1128/JVI.76.7.3125-3134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman MA, Myoung J, Mohindru M, Kim BS. Quantitative, not qualitative, differences in CD8(+) T cell responses to Theiler’s murine encephalomyelitis virus between resistant C57BL/6 and susceptible SJL/J mice. Eur J Immunol. 2004;34 (10):2730–2739. doi: 10.1002/eji.200324811. [DOI] [PubMed] [Google Scholar]
- Massa PT, Cowan EP, Levi BZ, Ozato K, McFarlin DE. Genetic regulation of class I major histocompatibility complex (MHC) antigen induction on astrocytes. J Neuroimmunol. 1989;24 (1–2):125–32. doi: 10.1016/0165-5728(89)90106-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDole J, Johnson AJ, Pirko I. The role of CD8+ T-cells in lesion formation and axonal dysfunction in multiple sclerosis. Neurol Res. 2006;28 (3):256–61. doi: 10.1179/016164106X98125. [DOI] [PubMed] [Google Scholar]
- Murray PD, McGavern DB, Lin X, Njenga MK, Leibowitz J, Pease LR, Rodriguez M. Perforin-dependent neurologic injury in a viral model of multiple sclerosis. J Neurosci. 1998;18 (18):7306–14. doi: 10.1523/JNEUROSCI.18-18-07306.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nansen A, Marker O, Bartholdy C, Thomsen AR. CCR2+ and CCR5+ CD8+ T cells increase during viral infection and migrate to sites of infection. Eur J Immunol. 2000;30 (7):1797–806. doi: 10.1002/1521-4141(200007)30:7<1797::AID-IMMU1797>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Nuovo GJ, Alfieri ML. AIDS dementia is associated with massive, activated HIV-1 infection and concomitant expression of several cytokines. Mol Med. 1996;2 (3):358–66. [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. 2003;3 (7):569–581. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- Rodriguez M, David CS. Demyelination induced by Theiler’s virus: influence of the H-2 haplotype. J Immunol. 1985;135:2145–2148. [PubMed] [Google Scholar]
- Schnitzer J, Schachner M. Expression of Thy-1, H-2, and NS-4 cell surface antigens and tetanus toxin receptors in early postnatal and adult mouse cerebellum. J Neuroimmunol. 1981;1 (4):429–56. doi: 10.1016/0165-5728(81)90022-9. [DOI] [PubMed] [Google Scholar]
- Shacklett BL, Cox CA, Wilkens DT, Karl Karlsson R, Nilsson A, Nixon DF, Price RW. Increased adhesion molecule and chemokine receptor expression on CD8+ T cells trafficking to cerebrospinal fluid in HIV-1 infection. J Infect Dis. 2004;189 (12):2202–12. doi: 10.1086/421244. [DOI] [PubMed] [Google Scholar]
- Skias DD, Kim DK, Reder AT, Antel JP, Lancki DW, Fitch FW. Susceptibility of astrocytes to class I MHC antigen-specific cytotoxicity. J Immunol. 1987;138:3254–3258. [PubMed] [Google Scholar]
- Sorensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA, Qin S, Rottman J, Sellebjerg F, Strieter RM, Frederiksen JL, Ransohoff RM. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest. 1999;103 (6):807–815. doi: 10.1172/JCI5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzumura A, Lavi E, Weiss SR, Silberberg DH. Coronavirus infection induces H-2 antigen expression on oligodendrocytes and astrocytes. Science. 1986;232 (4753):991–993. doi: 10.1126/science.3010460. [DOI] [PubMed] [Google Scholar]
- Thomsen AR, Nansen A, Madsen AN, Bartholdy C, Christensen JP. Regulation of T cell migration during viral infection: role of adhesion molecules and chemokines. Immunol Lett. 2003;85 (2):119–27. doi: 10.1016/s0165-2478(02)00236-5. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338 (5):278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Traugott U. Multiple sclerosis: relevance of class I and class II MHC-expressing cells to lesion development. J Neuroimmunol. 1987;16:283–302. doi: 10.1016/0165-5728(87)90082-8. [DOI] [PubMed] [Google Scholar]
- Trifilo MJ, Bergmann CC, Kuziel WA, Lane TE. CC chemokine ligand 3 (CCL3) regulates CD8(+)-T-cell effector function and migration following viral infection. J Virol. 2003;77 (7):4004–14. doi: 10.1128/JVI.77.7.4004-4014.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ure DR, Lane TE, Liu MT, Rodriguez M. Neutralization of chemokines RANTES and MIG increases virus antigen expression and spinal cord pathology during Theiler’s virus infection. Int Immunol. 2005;17 (5):569–79. doi: 10.1093/intimm/dxh236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ure DR, Rodriguez M. Preservation of neurologic function during inflammatory demyelination correlates with axon sparing in a mouse model of multiple sclerosis. Neuroscience. 2002;111 (2):399–411. doi: 10.1016/s0306-4522(02)00012-x. [DOI] [PubMed] [Google Scholar]
- Wong GHW, Bartlett PF, Clark-Lewis I, Battye F, Schrader JW. Inducible expression of H-2 and Ia antigens on brain cells. Nature. 1984;310:688–691. doi: 10.1038/310688a0. [DOI] [PubMed] [Google Scholar]
- Zheng L, Calenoff MA, Dal Canto MC. Astrocytes, not microglia, are the main cells responsible for viral persistence in Theiler’s murine encephalomyelitis virus infection leading to demyelination. J Neuroimmunol. 2001;118 (2):256–267. doi: 10.1016/s0165-5728(01)00338-1. [DOI] [PubMed] [Google Scholar]